

SUMMARY
The human microbiota greatly affects physiology and disease. However, the contribution of bacteria to the response to chemotherapeutic drugs remains poorly understood. Caenorhabditis elegans and its bacterial diet provide a powerful system to study host-bacteria interactions. Here, we use this system to study how bacteria affect the C. elegans response to chemotherapeutics. We find that different bacterial species can increase the response to one drug yet decrease the effect of another. We perform genetic screens in two bacterial species using three chemotherapeutic drugs, 5-fluorouracil (5-FU), 5-fluoro-2′-deoxyuridine (FUDR) and camptothecin (CPT). We find numerous bacterial nucleotide metabolism genes that affect drug efficacy in C. elegans. Surprisingly, we find that 5-FU and FUDR act through bacterial ribonucleotide metabolism to elicit their cytotoxic effects in C. elegans, rather than by thymineless death or DNA damage. Our study provides a blueprint for characterizing the role of bacteria in the host response to chemotherapeutics.
INTRODUCTION
The microorganisms that inhabit our intestine, known as our gut microbiota, greatly influence our development, health and propensity to get sick (Brennan and Garrett, 2016; Sommer and Backhed, 2013). More recently, it has become clear that the gut microbiota can also affect the response to anticancer immunotherapy (Sivan et al., 2015; Vetizou et al., 2015; Viaud et al., 2013). However, the involvement of the microbiota in the response to chemotherapeutics and the mechanisms involved remain to be elucidated.
Chemotherapeutics have been used for decades to treat a variety of human tumors. Some cancers, such as colorectal cancer, occur in physical proximity to the microbiota. Therefore, we reasoned that the efficacy of anti-colorectal cancer drugs, such as the anti-pyrimidine drugs 5-fluorouracil (5-FU) and 5-fluoro-2′-deoxyuridine (FUDR), and the topoisomerase I (topo-I) inhibitor camptothecin (CPT) may be affected by the microbiota. These drugs have different reported mechanisms of action, but are all thought to confer cytotoxicity through DNA damage (Figure S1). CPT directly inhibits the topo-I-DNA complex, which leads to single-stranded DNA breaks and cell death (Pommier, 2006), while 5-FU and FUDR produce FdUMP, which directly inhibits thymidylate synthase thereby causing thymineless cell death. In addition, products of 5-FU and FUDR may be converted into FUTP or FdUTP, leading to RNA or DNA damage, respectively (Longley et al., 2003).
It is extremely challenging to systematically delineate which anticancer drug efficacies are affected by the microbiota using patient data or mammalian model systems, such as mice, and to dissect the mechanisms involved. This difficulty is not only because of limitations in scale and cost, but also because of heterogeneity in human genotype, diet, and microbiota.
The nematode C. elegans and its bacterial diet have recently become a powerful interspecies model system to study the effects of diet and the microbiota on animal life history traits (Watson and Walhout, 2014; Watson et al., 2015; Yilmaz and Walhout, 2014). C. elegans has a short life cycle, is amenable to high-throughput screens, and is easily maintained in the laboratory. In the wild, C. elegans has been found associated with different bacteria, which function not only as diet but also as a microbiota (Berg et al., 2016; Dirksen et al., 2016; Felix and Duveau, 2013; Felix and Braendle, 2010). We, and others, have proposed that C. elegans and its bacterial diet provide an apt model system to study the effects of bacteria on the animal’s physiology and gene expression (Cabreiro et al., 2013; Gusarov et al., 2013; MacNeil et al., 2013; Watson et al., 2013; Watson et al., 2014). For instance, we previously found that, when fed a diet of Comamonas aquatica DA1877 (Comamonas), C. elegans develop faster, have reduced fecundity and live shorter than animals fed the standard laboratory diet of Escherichia coli OP50 (E. coli)(MacNeil et al., 2013). By high-throughput forward and reverse genetics, in both the animal and these bacteria, we found that the Comamonas effects on C. elegans development and fecundity can be explained, in a large part, by the fact that this bacterial species can synthesize vitamin B12, whereas E. coli cannot (Watson et al., 2013; Watson et al., 2014; Watson et al., 2016).
Here, we used the interspecies system of C. elegans and its bacterial diet to ask whether different bacteria have different effects on the host response to chemotherapeutics. We exposed C. elegans to 11 drugs while feeding them either E. coli or Comamonas and found that four chemotherapeutics cause abnormal phenotypes in C. elegans at the administered dose. We tested the effects of three drugs, 5-FU, FUDR, and CPT, at a range of concentrations and used progeny production — no progeny (sterile), dead progeny (dead embryos), or live progeny — as a proxy for drug efficacy. We found that E. coli and Comamonas oppositely affect the C. elegans response to FUDR and CPT. By performing genetic screens in both bacterial species, we identified numerous bacterial genes that modulate 5-FU, FUDR, and/or CPT efficacy in C. elegans. Altogether, we found that bacterial nucleotide metabolism networks modulate drug efficacy in C. elegans. Mutations in E. coli or Comamonas upp, which generates 5-fluorouridine monophosphate (FUMP) from 5-FU, reduce the efficacy of 5-FU and FUDR. However, mutations in bacterial tdk, tmk, or thyA, which are involved in dTMP synthesis, do not. These observations suggest that these drugs elicit their effects on C. elegans fecundity through bacterial production of FUMP, and RNA metabolism, and not through the generation of FdUMP, DNA metabolism or thymineless death. Indeed, uracil rescues 5-FU and FUDR toxicity in C. elegans while thymine had no effect. Altogether, these results demonstrate that bacterial metabolism can greatly affect chemotherapeutic drug efficacy in the host.
RESULTS
The Effects of Diverse Chemotherapeutic Drugs on C. elegans Fed Different Bacteria
We selected drugs from major classes of chemotherapeutics: alkylating agents (temozolomide), antimetabolites (5-FU; FUDR; methotrexate), anti-tumor antibiotics (doxorubicin; neocarzinostatin), a mitotic inhibitor (paclitaxel), topoisomerase inhibitors (CPT; etoposide), a hormone receptor antagonist (tamoxifen), and a receptor tyrosine kinase small-molecule inhibitor (sunitinib). For bacterial diets, we used E. coli and Comamonas, because large mutant collections are available for each species, enabling genetic screens to identify bacterial genes that may modulate drug efficacy in the nematode (Baba et al., 2006; Watson et al., 2014). We tested each drug at a single dose of 10 μM in animals fed each bacteria. Briefly, bacteria were seeded onto solid drug-containing media and incubated overnight. The next morning, 5–10 L4 larvae were transferred to drug or control plates, and phenotypic differences between drug-exposed and control animals were assayed after 72 hours of growth at 20°C.
Four of the 11 drugs tested caused abnormal phenotypes in C. elegans. CPT, 5-FU, and FUDR all result in impaired fecundity, from sterility (5-FU) to the production of few dead embryos (CPT) (Figure 1A). Paclitaxel did not affect fecundity but rather resulted in a developmental delay (Figure 1A). Interestingly, the C. elegans response to FUDR was different between animals fed the two bacteria: on E. coli the animals were completely sterile, while on Comamonas they produced live progeny (Figure 1A). This observation indicates that bacteria can affect chemotherapeutic drug efficacy in C. elegans.
Figure 1. Screen of Cancer Chemotherapeutics in C. elegans Fed Two Different Bacteria.
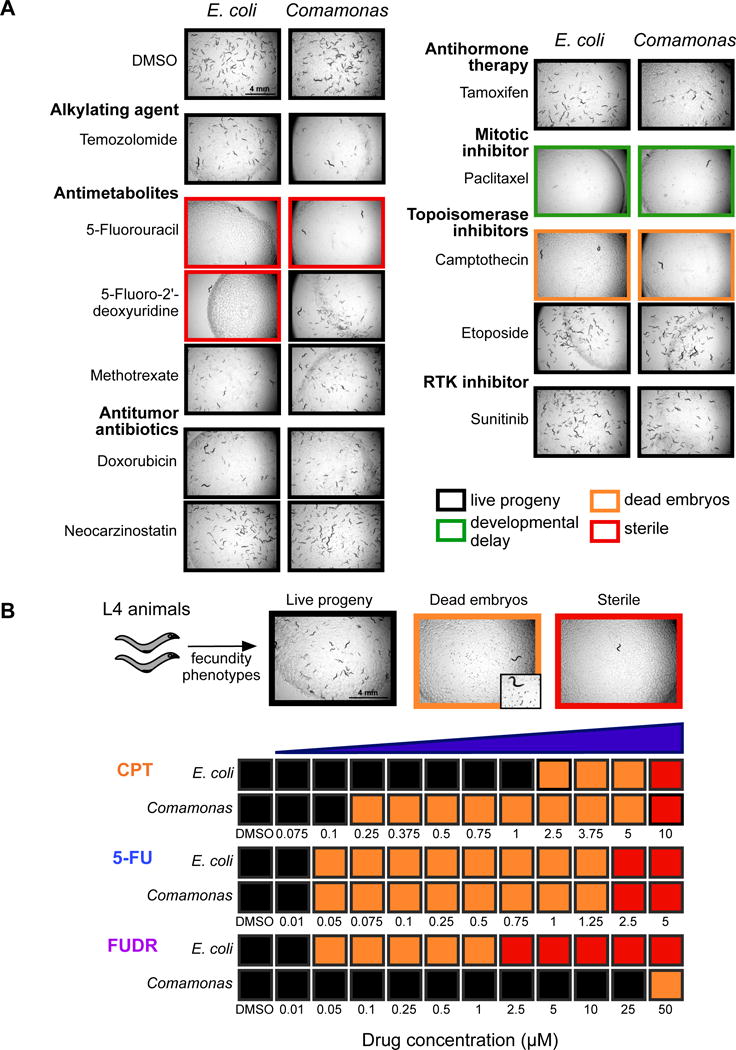
(A) Fecundity and developmental phenotypes in C. elegans in response to chemotherapeutic drugs when fed either E. coli OP50 or Comamonas. All drugs were tested at a concentration of 10 μM. Brightfield images were taken at 2× magnification. RTK: receptor tyrosine kinase.
(B) Chemotherapeutic drug titrations in C. elegans L4 animals fed either E. coli OP50 or Comamonas. Colors correspond to phenotypes observed after 72 hours in three independent experiments, as indicated in (A). Representative images of fecundity phenotypes in C. elegans are shown. All images were taken at 2× or 4× (inset) total magnification. See also Figure S1.
E. coli and Comamonas Oppositely Modulate Drug Efficacy of CPT and FUDR
Next, we titrated the three drugs that impaired C. elegans fecundity- CPT, 5-FU and FUDR- to determine if, and to what extent, animals display a difference in the response to these drugs on either bacterial diet. We compared fecundity of L4 animals fed each bacteria and drug combination after 72 hours and scored three traits: viable progeny, dead embryos, and sterility (Figure 1B). For 5-FU, we did not observe any differences in drug efficacy between animals fed E. coli OP50 or Comamonas (Figure 1B). In agreement with the initial screen (Figure 1A), animals fed Comamonas fared much better on FUDR than animals fed E. coli OP50. On E. coli, animals laid dead embryos when grown on 50 nM FUDR and were sterile when grown on 2.5 μM FUDR. By contrast, dead embryos were laid by animals grown on the highest concentration of FUDR tested (50 μM) when fed Comamonas (Figure 1B). Therefore, we observed a large difference in FUDR efficacy in animals fed E. coli OP50 versus those fed Comamonas. Remarkably, we observed an opposite, albeit milder, difference in the C. elegans response to CPT: dead embryos were laid by animals exposed to 2.5 μM CPT on the E. coli OP50 diet and by animals exposed to 250 nM on Comamonas. Taken together, by testing two bacteria and three chemotherapeutic drugs, we observed that the same bacteria can lead to increased efficacy of one drug but decreased efficacy of a different drug.
Active Bacterial Metabolism is Required to Modify FUDR Efficacy in C. elegans
An important question is whether differential drug efficacy is due to one bacterial species increasing it, the other one reducing it, or both. To start addressing this question, we mixed the two bacterial species in different ratios and compared the drug response in C. elegans. We found that the increase in drug efficacy elicited by Comamonas for CPT and by E. coli OP50 for FUDR is ‘dominant’, because even when either species was greatly diluted into the other, we observed enhanced drug efficacy in C. elegans (Figure 2A). This result suggests that E. coli OP50 actively increases drug efficacy of FUDR, while Comamonas increases drug efficacy of CPT.
Figure 2. Active Bacterial Metabolism is Required to Modulate FUDR Efficacy in C. elegans.
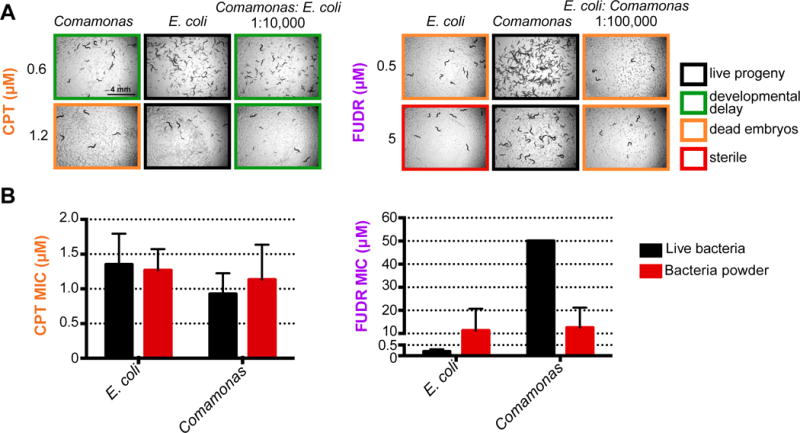
(A) Bacteria mixing experiment. Brightfield images at 2× magnification (right).
(B) Graph of MIC = minimum inhibitory concentration, or the lowest concentration tested at which complete embryonic lethality was observed 96 hours after L4 animals were added to drug plates. The mean and standard deviation of three independent experiments are shown.
There are two possible mechanisms by which bacteria can increase drug efficacy. The first is a passive mechanism by which bacteria supplement metabolites that, when ingested by the host, increase drug efficacy. The second is an active mechanism in which bacteria metabolize the drug, or convert a metabolite in response to drug exposure, thereby affecting drug efficacy in the animal. We tested whether we could discriminate between these possibilities by comparing drug efficacy between C. elegans fed live bacteria (active metabolism) and dead bacteria (no active metabolism). We reasoned that the traditional method of UV-killing of bacteria may not be most suitable because, even though bacteria cannot proliferate after high doses of UV they may remain metabolically active, and because UV causes DNA damage, which is related to the reported mechanism of action of these drugs (Figure S1). Instead, we generated bacterial powder from each species by disrupting the cells at high-pressure followed by lyophilization. To quantify drug efficacy in C. elegans fed live bacteria versus bacterial powder, we adopted the term minimum inhibitory concentration (MIC), which is commonly used to describe antimicrobials (Andrews, 2001). Here, MIC refers to the minimum concentration of the chemotherapeutic drug at which we observed no live progeny. Instead of examining phenotypes after 72 hours as described above, we did so after 96 hours of placing L4 animals on each bacteria and drug combination to avoid differences caused by potential egg-hatching delays. For CPT, we did not observe a significant difference between animals fed live bacteria versus those fed bacterial powder (Figure 2B, left). This result indicates that bacteria mostly modulate CPT efficacy through a passive mechanism.
In contrast, the bacterial influence on drug efficacy of FUDR is completely dependent on active bacterial metabolism. First, we found no difference in drug efficacy between C. elegans fed the two bacterial species when fed as metabolically inactive powder (Figure 2B, right). Second, we observed both a decrease in drug efficacy (higher MIC) with E. coli OP50 powder and an increase in drug efficacy with Comamonas powder (lower MIC), relative to either live bacteria. From this finding, we conclude that at least two mechanisms are involved: E. coli metabolism increases FUDR efficacy in C. elegans, while Comamonas metabolism reduces it. The latter effect indicates that bacterial metabolism not only increases drug efficacy but that it can also be protective.
Genetic Screens with Two Bacterial Species and Three Drugs
In order to gain insight into the mechanisms by which bacteria modulate the C. elegans response to chemotherapeutics, we performed genetic screens in both E. coli and Comamonas, and using all three drugs: CPT, 5-FU and FUDR. First, we used the E. coli Keio collection that comprises 3,985 strains, each with a deletion in a single, non-essential gene (Baba et al., 2006). We found that the parental strain of this collection, E. coli K-12 BW25113, modulates drug efficacy in a similar manner as E. coli OP50, although there was a relatively small difference between animals fed E. coli OP50 and E. coli K-12 BW25113 with 5-FU (Figure S2). Second, we used a collection of 5,760 Comamonas mutants we generated previously (Watson et al., 2014). Each of these mutant strains harbors a single transposon insertion into its genome.
For each of the six genetic screens, we used a single dose of drug where animals fed the parental strain produced dead embryos. This dose allowed us to simultaneously screen for bacterial mutants that increased drug efficacy (when we observe sterility) or decreased drug efficacy (when we observe live progeny) (Figure 3A, top). In each of the primary screens, between two and 20 percent of the bacterial mutants tested scored positively, either increasing or decreasing drug efficacy in C. elegans. These mutants were retested in a secondary screen with three doses of the drug (Figure 3A, middle), and only those mutants that scored in at least two of the three doses were tested in a tertiary screen (triplicate and with six concentrations of each drug, Figure 3A, bottom). This process resulted in a final set of mutants that range between a single Comamonas mutant that increases FUDR efficacy to 16 E. coli mutants that reduce the efficacy of CPT (Figure 3A, bottom). The identity of the disrupted genes in the Comamonas mutants was determined by sequencing (Watson et al., 2014). The E. coli gene deletions were identified according to their coordinate in the collection and all were confirmed by sequencing.
Figure 3. Genetic Screens Identify Bacterial Genes that Affect Drug Efficacy in C. elegans.
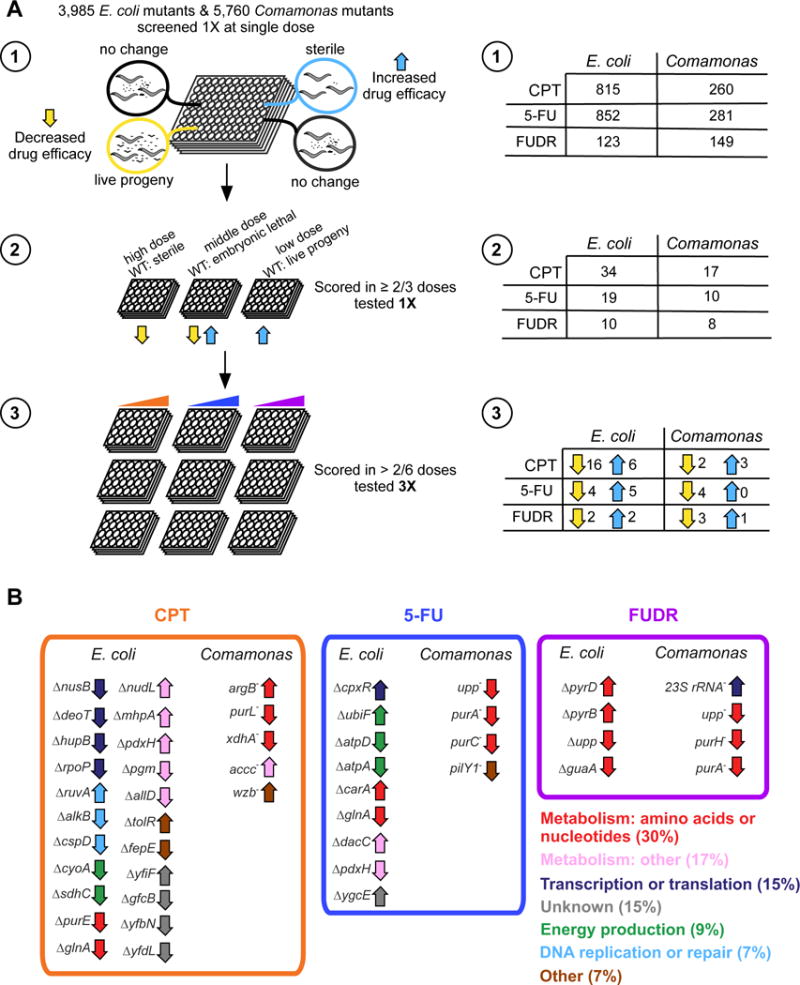
(A) (Left) Schematic of genetic screens. (Right) Number of mutants, for each bacteria and drug combination, identified in each round of screening, as indicated. See also Figure S2 and Table S1.
(B) Unique bacterial mutants identified in each genetic screen. Up/downward arrows correspond to in/decreased drug efficacy in animals fed the mutant bacteria. Arrows are colored based on listed functional categories.
Several observations support the high quality of the different screens. First, we found bacterial mutants for each drug/bacteria combination and, for each combination, except for Comamonas and 5-FU, we identified strains that increased or decreased drug efficacy in C. elegans. Second, two genes were each found twice in the Comamonas screens, including two independent mutations in upp found in the 5-FU and FUDR screens, and three independent mutations in purH, found in the FUDR screen (Table S1). Third, a mutant in Comamonas purA was found in both the 5-FU and FUDR screens, while E. coli mutations in glnA were found in both the CPT and 5-FU screens. Strikingly, E. coli mutations in pdxH were also found with both CPT and 5-FU, but a deletion in this gene increased efficacy of CPT and decreased efficacy of 5-FU (Figure 3B). Finally, one gene was found with both bacterial species and with multiple drugs: mutations in upp decreased the efficacy of 5-FU in Comamonas and FUDR in both bacteria (Figure 3B). Altogether, we identified 44 unique bacterial genes that can be grouped into several broad functional categories, including different types of metabolism, most notably amino acid and nucleotide metabolism (Figure 3B).
Different Classes of Bacterial Mutants
Due to the stringency of the six bacterial genetic screens, it is unlikely that we uncovered all genes that contribute to the C. elegans response to each of the three drugs, i.e., we expect a considerable rate of false negatives. Therefore, we next performed a matrix experiment in which we tested animals fed each of the mutants that scored positively in the second retest (n = 98, Figure 3A) against six concentrations of each of the three drugs in quadruplicate (Figure S3). We reasoned that this experiment would enable us to define different classes of genes, including those genes that specifically affect the response to a single drug, and other genes that may elicit a more general change in drug response. Indeed, this experiment uncovered additional gene/drug interactions with the same set of mutants. In total, we identified 98 interactions involving 44 bacterial mutants, including 48 interactions that were observed previously (Figure 3A) and 50 additional interactions.
Bacterial mutants can be grouped into four broad classes according to their effect on drug efficacy (Figure 4A). Class I mutants (13 genes) have drug-specific effects on chemotherapeutic efficacy. Most of these (n = 11) specifically affect the response to CPT, which may not be surprising given that 5-FU and FUDR are thought to function through the same pathway (Figure S1) (Longley et al., 2003). Of these 11 genes, eight increase CPT efficacy and three decrease it. Class II mutants involve eight genes that affect the response to two of the three drugs. With the exception of one (purC), these genes affect the response to both anti-pyrimidine drugs: five increase their efficacy while two reduce it. Class III involves six genes that decreased the efficacy of all three drugs. Finally, class IV genes (n = 17) affect all three drugs but in an opposite way between the anti-pyrimidine drugs, 5-FU and FUDR, and the topo-I inhibitor CPT. Most of these mutants increase the efficacy of the anti-pyrimidine drugs and decrease the C. elegans response to CPT.
Figure 4. Four Classes of Bacterial Mutants Affect Drug Efficacy in C. elegans.
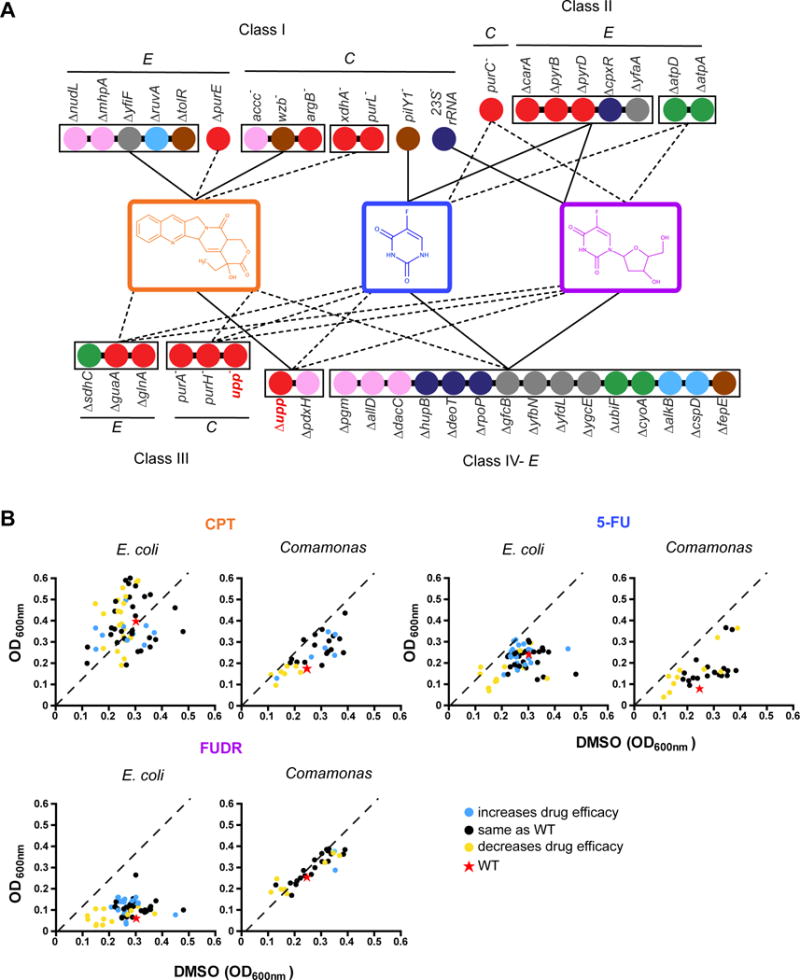
(A) Network diagram for bacterial mutant drug interactions identified in matrix experiment, in which each mutant was used with each drug. Each node indicates a bacterial mutant that affects drug efficacy in C. elegans. Nodes are colored based on functional categories listed in Figure 3B. Bacterial mutants are connected when they share the same effect on drug efficacy; edges are as follows: solid lines indicate increased drug efficacy and dashed lines indicate decreased drug efficacy. Bacterial mutant-drug interactions were grouped into four classes: class I- mutants that affect drug efficacy of only one drug; class II- mutants that increase OR decrease drug efficacy of two drugs; class III- mutants that increase OR decrease drug efficacy of all three drugs; class IV- mutants that increase drug efficacy of one or more drugs AND decrease efficacy of other drugs. E = E. coli; C = Comamonas. See also Figure S3.
(B) Bacterial growth assays in the presence or absence of drug for all 98 bacterial mutants in round 2 of screening, as indicated in Figure 3A. All drugs were tested at a concentration of 50 μM. Each value represents OD600nm after eight hours of incubation with DMSO (x-axis) or drug (y-axis). Data points are colored according to host drug phenotype.
One explanation for the effects of bacterial mutants on drug efficacy in C. elegans could be that the growth of the microbes is affected by the mutation and that this in turn affects the response in the animal. To test this, we compared growth of all 98 bacterial mutants that passed the second round of screening (Figure 3A) in the presence or absence of drug. Overall, we did not find a strong relationship between bacterial growth in the presence of drug and an increase in drug efficacy in C. elegans. We did observe that several mutants that decrease drug efficacy are slow growing, but that this is generally not affected by the presence of the drug (Figure 4B).
Bacterial Nucleotide Metabolism Affects Chemotherapeutic Drug Efficacy
The set of bacterial mutants that we identified contains numerous genes involved in nucleotide metabolism (Figure 5A). Given the considerable level of false negatives in our screens, we selected all available E. coli mutants representing the genes in these pathways (e.g., including those mutants that were not recovered in the primary screen) and systematically tested them in triplicate as described for the matrix experiment (Figure 4A). We tested two independent deletion copies for each gene from the E. coli mutant collection (Baba et al., 2006). Altogether, we tested 37 duplicate mutant strains. The interactions are summarized in Figure 5A, and include 42 that were not observed before.
Figure 5. Bacterial Nucleotide Metabolism Affects Chemotherapeutic Drug Efficacy in C. elegans.
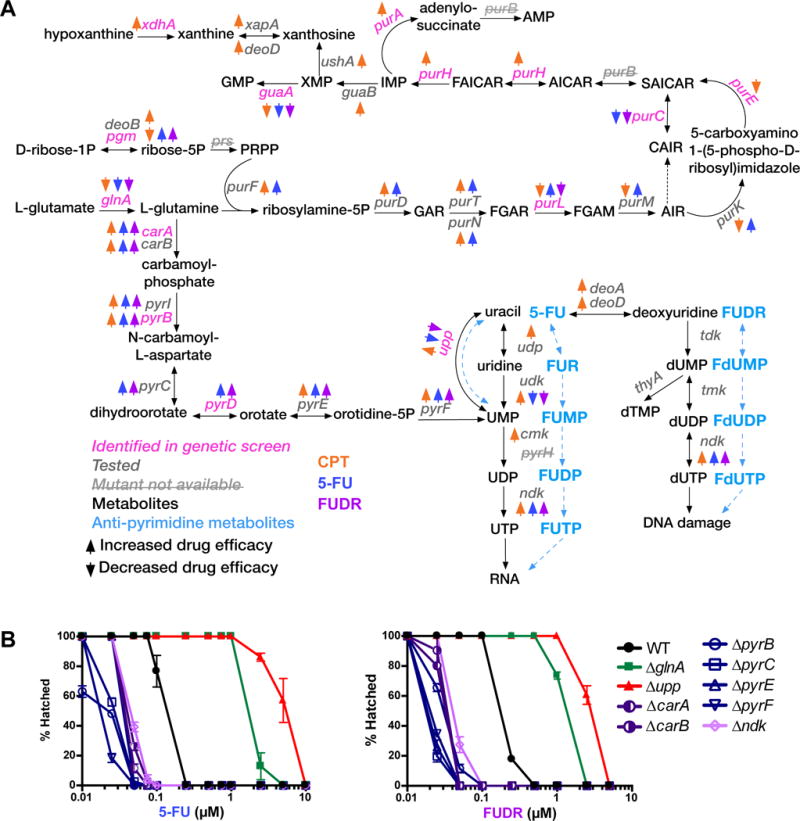
(A) Summary of the E. coli nucleotide biosynthesis network. Colored arrows indicate increase (upward) or decrease (downward) efficacy with CPT, 5-FU, and FUDR. Conversion products from reactions involving 5-FU and FUDR and indicated in light blue.
(B) Dose-response curves of selected E. coli mutants. Mean and standard deviation of technical replicates in one experiment are shown. The other biological replicate experiments are provided in Figure S4.
Several observations can be gleaned from this experiment. First, we observed striking differences between the three drugs: the upper branch of the network shown in Figure 5A, which depicts the final biosynthetic reactions of purine metabolism, is relatively specific for CPT, with most mutants increasing the efficacy of this drug in C. elegans. An exception is guaA, which encodes guanine monophosphate (GMP) synthase that catalyzes the final step in GMP synthesis. A deletion in guaA results in slow bacterial growth (Baba et al., 2006) and decreases the efficacy of all three drugs. Second, the de novo pyrimidine biosynthesis branch of nucleotide metabolism, which converts L-glutamine into uridine monophosphate (UMP, Figure 5A), increases the efficacy of all three drugs. Third, a deletion in the E. coli pyrimidine salvage pathway gene upp, which encodes uracil phosphoribosyltransferase that generates UMP from uracil, decreases drug efficacy for 5-FU and FUDR, but increases the efficacy of CPT (Figure 5A).
Next, we quantitatively compared the modulation of 5-FU and FUDR efficacy by each pyrimidine synthesis pathway. The dose-response curves show a ~10-fold increase in drug efficacy upon mutation of genes in the de novo pyrimidine biosynthesis pathway and a ~10-fold decrease in efficacy upon deletion of upp (Figure 5B and Figure S4). These observations suggest that the bacterial conversion of 5-FU and FUDR into FUMP, an analog of UMP, by upp, is critical for the cytotoxic effects of both anti-pyrimidine drugs in C. elegans. When upp is deleted, this conversion is blocked or reduced, and the bacteria depend on the de novo UMP biosynthesis pathway. When the latter pathway is perturbed, however, the flux through the salvage pathway may be increased leading to higher levels of FUMP.
Modulation of 5-FU and FUDR Efficacy by Bacterial RNA Rather than DNA Nucleotide Metabolism
Remarkably, deletions in either E. coli upp or udk decreases anti-pyrimidine drug efficacy, while deletions in tdk, tmk, or thyA have no effect (Figure 5A). This observation suggests that 5-FU and FUDR predominantly affect C. elegans fecundity via bacterial ribonucleotide metabolism (e.g., by generating FUMP), and not DNA metabolism (e.g., by generating FdUMP which inhibits thymidylate synthase). To test this prediction, we supplemented the animals fed different bacteria and exposed to the two drugs with each of the five nucleobases. We found that only supplementation with uracil rescues the effect of 5-FU on both E. coli and Comamonas. Uracil supplementation also rescues the effect of FUDR on E. coli, with no detrimental effect on Comamonas-fed animals (Figure 6A and Figure S5). The rescue by uracil, but not thymine, supports a model in which bacteria modulate 5-FU and FUDR efficacy through ribonucleotide- rather than DNA metabolism. The observation that thymine fails to rescue the anti-pyrimidine drug effects indicates that inhibition of thymidylate synthase may not be the principal mechanism of action (Longley et al., 2003).
Figure 6. Nucleobase supplementation and flux balance analysis of E. coli mutants.
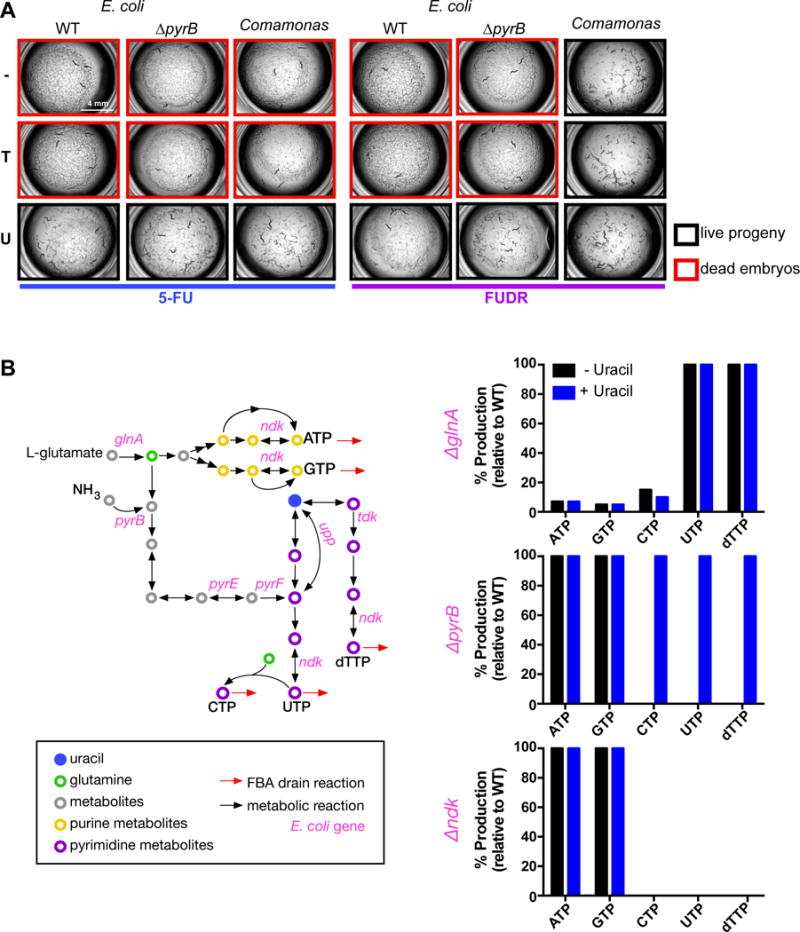
(A) Nucleobase supplementation. WT = wild type E. coli. Adenine, Guanine and Cytosine did not rescue (Figure S5). Representative images of three independent experiments for each bacterial genotype, drug, and nucleobase are shown. 5-FU and FUDR were tested at 0.5 μM. Each nucleobase was added at a concentration of 200 μM (− = vehicle control, T = thymine, U = uracil). See also Figure S5.
(B) Flux balance analysis (FBA). FBA was performed on the E. coli metabolic network model iJO1366 (Orth et al., 2011) with an objective function that maximized the production of a nucleoside triphosphate (represented by the flux of drain reactions shown as red arrows) in each simulation. Production fluxes were recalculated upon the deletion of reactions associated with each gene on the right panel. The analysis was repeated with the addition of uracil as represented by an allowed negative flux in a uracil exchange reaction. Bar graphs show production rates during gene deletion simulations as a percentage of wild type bacteria. The production rate of ribonucleotides and deoxyribonucleotides varied by less than 2%, therefore, only ribonucleotides and dTTP are shown for simplicity.
Flux Balance Analysis of Bacterial Nucleotide Metabolism
Deletion of any gene in the de novo pyrimidine biosynthesis pathway increases efficacy of 5-FU and FUDR, with the exception of glnA, which encodes glutamine synthetase that converts L-glutamate into L-glutamine (Figure 5A). One explanation is that this reaction is also upstream of purine biosynthesis. We used flux balance analysis (FBA) to computationally model nucleotide production in E. coli ΔglnA. Interestingly, glnA deletion greatly affects purine nucleotide production but has little effect on the production of pyrimidines (Figure 6B). As a control, we also performed FBA with mutants of pyrB, which functions downstream of glnA in de novo pyrimidine biosynthesis (Figure 5A, 6B). With these mutations, FBA predicts a reduction in pyrimidine biosynthesis, while having no effect on purines, and further predicts that uracil supplementation rescues this mutation, which is indeed what we observed (Figure 6A). Altogether, these observations help to explain why glnA deletion does not mimic a deletion of downstream genes in de novo pyrimidine biosynthesis. However, it does not explain why glnA deletion results in a decreased drug efficacy. Possibilities include a severe reduction in bacterial growth or an overall nucleotide imbalance.
A deletion in ndk, which encodes nucleotide diphosphate kinase, results in an increase in 5-FU and FUDR efficacy (Figures 5). This enzyme is involved in the biosynthesis of all nucleotides (Figure 6B). However, FBA indicates that ndk mutant bacteria only affect the biosynthesis of pyrimidine nucleotides (Figure 6B). This is because, according to the metabolic network model, ATP and GTP can be generated by alternative pathways (Orth et al., 2011). Thus, an increase in drug efficacy upon ndk deletion agrees with the model that bacterial synthesis of FUMP from 5-FU and FUDR is a primary mechanism of eliciting cytotoxicity in C. elegans: when ndk is deleted, FUMP can no longer be converted into FUTP and accumulates.
A Comamonas Mutant that Increases C. elegans Sensitivity to FUDR
So far, we have found that bacterial ribonucleotide metabolism modulates the C. elegans response to 5-FU and FUDR. Further, we found that FUDR efficacy is much lower when the animals are fed the Comamonas diet relative to a diet of E. coli, whereas we did not observe such a difference for 5-FU. Why does E. coli increase drug efficacy of FUDR relative to a diet of Comamonas? Or, conversely, how does Comamonas exert its protective effect on FUDR toxicity? We found only one Comamonas mutant that specifically increases FUDR efficacy, but that does not modulate the efficacy of 5-FU. This mutant harbors a transposon insertion in one of the 23S rRNA genes (Figure 7A). Dose-response curves show that this mutant increases the efficacy of FUDR to the level found with E. coli (Figure 7B), which cannot be explained by changes in bacterial growth (Figure 7C). As with E. coli, uracil supplementation rescues progeny production in animals fed this mutant and supplemented with FUDR (Figure 7D). Taken together, the protective effect of Comamonas on animals treated with FUDR has a genetic basis. However, the precise mechanism by which the protective effect occurs and why it is specific to FUDR (and not 5-FU) remains to be elucidated.
Figure 7. Model for Anti-Pyrimidine Efficacy Modulation by Bacteria.
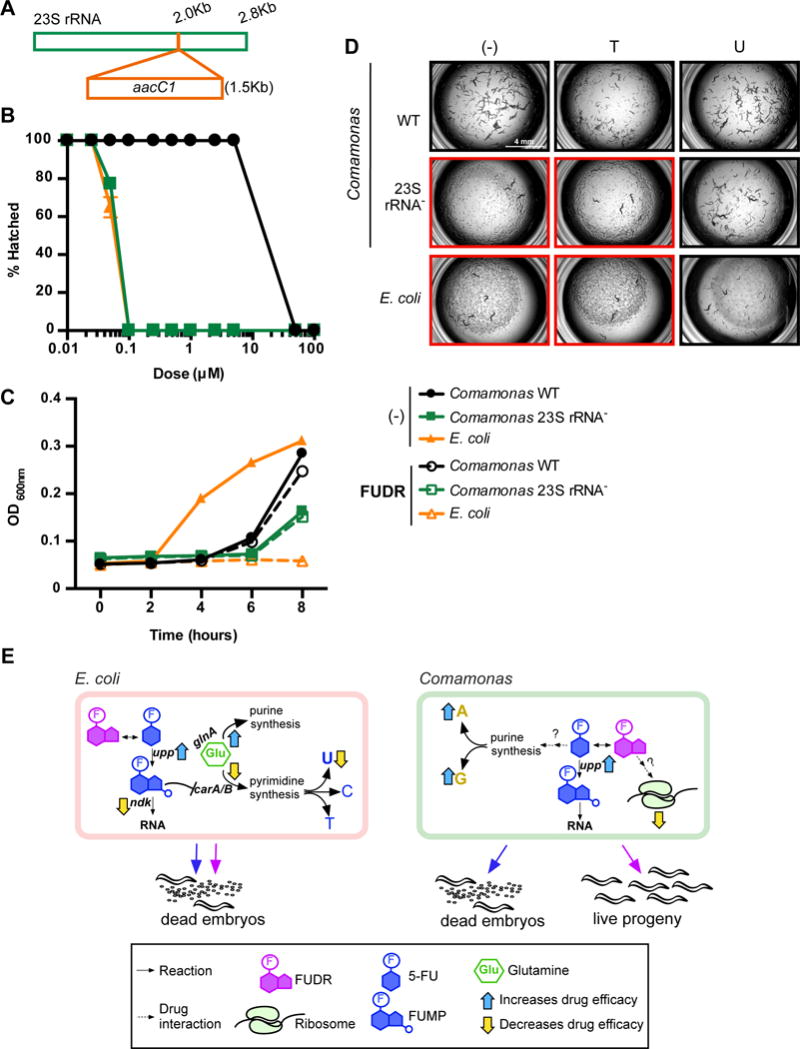
(A) Cartoon of transposon-insertion site in the Comamonas 23S rRNA- mutant.
(B) Dose-response curves for FUDR. A representative experiment out of three biological replicates is shown.
(C) Representative experiment of bacterial growth in the presence or absence of 50 μM FUDR.
(D) Nucleobase supplementation; (−) = vehicle control, T = thymine, U = uracil.
(E) Model for Anti-Pyrimidine Efficacy Modulation by Bacteria
DISCUSSION
Bacterial Modulation of Chemotherapeutic Drug Efficacy
In this study, we have shown that different bacteria can affect chemotherapeutic drug efficacy in C. elegans. For two of the three drugs examined in detail, we found that E. coli and Comamonas oppositely affect the response to CPT and FUDR: animals do better on Comamonas when challenged with FUDR, but worse when supplemented with CPT. For the third drug, 5-FU, we did not observe a difference between the two bacterial species. However, our data show that the response to each drug is affected by bacterial metabolism because we identified bacterial mutants that increase or decrease the efficacy of each of the three drugs, including 5-FU. Although we cannot yet fully explain the mechanisms that cause the bacterial differences in modulation of drug efficacy, our experiments provide insights into the mechanism of action of the different chemotherapeutic drugs.
Bacterial Modulation of CPT Efficacy
Our data suggest that multiple complex mechanisms in bacteria modulate drug efficacy in C. elegans. Some of these mechanisms are drug-specific, while others are more generic, at least for the drugs tested here. First, the efficacy of CPT is affected by bacteria, but likely in a passive way that does not require bacterial metabolism, for instance through nutritional support of nucleotides to the host. We did observe a rather striking difference between the two bacteria with CPT where E. coli deletions in upp, purH, and purA increase CPT efficacy, while the corresponding Comamonas mutant strains cause a decrease (Figure 4A, S3). However, supplementing nucleobases had no effect on CPT efficacy in either E. coli or Comamonas-fed animals (data not shown). Therefore, differences in the pools of individual nucleotides do not directly affect the response to CPT, nor explain the difference in CPT efficacy between animals fed E. coli and Comamonas. The phenotypes we observed in E. coli and Comamonas nucleotide synthesis mutants likely reflect complex relationships between bacterial metabolic networks and host physiology. However, the precise mechanism by which this difference occurs remains to be elucidated.
Bacterial Modulation of 5-FU and FUDR Efficacy: Ribonucleotide Rather than DNA Metabolism
The efficacy of FUDR is differently modulated by E. coli and Comamonas and this difference requires active metabolism in both bacteria: E. coli metabolism to increase and Comamonas metabolism to decrease drug efficacy in C. elegans (Figure 7). With 5-FU, however, we did not observe a difference between animals fed these two bacterial species. This observation indicates that 5-FU and FUDR are differentially affected by bacteria and may not solely function in a linear pathway.
Genetic screens in bacteria revealed that both anti-pyrimidine drugs act by affecting ribonucleotide, rather than DNA metabolism, and the bacterial powder experiments demonstrate that this requires active bacterial metabolism. Our genetic data indicate that both drugs may mainly act through the production of FUMP, which is analogous to UMP. E. coli has two pathways that can synthesize UMP. The first pathway involves the conversion of glutamine into carbamoylphospate by the carA protein and the subsequent multistep generation of UMP. The second pathway involves conversion of uracil into UMP, either via uridine as an intermediate, or directly by the upp enzyme. Mutations in the first pathway increase the efficacy of 5-FU and FUDR, while mutations in the second pathway upstream of UMP decrease efficacy and downstream (in ndk) increase efficacy. Altogether, these findings suggest that bacteria use the first pathway to generate pyrimidines that support C. elegans fecundity in the presence of anti-pyrimidines and use the second pathway to convert the anti-pyrimidine drugs into FUMP, which is then delivered to C. elegans. The natural analog of FUMP, UMP, has been shown to inhibit the bacterial carA/B enzyme (Boettcher and Meister, 1981; Braxton et al., 1999), thereby blocking the de novo pyrimidine biosynthesis pathway, in bacteria, C. elegans, or both (Figure 7E). Unfortunately, it is not feasible to test this hypothesis directly by supplementing FUMP to C. elegans fed Δupp bacteria due to limited availability and high cost. Further, the nucleotide may not be taken up as efficiently by the animal as the nucleobase, which would make any results difficult to interpret. Interestingly, the food and drug administration recently approved the use of uridine triacetate to mitigate 5-FU toxicity (Ison et al., 2016). This change supports our findings that ribonucleotide metabolism is a central mechanism of action of anti-pyrimidine drugs rather than the inhibition of thymidylate synthase by FdUMP.
Implications for Microbiotal Effects on Chemotherapeutic Drug Efficacy in Humans
The human gut microbiota is highly complex and contains many bacterial species. These bacteria may affect drug efficacy even when present in small proportions, and may affect each other as well, further illustrating the complexity of potential bacteria-drug interactions. Additionally, when bacterial growth of some species is affected by chemotherapeutic drugs, the microbiota composition may change, which can further affect drug efficacy and overall health. While our study focused on a simpler metazoan system with only two bacterial species, our findings illuminate the possible effects the microbiota may have on the efficacy of some chemotherapeutics used to treat colorectal cancer in humans. It is tempting to speculate that the treatment of such cancers may benefit from perturbations of the microbiota prior to or after administration of the drugs, such as with probiotics or with fecal transplants. Longer term, studies using additional drugs, as well as multiple bacterial species can be used to further examine the spectrum of bacteria-drug interactions. C. elegans and its bacterial diet provide a fruitful interspecies model system to characterize these interactions and the mechanisms involved.
STAR * METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author A.J.M. Walhout (marian.walhout@umassmed.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans
N2 (Bristol) was used as the wild-type strain, and animals were maintained with E. coli OP50 as diet on Nematode Growth Media (NGM), as previously described (Brenner, 1974).
Bacterial Strains
Bacterial strains E. coli OP50, E. coli BW25113, and Comamonas aquatica DA1877 were grown as described (Watson et al., 2014). E. coli deletion mutants (Baba et al., 2006) were grown in Luria Broth (LB) with 50 μg/mL kanamycin. Comamonas aq. DA1877 mutants (Watson et al., 2014) were grown in LB containing 10 μg/mL gentamicin. One mutant in our screens, E. coli ΔthyA, is a thymine auxotroph, and this mutant was grown in the presence of 20 μg/mL thymine (Figure 5A).
METHOD DETAILS
C. elegans Phenotypic Assays
All animals were fed the standard laboratory diet E. coli OP50 until transferred to plates containing the respective bacteria and drug combination. For all fecundity assays, embryos were harvested from gravid animals by bleaching in sodium hypochlorite solution, washing embryos repeatedly with M9 buffer and then incubating overnight in M9 to allow hatching. Then, synchronized L1 animals were grown on NGM agar plates with the E. coli OP50 diet for ~48 hours until the animals reached the L4 stage. Animals were then transferred to NGM agar drug plates and incubated at 20°C. Approximately 72 hours later, fecundity phenotypes were visually identified by microscopy, and brightfield images were collected using an Invitrogen™ EVOS™ FL microscope set at 2× magnification.
Chemotherapeutic Drugs
Camptothecin (CPT) was purchased from Sigma Aldrich (C9911) and dissolved in dimethyl sulfoxide (DMSO) to 10 mM for a stock solution. 5-Fluorouracil (5-FU) was purchased from Sigma Aldrich (F6627) and dissolved at 300 mM in DMSO. 5-fluoro-2′-deoxyuridine (FUDR) was purchased from bioWORLD (40690016-1) and Sigma Aldrich (F0503) and dissolved in sterile water to 200 mM. All drug solutions were filter-sterilized using a Millex® GP filter unit of 0.22 μm and then stored at −20°C. At the start of each experiment, each stock solution was diluted as needed to 1000X the final concentration. To reach the final drug concentration, all drugs were added to a final volume of 0.1% in NGM agar kept at 55°C. All NMG + drug plates were prepared a day before use and allowed to dry overnight at room temperature.
Bacterial Powder
For each experiment, ~250 mg of bacterial powder was generated from 2 L of bacterial culture. We started with a single colony, grown overnight and subsequently diluted 1:100 in 2 L of LB with no antibiotics and grown to log phase (OD600nm 0.8–1.0). Then, cultures were centrifuged at 34,000 rpm for 30 min and washed three times with sterile water. Each bacterial pellet was weighed and sterile water was added to obtain approximately 1 g of wet weight per 25 mL. Bacteria were then disrupted using a Microfluidics™ M-110P lab homogenizer set to ~22,000 psi for five cycles. Disrupted cells were flash-frozen and lyophilized using a Labconco® FreeZone 2.5L benchtop freeze dryer/lyophilizer set to ~0.133 mBar and −80°C for approximately 60 hours until completely dry. Bacterial powder was dissolved in sterile water to achieve a concentration of 50 mg/mL. Plating the powder confirmed that there was no remaining growth.
Bacterial Mixing Experiments
Cultures of E. coli OP50 and Comamonas aq. DA1877 were grown overnight from a single colony and diluted to OD600nm of 0.5. Colony forming units (CFUs)/μL were determined by plating the diluted cultures. Cultures were mixed as ratios of CFUs.
Bacterial Mutant Drug Screens
The following drug doses were used for the primary E. coli deletion collection screens (Baba et al., 2006): CPT 4 μM, 5-FU 4 μM, FUDR 4 μM. For the Comamonas aq. DA1877 mutant collection screens the doses were: CPT 4 μM, 5-FU 4 μM, FUDR 65 μM. All primary screens were performed in 96-well NGM agar plates containing drug and antibiotics as described above. All bacterial mutants were grown from frozen glycerol stocks into one mL LB, incubated for ~16–18 hours at 37°C shaking at 200 rpm. Fifty μL of bacteria from overnight culture was added to each NGM plus drug well and plates were incubated overnight at room temperature. Approximately ten L4 C. elegans were added to each well and plates were incubated at 20°C for 72 hours. In the E. coli collection, two independent mutant clones are present for each gene, one mutant in an even number plate and the other mutant in the consecutive plate (Baba et al., 2006). We screened each plate of the Keio collection, thus screened each mutant twice. For the second and third rounds of screening, 24-well NGM agar plates with drug and without antibiotics were used.
Genotyping Bacterial Mutants
All mutant hits from the Keio collection were sequence-confirmed using gene-specific primers at −200 bp and +200 bp from the start and the stop codons, respectively (Supplementary Table S1). All mutant hits from the Comamonas aq. DA1877 collection were sequenced to identify the disrupted gene, as described previously (Watson et al., 2014). Gene names for the Comamonas aq. DA1877 mutants listed in Figures 3–4 were derived from the closest E. coli gene matching the gene function.
Bacterial Growth Assay
All mutants were growth overnight at 37°C in 96-well deep well dishes with LB medium. Each bacterial culture was diluted 1,000-fold in 1.2 mL of Sugar Optimal Broth media containing 50 μM of each drug. 150 μL of bacterial drug culture was added to flat-bottom 96-well plates, one for each time point. All plates were incubated at 37°C, shaking at 200 rpm. The OD 600nm was measured every two hours for 10 hours using an Infinite® M1000 PRO plate reader.
Dose-Response Curves
For each experiment, each drug dose and bacteria combination was tested in duplicate. We used the following doses (μM): 5-FU (0.01, 0.025, 0.05, 0.075, 0.1, 0.25, 0.5, 0.75, 1.0, 2.5, 5.0, 10.0); and FUDR (0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 1.0, 2.5, 5.0, 50, 100, 200). At the start of each experiment, two to five animals were placed in each well. Approximately 72 hours later, brightfield images of the entire well were collected using an an Invitrogen™ EVOS™ FL microscope set at 2× magnification. We used the Cell Counter Plugin from Fiji (Schiff et al., 2011) to manually count dead embryos and live progeny.
Nucleobase Supplementation
Each nucleobase was obtained from Sigma and diluted to 150 mM as follows: adenine was dissolved in 0.5 M HCl, guanine, thymine and uracil were dissolved in 1 M NaOH, and cytosine was dissolved in 0.1 M HCl. Each stock solution was stored in aliquots at −20°C. Forty μL of each base or solvent control was added to 30 mL NGM agar kept at 55°C before pouring for a final concentration of 200 μM of each nucleobase.
E. coli Flux Balance Analysis
To predict the effect of gene deletions and uracil supplementation on the production of nucleotides by E. coli K-12 BW25113 (the parent strain in the Keio collection)(Baba et al., 2006), flux balance analysis (FBA) was performed with a slightly modified version of the genome-scale E. coli metabolic network model iJO1366 (Orth et al., 2011). This model was originally developed for E. coli K-12 MG1655. Based on the differences between the BW25113 and MG1655 strains (Baba et al., 2006), eight genes used in iJO1366 are absent from E. coli K-12 BW25113 (gatC, lacZ, mhpC, rhaA, rhaB, rhaD, araA, araB, araD). To convert iJO1366 into a better representative model of E. coli K-12 BW25113, a total of ten reactions each of which is strictly dependent on the presence of one of these genes were eliminated from the network.
FBA requires a defined input based on nutrient exchange reactions, additional reaction constraints that describe the tested condition, and an objective function to maximize or minimize depending on the biological question. Here, metabolite uptake rates were obtained from previous modeling studies that characterized the growth of E. coli in LB medium (Tawornsamretkit et al., 2012). To simulate uracil availability, the uracil uptake flux in the model was set to 9 mmol/g dW/hr, which corresponds to the maximum uracil production that can be achieved by the metabolic network from LB medium, and therefore represents a saturation condition that requires no de novo biosynthesis of uracil for any objective function. Gene deletions were represented by constraining all reactions associated with the deleted gene (glnA, pyrB, or ndk) to zero flux. The existence of paralogs encoding an enzyme with the same function was ignored, i.e., redundancy in gene-reaction associations was not taken into account. In each FBA run, the objective was set to maximize the production of one of the eight nucleotides tested (ribonucleotides: ATP, GTP, CTP, and UTP; deoxyribonucleotides: dATP, dGTP, dCTP, and dTTP) as represented by the flux in an artificially added reaction that consumed the nucleotide. Thus eight optimizations were carried out for each one of the two uracil conditions, absent or present, resulting in 16 optimizations total. Results from gene deletions were compared with wild type results, i.e., when no reactions were constrained to zero flux. The maximum achievable production of each nucleotide was calculated and reported as a percentage of wild type production.
Supplementary Material
Schematic of reported mechanisms of action for the topo-I inhibitor CPT (left), and the anti-pyrimidines 5-FU and FUDR (right). Solid dark arrows indicate main reported mechanisms of action and gray dashed arrows indicate side pathways. TYMS — thymidylate synthase.
C. elegans phenotypes in animals fed E. coli BW25113, the background strain for the Keio collection (Baba et al., 2006), compared to E. coli OP50.
Each colored block represents bacterial mutant-drug interactions identified in ≥ 2/3 independent experiments. All doses listed are in μM.
Each plot represents an independent experiment, plotted are the mean and standard deviation between technical replicates.
(A) Effect of nucleotide base supplementation at 200 μM in three independent experiments. (− = vehicle control, A = adenine, G = guanine, C = cytosine, T = thymine, U = uracil). (B) Representative brightfield images at 2X magnification.
Table S1. Bacterial mutants identified in genetic screens. Related to Figure 3.
HIGHLIGHTS.
Bacteria differentially affect the C. elegans response to FUDR and camptothecin
Bacterial metabolism is required for the C. elegans chemotherapeutic response
Genetic screens with two bacterial species and three drugs to unravel mechanism
5-FU and FUDR affect C. elegans through bacterial RNA rather than DNA metabolism
Bacteria modulate the efficacy of chemotherapeutic drug responses of the host through distinct mechanisms as established by genetic screens in C. elegans.
Acknowledgments
We thank members of the Walhout lab, Beth McCormick, David Bates, Abraham L. Sonenshein and Job Dekker for discussion and critical reading of the manuscript. This work was supported by grants from the National Institutes of Health DK068429 and GM082971 to A.J.M.W., and GM107227 and AI121836 to E.C.A., and GM122393 to A.G.G. Some bacterial and nematode strains used in this work were provided by the CGC, which is funded by the NIH Office or Research Infrastructure Programs (P40 OD010440).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes five Supplemental Figures and one Supplemental Table.
AUTHOR CONTRIBUTIONS
A.G.G., A.D.R. and A.J.M.W. conceived the study. A.D.R. performed initial experiments. A.G.G. performed all experiments included in the paper with technical help from S.S. L.S.Y. performed the flux balance analysis. The unpublished bacterial lysate powder protocol was provided by E.C.A. The paper was written by A.G.G. and A.J.M.W. with editing by all other authors.
References
- Andrews JM. The development of the BSAC standardized method of disc diffusion testing. J Antimicrob Chemother. 2001;48(Suppl 1):29–42. doi: 10.1093/jac/48.suppl_1.29. [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology. 2006;2:2006–0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M, Stenuit B, Ho J, Wang A, Parke C, Knight M, Alvarez-Cohen L, Shapira M. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 2016;10:1998–2009. doi: 10.1038/ismej.2015.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher B, Meister A. Conversion of UMP, an allosteric inhibitor of carbamyl phosphate synthetase, to an activator by modification of the UMP ribose moiety. J Biol Chem. 1981;256:5977–5980. [PubMed] [Google Scholar]
- Braxton BL, Mullins LS, Raushel FM, Reinhart GD. Allosteric dominance in carbamoyl phosphate synthetase. Biochemistry. 1999;38:1394–1401. doi: 10.1021/bi982097w. [DOI] [PubMed] [Google Scholar]
- Brennan CA, Garrett WS. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu Rev Microbiol. 2016;70:395–411. doi: 10.1146/annurev-micro-102215-095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cocheme HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen P, Marsh SA, Braker I, Heitland N, Wagner S, Nakad R, Mader S, Petersen C, Kowallik V, Rosenstiel P, et al. The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol. 2016;14:38. doi: 10.1186/s12915-016-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MA, Duveau F. Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC Biology. 2013;10:59. doi: 10.1186/1741-7007-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MA, Braendle C. The natural history of Caenorhabditis elegans. Curr Biol. 2010;20:R965–969. doi: 10.1016/j.cub.2010.09.050. [DOI] [PubMed] [Google Scholar]
- Gusarov I, Gautier L, Smolentseva O, Shamovsky I, Eremina S, Mironov A, Nudler E. Bacterial nitric oxide extends the lifespan of C. elegans. Cell. 2013;152:818–830. doi: 10.1016/j.cell.2012.12.043. [DOI] [PubMed] [Google Scholar]
- Ison G, Beaver JA, McGuinn WD, Jr, Palmby TR, Dinin J, Charlab R, Marathe A, Jin R, Liu Q, Chen XH, et al. FDA Approval: Uridine Triacetate for the Treatment of Patients Following Fluorouracil or Capecitabine Overdose or Exhibiting Early-Onset Severe Toxicities Following Administration of These Drugs. Clin Cancer Res. 2016;22:4545–4549. doi: 10.1158/1078-0432.CCR-16-0638. [DOI] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- MacNeil LT, Watson E, Arda HE, Zhu LJ, Walhout AJM. Diet-induced developmental acceleration independent of TOR and insulin in C. elegans. Cell. 2013;153:240–252. doi: 10.1016/j.cell.2013.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Conrad TM, Na J, Lerman JA, Nam H, Feist AM, Palsson BO. A comprehensive genome-scale reconstruction of Escherichia coli metabolism--2011. Molecular Systems Biology. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Schiff M, Benoist JF, Tilea B, Royer N, Giraudier S, Ogier de Baulny H. Isolated remethylation disorders: do our treatments benefit patients? Journal of inherited metabolic disease. 2011;34:137–145. doi: 10.1007/s10545-010-9120-8. [DOI] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Backhed F. The gut microbiota-masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- Tawornsamretkit I, Thanasomboon R, Thaiprasit J, Waraho D, Cheevadhanarak S, Meechai A. Analysis of metabolic network of synthetic Escherichia coli producing linalool using constraint-based modeling. Procedia Computer Science. 2012;11:24–35. [Google Scholar]
- Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, MacNeil LT, Arda HE, Zhu LJ, Walhout AJM. Integration of metabolic and gene regulatory networks modulates the C. elegans dietary response. Cell. 2013;153:253–266. doi: 10.1016/j.cell.2013.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, MacNeil LT, Ritter AD, Yilmaz LS, Rosebrock AP, Caudy AA, Walhout AJM. Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell. 2014;156:759–770. doi: 10.1016/j.cell.2014.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, Olin-Sandoval V, Hoy MJ, Li CH, Louisse T, Yao V, Mori A, Holdorf AD, Troyanskaya OG, Ralser M, et al. Metabolic network rewiring of propionate flux compensates vitamin B12 deficiency in C. elegans. Elife. 2016;5 doi: 10.7554/eLife.17670. pii:e17670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, Walhout AJ. Caenorhabditis elegans metabolic gene regulatory networks govern the cellular economy. Trends Endocrinol Metab. 2014;25:502–508. doi: 10.1016/j.tem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, Yilmaz LS, Walhout AJM. Understanding metabolic regulation at a systems level: metabolite sensing, mathematical predictions and model organisms. Annu Rev Genet. 2015;49:553–575. doi: 10.1146/annurev-genet-112414-055257. [DOI] [PubMed] [Google Scholar]
- Yilmaz LS, Walhout AJM. Worms, bacteria and micronutrients: an elegant model of our diet. Trends Genet. 2014;30:496–503. doi: 10.1016/j.tig.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic of reported mechanisms of action for the topo-I inhibitor CPT (left), and the anti-pyrimidines 5-FU and FUDR (right). Solid dark arrows indicate main reported mechanisms of action and gray dashed arrows indicate side pathways. TYMS — thymidylate synthase.
C. elegans phenotypes in animals fed E. coli BW25113, the background strain for the Keio collection (Baba et al., 2006), compared to E. coli OP50.
Each colored block represents bacterial mutant-drug interactions identified in ≥ 2/3 independent experiments. All doses listed are in μM.
Each plot represents an independent experiment, plotted are the mean and standard deviation between technical replicates.
(A) Effect of nucleotide base supplementation at 200 μM in three independent experiments. (− = vehicle control, A = adenine, G = guanine, C = cytosine, T = thymine, U = uracil). (B) Representative brightfield images at 2X magnification.
Table S1. Bacterial mutants identified in genetic screens. Related to Figure 3.