

Abstract
Background
Relative risk reduction with statin therapy has been consistent across nearly all subgroups studied to date. However, in analyses of two randomized controlled primary prevention trials (ASCOT and JUPITER), statin therapy led to a greater relative risk reduction among a subgroup at high genetic risk. Here, we sought to confirm this observation in a third primary prevention randomized controlled trial. Additionally, we assessed if those at high genetic risk had a greater burden of subclinical coronary atherosclerosis.
Methods
We studied participants from a randomized controlled trial of primary prevention with statin therapy (WOSCOPS, n=4,910) and two observational cohort studies (CARDIA and BioImage, n=1,154 and 4,392). For each participant, we calculated a polygenic risk score (PRS) derived from up to 57 common DNA sequence variants previously associated with coronary heart disease (CHD). We compared the relative efficacy of statin therapy in those at high genetic risk (top quintile of PRS) versus all others (WOSCOP)S as well as the association between the PRS and coronary artery calcification (CARDIA) and carotid artery plaque burden (BioImage).
Results
Among WOSCOPS trial participants at high genetic risk, statin therapy was associated with a relative risk reduction of 44% (95% CI, 22%–60%; P < 0.001) whereas in all others, relative risk reduction was 24% (95% CI 8%–37%; P = 0.004) despite similar LDL cholesterol lowering. In a study-level meta-analysis across the WOSCOPS, ASCOT, and JUPITER primary prevention, relative risk reduction in those at high genetic risk was 46% versus 26% in all others (P for heterogeneity = 0.05). Across all three studies, the absolute risk reduction with statin therapy was 3.6% (95% CI, 2.0%–5.1%) among those in the high genetic risk group and was 1.3% (95% CI, 0.6%–1.9%) in all others. Each standard deviation increase in the polygenic risk score was associated with 1.32-fold (95% CI, 1.04–1.68) greater likelihood of having coronary artery calcification and 9.7% higher (95% CI, 2.2–17.8%) burden of carotid plaque.
Conclusions
Those at high genetic risk have a greater burden of subclinical atherosclerosis and derive greater relative and absolute benefit from statin therapy to prevent a first CHD event.
Clinical Trial registration
WOSCOPS was carried out and completed prior to the requirement for clinical trial registration. BioImage: NCT00738725 (https://www.clinicaltrials.gov/ct2/show/NCT00738725). CARDIA: NCT00005130 (https://clinicaltrials.gov/ct2/show/NCT00005130).
Keywords: primary prevention, statin, human genetics, genetic polymorphism, and coronary artery calcification
INTRODUCTION
Coronary heart disease (CHD) is a complex, chronic disease responsible for about 7 million deaths worldwide in 2010.1 Statin therapy reduces the risk of a first coronary event.2, 3 Effect size as measured by relative risk reduction is approximately 20% per 1.0 mmol/L reduction of low density lipoprotein (LDL) cholesterol and has been consistent across nearly all subgroups defined by clinical and biochemical measures.2, 4 However, if statin therapy was more efficacious in one subgroup versus another, this might impact decisions regarding who gets prescribed statin therapy in the primary prevention setting.
We recently reported that those at high genetic risk, defined as the top quintile of a 27-SNP polygenic risk score for CHD, derived greater relative risk reduction from statin therapy compared with all others.5 In two primary prevention trials (ASCOT-LLA: Anglo-Scandinavian Cardiac Outcomes Trial – Lipid-Lowering Arm6 and JUPITER: Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin7), this higher relative benefit from statin therapy was observed despite similar levels of LDL cholesterol lowering between those at high genetic risk and all others.
The present study had two main goals: first, to test in a third statin trial if statin treatment confers a greater relative risk reduction for a first coronary event in those at high genetic risk, as assessed by an expanded 57-SNP polygenic risk score, compared to all others; second, to test if there is a greater burden of subclinical coronary atherosclerosis present in those at high genetic risk compared to all others.
METHODS
Cohort Descriptions
The WOSCOPS (West of Scotland Coronary Prevention Study) trial has been described previously.8, 9 In brief, WOSCOPS was a randomized controlled trial of 6,595 men (45–64 years) with hypercholesterolemia but without a history of myocardial infarction, allocated to pravastatin 40mg daily versus placebo to prevent coronary events. Genetic data were available in 4,892 men. Long-term WOSCOPS results beyond the study’s end were included to assess the durable effects of primary preventive statin therapy by genetic risk. Results were also available from a prior analysis5 that included a subset of 6,978 individuals with genetic data from ASCOT-LLA, a randomized trial of atorvastatin 10mg daily versus placebo in those with hypertension but without cardiovascular disease, and 8,769 individuals with genetic data from JUPITER, a randomized trial of rosuvastatin 20mg daily versus placebo in those with no history of cardiovascular events but elevated C-reactive protein.
In two observation cohorts (CARDIA and BioImage), we explored a potential reason for the greater clinical benefit of statin therapy in those at high genetic risk. We hypothesized that individuals at high genetic risk carried a greater burden of subclinical atherosclerosis. We assessed the association of a high genetic risk status with subclinical atherosclerosis in two vascular beds in those without clinical CHD. The CARDIA (Coronary Artery Risk Development in Young Adults) study (NCT00005130) is an observational study of cardiovascular risk factors in 5,115 young adults (18–30 years at baseline, 1989–1990) as previously described.10, 11 Of these participants, 1,154 European ancestry individuals without CHD at baseline, available genetic data and coronary arterial calcification (CAC) assessment at the 15-year follow-up were included in analyses. The BioImage study (NCT00738725) is a multi-ethnic, observational study aimed at characterizing subclinical atherosclerosis in 6,699 US adults (55–80 years at baseline, 2008–2009) at risk for, but without, clinical atherosclerotic cardiovascular disease.12, 13 As current genome-wide association effect estimates for coronary heart disease are most robust in those of European ancestry14, we focused on the 4,929 individuals of European ancestry. Of those individuals, 4,392 with both genetic data available and carotid plaque assessment were included in analyses. In WOSCOPS, CARDIA, and BioImage, participants were not screened for familial hypercholesterolemia, a monogenic disorder associated with increased risk of premature CHD events. Each trial was approved by institutional review boards, all subjects gave their informed consent, and the procedures that were followed were in accordance with institutional guidelines.
Polygenic Risk Score
Genome-wide association analyses have identified 67 single nucleotide polymorphisms (SNPs) across the genome independently associated with CHD (Supplementary Table 1).14–16 We recently showed that an expanded set of SNPs compared to a polygenic risk score comprised of 27 SNPs provided modestly improves risk discrimination.17 Fifty-seven of these variants were genotyped among WOSCOPS participants using the Illumina Metabochip18 and included in analyses. Thirteen variants were directly genotyped among CARDIA participants using the Affymetrix Human SNP Array 6.0 and another 25 proxy variants available through statistical imputation. CARDIA genotypes were downloaded from the NIH dbGAP data repository (accession phs000613.v1.p2). Fifty-nine variants were directly genotyped among BioImage participants using the Illumina HumanExome Beadchip19 and an additional four proxy variants (r2 > 0.8) available through statistical imputation.
A polygenic risk score was constructed by weighting the total number of risk alleles by their effects (log of the odds ratios) of CHD risk from the published literature. Incremental scores from missing genotypes in individuals were imputed based on the allele frequency in each cohort. To account for the differences in the numbers of variants per cohort, a normalized polygenic risk score (mean = 0, standard deviation = 1) was created per cohort (Supplementary Figure 1).
Outcomes
The primary outcome for the WOSCOPS analysis was nonfatal myocardial infarction or death from CHD.8 In WOSCOPS we also studied change in LDL cholesterol from baseline; on-treatment LDL cholesterol was obtained one year after study drug initiation.
An exploratory analysis focused on subclinical atherosclerosis - total CAC quantity by the Agatston method in CARDIA and total carotid plaque burden in BioImage.20 In the CARDIA study, CAC was assessed by electrocardiographic-gated electron beam CT (Imatron C-150, GE Imatron, San Francisco, CA) or multidetector CT (GE LightSpeed, GE Healthcare, Little Chalfont, UK; or Siemens VZ, Siemens Healthcare, Erlangen, Germany) at the 15 year-follow-up (33–45 years, 2000–2001).11 In BioImage, carotid plaque was ascertained using the Philips iU22 carotid ultrasound system (Philips Healthcare, Bothell, Washington) interpreted by at the University of Copenhagen (Copenhagen, Denmark) as described previously.12, 21 If carotid plaque was present based on local carotid intima media thickness, it was quantified using the Philips QLAB-VPQ software and carotid plaque burden was the sum of all areas of carotid plaque from the proximal common carotid artery to the distal internal carotid artery.
Statistical Analysis
Within each cohort, we defined high genetic risk as individuals in the top quintile of the distribution of polygenic risk score. Among placebo-treated participants in WOSCOPS, we first used a Cox proportional hazards model to determine if polygenic risk score (per standard deviation, and high genetic risk versus all others) associated with risk of developing incident nonfatal myocardial infarction or death due to CHD. The models were adjusted for age, sex, diabetes mellitus status, smoking status, baseline LDL cholesterol, baseline HDL cholesterol, systolic blood pressure, antihypertensive medication status, and family history of myocardial infarction or stroke. Sex was not used as a covariate in WOSCOPS as all participants were male. Higher order terms for PRS were not significantly associated with outcome and the linear assumption was not violated. Next, we stratified participants in two groups (high genetic risk, all others) and tested the difference in relative risk reduction with statin therapy versus placebo in each subgroup. Analysis of Schoenfeld residuals demonstrated similar proportionality across the follow-up time.
To determine the confidence interval of the absolute risk reduction from statin therapy for each polygenic risk score group, we calculated the standard error of the absolute reduction for each group as where a is statin-treated individuals who had events, n1 is all statin-treated individuals, c is placebo-treated individuals who had events, and n2 is all placebo-treated individuals. A chi square test for heterogeneity was used to test the differences in absolute risk reduction from statins between those at high genetic risk versus all others.
In order to summarize all currently available data across primary prevention trials, we performed a study-level meta-analysis combining the present study results with those published earlier from the JUPITER and ASCOT-LLA trials5. The analyzed outcome was the primary outcome for each primary prevention trial. Effect estimates for high genetic risk versus all others was combined using a fixed-effects meta-analysis. A chi-square test for heterogeneity was used to compare the proportional risk reductions between the meta-analyzed effect estimates as previously described.22
In two population-based cohort studies, we tested whether prevalent subclinical atherosclerosis differed between those at high genetic risk versus all others. Given the younger age of CARDIA participants and a consequential lower prevalence of individuals with any CAC, a dichotomous outcome variable (CAC>0 versus CAC=0) was used in in CARDIA. We determined whether two predictors (polygenic risk score as a continuous variable or a dichotomized high genetic risk versus all others) was associated with CAC using multivariate logistic regression in CARDIA. We similarly tested whether polygenic risk score was associated with prevalent carotid plaque in BioImage. The outcome variable was the natural log transformation of total bilateral carotid plaque + 1. The models were adjusted for age, sex, diabetes mellitus status, smoking status, LDL cholesterol, HDL cholesterol, systolic blood pressure, antihypertensive medication status, and family history of CHD.
All tests were two-tailed with alpha threshold of 0.05. Statistical analyses were conducted in R v3.2.1 (R Foundation, Vienna, Austria).
RESULTS
The mean length of follow-up for WOSCOPS participants in trial was 4.8 years (SD, 0.7 years) for both placebo and statin groups, and out of trial was 8.7 years (SD, 2.6 years) in the placebo group and 8.9 years (SD, 2.4 years) in the statin-treated group; baseline characteristics by genetic risk (Table 1) and by randomized treatment groups (Supplementary Table 2) are presented. Individuals at high genetic risk (top quintile of polygenic risk scores) were more likely to report a family history of CHD (7% vs 5%; P = 0.004) and were less likely to be current smokers (40% vs 45%, P = 0.006) compared to all others whereas there was no difference in other baseline characteristics including treatment allocation. In placebo-treated WOSCOPS participants, predicted 10-year risks for atherosclerotic cardiovascular disease (calculated using ACC/AHA pooled cohort equations) were similar across quintiles of polygenic risk score (ACC/AHA pooled cohort equations) were similar (Supplementary Table 3).
Table 1.
Characteristics of WOSCOPS Participants by Genetic Risk Group
| High Genetic Risk (≥80th percentile polygenic risk score) (n = 979) |
All Others (<80th percentile polygenic risk score) (n = 3,913) |
P | |
|---|---|---|---|
| Age, years | 54.9 (5.5) | 55.2 (5.5) | 0.17 |
| Male, % | 100 | 100 | - |
| BMI, kg/m2 | 26.1 (3.1) | 26.0 (3.2) | 0.35 |
| Family history of CHD, % | 7 | 5 | 0.004 |
| Smoking, % | 40 | 45 | 0.006 |
| Diabetes mellitus, % | 1 | 1 | 0.61 |
| Systolic blood pressure, mmHg | 135.6 (17.1) | 135.5 (17.3) | 0.95 |
| Antihypertensive therapy, % | 15 | 14 | 0.38 |
| Total cholesterol, mg/dl | 272 (22.9) | 272 (22.6) | 0.74 |
| LDL cholesterol, mg/dl | 192 (17.5) | 192 (17.3) | 0.20 |
| HDL cholesterol, mg/dl | 44 (9.5) | 44 (9.4) | 0.41 |
| Triglycerides, mg/dl | 160 (70.0) | 160 (66.8) | 0.92 |
| Statin, % | 48 | 51 | 0.11 |
| Follow Up, years | 13.6 (2.8) | 13.6 (2.7) | 0.49 |
| Follow Up within trial, years | 4.9 (0.7) | 4.8 (0.7) | 0.17 |
| Follow Up after trial, years | 8.7 (2.6) | 8.3 (2.5) | 0.25 |
Values are presented as mean (standard deviation) or %. High genetic risk is defined as the top quintile of polygenic risk score. Differences between continuous variables were tested with Student t-tests and categorical variables with chi-square tests.
BMI = body-mass index; CHD = coronary heart disease; HDL = high-density lipoprotein; LDL = low-density lipoprotein.
Among those allocated to placebo, those at high genetic risk were at increased risk for a first CHD event (HR 1.62; 95% CI, 1.29–2.05; P < 0.001) after adjustment for traditional cardiovascular risk factors (Table 2) (Supplementary Figure 2, Supplementary Table 4). Furthermore, among placebo-treated participants in WOSCOPS, a one SD increase in polygenic risk score was associated with a 25% increased risk in incident CHD (HR 1.25; 95% CI, 1.20–1.35, P <0.001). Association of polygenic risk score with CHD did not vary between those with and without a self-reported family history of CHD (P for interaction = 0.47) (Supplementary Table 5). The mean baseline LDL cholesterol was 192 (SD, 17.5) mg/dl in the high genetic risk group and 192 (SD, 17.3) mg/dl among all others (P = 0.4).
Table 2.
Incident CHD event risk, LDL cholesterol lowering and relative risk reduction of CHD across quintiles of polygenic risk score in WOSCOPS
| Polygenic risk score quintile | CHD event risk* HR (95% CI, P) |
LDL cholesterol reduction after statin mean mg/dl (SD) |
Relative risk reduction with statin therapy within each quintile of polygenic risk HR (95% CI, P) |
|---|---|---|---|
| Q1 | – | −44.7 (1.8) | 0.65 (0.44–0.97, P =0.035) |
| Q2 | 0.83 (0.57–1.20, P =0.33) | −44.8 (1.7) | 1.00 (0.67–1.48, P =0.99) |
| Q3 | 1.22 (0.86–1.71, P =0.26) | −43.4 (1.8) | 0.68 (0.48–0.97, P =0.04) |
| Q4 | 1.06 (0.74–1.51, P =0.77) | −43.7 (1.7) | 0.77 (0.54–1.11, P =0.16) |
| Q5 (High) | 1.66 (1.21–2.29, P =0.0019) | −42.5 (1.8) | 0.56 (0.40–0.78, P <0.001) |
Adjusted for age, sex, diabetes mellitus status, smoking status, LDL cholesterol, HDL cholesterol, systolic blood pressure, antihypertensive medication status, and family history of CHD.
CHD = coronary heart disease; CI = confidence interval; HR = hazard ratio; Q = quintile
Placebo-treated participants
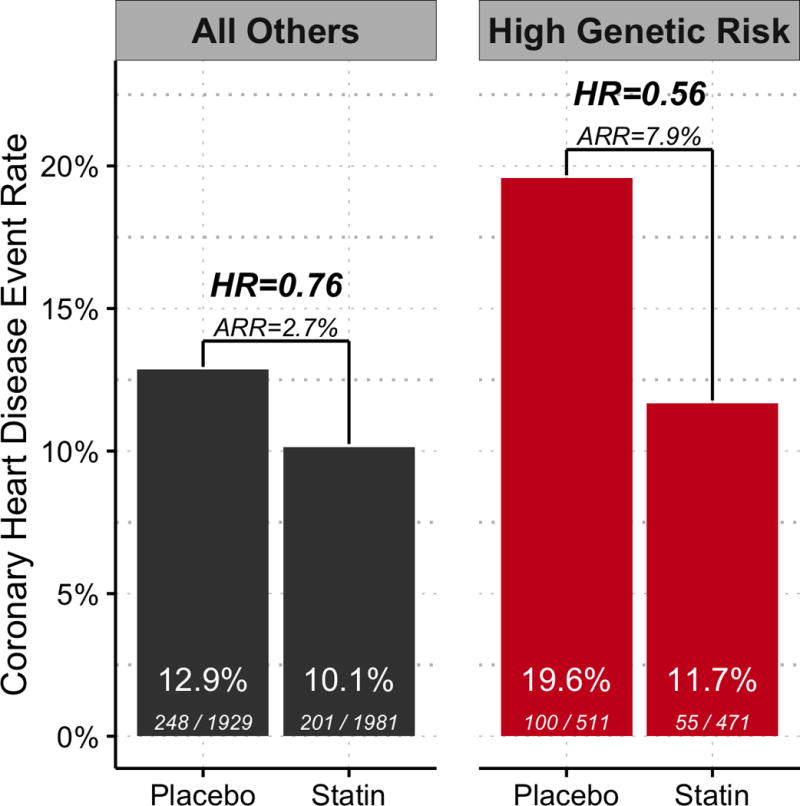
Among those at high genetic risk, statin therapy reduced risk for a first CHD event by 44% (HR 0.56; 95% CI, 0.40–0.78; P < 0.001) whereas statin therapy reduced risk by 24% among all others (HR 0.76; 95% CI, 0.63–0.92; P = 0.004). Absolute risk reduction with statin therapy was 7.9% (95% CI, 3.4%–12.4%) among those at high genetic risk whereas it was 2.7% (95% CI, 0.7%–4.7%) among all others during the 13-year follow-up (P for heterogeneity = 0.04) (Figure 1). Thus, the number needed to treat (NNT) to prevent one coronary event was 13 among high genetic risk participants and 38 among all others (Table 3). The degree of LDL cholesterol reduction achieved with statin treatment was similar in the high genetic risk group (−44 mg/dL, 22.9% reduction) compared to all others (−43 mg/dL, 22.2% reduction) (P = 0.52).
Figure 1. Incident Coronary Heart Disease Events by Statin Therapy and Genetic Risk Group in WOSCOPS.
Nonfatal myocardial infarction or death from coronary heart disease rate by randomized treatment group and polygenic risk group in the WOSCOPS trial. Absolute events (and percentage) per individuals in each group is shown at the bottom of the bars. This represents 604 events over 64,031 total patient-years of follow up. The follow up period was 4.8 years (SD 0.7 years) within the trial for both placebo and statin groups, and out of trial was 8.1 years (SD 3.4 years) in the placebo group and 8.4 years (SD 3.0 years) in the statin-treated group.
Table 3.
Coronary heart disease event rates by genetic risk and treatment allocation
| Trial / Polygenic risk score subgroup | Placebo
|
Statin Treated
|
ARR (%) | NNT | ||||
|---|---|---|---|---|---|---|---|---|
| Events (n) | Individuals (n) | Event rate (%) | Events (n) | Individuals (n) | Event rate (%) | |||
| WOSCOPS (604 events, 8.1 years of follow-up) | ||||||||
| All Others | 248 | 1,929 | 12.9% | 201 | 1,981 | 10.1% | 2.7% | 38 |
| High | 100 | 511 | 19.6% | 55 | 471 | 11.7% | 7.9% | 13 |
| JUPITER (108 events, 2.4 years of follow-up) | ||||||||
| All Others | 53 | 3,486 | 1.5% | 35 | 3,483 | 1.0% | 0.5% | 200 |
| High | 14 | 864 | 1.6% | 6 | 878 | 0.7% | 0.9% | 112 |
| ASCOT-LLA (149 events, 6.1 years of follow-up) | ||||||||
| All Others | 61 | 1,619 | 3.8% | 45 | 1,756 | 2.6% | 1.2% | 84 |
| High | 28 | 426 | 6.6% | 15 | 418 | 3.6% | 3.0% | 34 |
ARR = absolute risk reduction; NNT = number needed to treat with statin to prevent one event.
JUPITER and ASCOT-LLA data from Mega J*, Stitziel NO*, et al. Lancet. 20155 *Denotes equal contribution
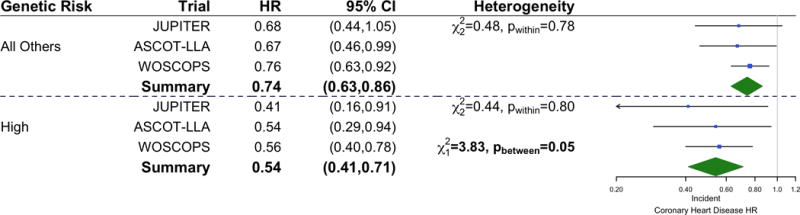
We performed a meta-analysis combining the results of this study with those published earlier from JUPITER and ASCOT-LLA (5). With all three studies combined, statin therapy reduced risk for a first CHD event by 46% (HR 0.54; 95% CI, 0.41–0.71; P <0.001) whereas statin therapy reduced risk by 26% among all others (HR 0.74; 95% CI, 0.63–0.86; P <0.001) (P for heterogeneity = 0.05) (Figure 2). Across all three studies, the absolute risk reduction with statin therapy was 3.6% (95% CI, 2.0%–5.1%) among those in the high genetic risk group and was 1.3% (95% CI, 0.6%–1.9%) in all others. This translates to a NNT to prevent one coronary event of 28 (95% CI, 20–50) in the high genetic risk score group and of 80 (95% CI, 52–175) in all others.
Figure 2. Forest Plot of Incident Coronary Heart Disease After Statin Therapy by Genetic Risk Group in Statin Primary Prevention Trials.
The multi-variable adjusted hazard ratios of incident coronary heart disease after statin therapy by genetic risk group are presented for three primary prevention trials. Data from JUPITER and ASCOT-LLA were obtained from prior analyses.5 Fixed effects meta-analysis was used to estimate the relative effect of statin therapy on incident coronary heart disease across trials for each genetic risk group (P for difference = 0.05). CI = confidence interval; HR = hazard ratio.
We tested if individuals at high genetic risk for CHD were more predisposed to developing subclinical atherosclerosis. Baseline characteristics of the CARDIA Study (ages 32–47 years at the time of CAC ascertainment) and the BioImage Study (ages 55–80 years) are presented in Supplementary Table 6. In CARDIA, for every standard deviation increase in polygenic risk score, the multi-variable adjusted odds ratio for CAC presence was 1.32 (95% CI, 1.04–1.68; P = 0.02) (Table 4).
Table 4.
CAC burden by polygenic risk score quintile in CARDIA
| Polygenic risk score quintile | CAC > 0 % |
CAC > 0 OR (95% CI, P) * |
|---|---|---|
| Q1 | 8.7 | 1 |
| Q2 | 12.1 | 2.08 (0.89–4.83, P =0.09) |
| Q3 | 10.9 | 2.08 (0.87–4.98, P =0.10) |
| Q4 | 14.3 | 3.02 (1.31–7.00, P =0.01) |
| Q5 (High) | 15.6 | 2.51 (1.08–5.85, P =0.04) |
Relative to Q1. Adjusted for age, sex, diabetes mellitus status, smoking status, LDL cholesterol, HDL cholesterol, systolic blood pressure, antihypertensive medication status, and family history of CHD
CAC = coronary artery calcification; CHD = coronary heart disease; CI = confidence interval HR = hazard ratio; PCE = Pooled Cohorts Equation; PRS = polygenic risk score; Q = quintile
In BioImage, for every standard deviation increase in polygenic risk score, there was a 9.7% increase (95% CI, 2.2% to 17.8%; P =0.01) in carotid artery plaque burden. The median carotid plaque burden among those at high genetic risk was 215 mm2 (IQR, 52 mm2 to 618 mm2) compared to 208 mm2 (IQR: 39 mm2 to 581 mm2) among all other participants (P = 0.02) (Supplementary Table 7, Supplementary Table 8). Unlike this expanded 57-SNP score, we did not observe an association of a prior restricted 27-SNP score5 with carotid artery plaque burden (Supplementary Table 9).
DISCUSSION
Among men with hyperlipidemia enrolled in a randomized controlled trial of primary prevention of CHD, statin therapy conferred greater relative benefit among those at high genetic risk when compared with all others. Relative risk reduction with statin therapy was 46% in those at high genetic risk and 26% among all others; this greater relative benefit was seen despite similar levels of LDL lowering by statin therapy in the high genetic risk subgroup compared to all others. Additionally, an expanded 57-SNP score was associated with subclinical atherosclerosis in two vascular beds.
These results permit several conclusions. First, on average, prior trials have shown that degree of LDL cholesterol lowering linearly associates with degree of coronary event risk reduction;22, 23 however, our data suggests that statins might confer greater relative risk reduction in one subgroup - those at high genetic risk. Across three primary prevention trials, those at high genetic risk have a nearly three-fold lower number needed to treat to prevent one CHD event. In those at high genetic risk, the lower number needed to treat to prevent one CHD event is driven by both an elevated baseline rate of events (1.6-fold greater) as well as a greater relative risk reduction of events from statin therapy.
Large-scale genetic association analyses have expanded the number of SNPs associated with CHD.14, 16 Compared to initial reports,24, 25 polygenic risk scores using expanded sets of SNPs show improved discrimination for incident CHD events.17, 26 We now show that, in the setting of hyperlipidemia, the 57-SNP score remains associated with incident CHD in those with or without a self-reported family history of CHD. Furthermore, although the 57-SNP score does not associate LDL cholesterol level or extent of LDL cholesterol lowering from statins, those with the highest scores still are more likely to experience clinical benefit among those with at least moderate hyperlipidemia.
Second, young and middle-aged asymptomatic individuals at high genetic risk for CHD have a greater burden of subclinical atherosclerosis. An increased number of CHD variants is linked to subclinical atherosclerosis in two vascular beds even after accounting for traditional cardiovascular risk factors. We recently demonstrated a step-wise increase in CAC among middle-aged asymptomatic adults in BioImage.27 We now extend these findings to a low-risk young cohort of essentially statin-ineligible individuals.
CAC and carotid plaque are both strong predictors of CHD events independent of traditional risk factors.12, 28, 29 Subclinical atherosclerosis is a highly heritable trait.30, 31 We and others showed that the genetic architectures of CHD and subclinical coronary atherosclerosis are highly concordant.32–35 Non-coding genomic variants at 9p21 and 6p24 are strongly associated with both CAC and CHD but do not appear to be associated with traditional risk factors.14, 32, 35 Furthermore, CHD polygenic risk score is strongly associated with CAC in both a cohort with the presence of traditional risk factors (BioImage)27 and a younger cohort with a paucity of traditional risk factors (CARDIA). This indicates that lifelong exposure to CHD risk alleles predisposes to both the development of subclinical and clinical atherosclerosis. The association with subclinical atherosclerosis burden may highlight a potential reason why those at high genetic risk derive enhanced clinical benefit from primary preventive statin therapy. Further study is required to compare genetic versus atherosclerosis imaging markers to refine decision-making for initiation of primary preventive therapy with statins.
Third, increased CHD risk conferred by genetics seems to be modifiable. We recently showed that adherence to a healthy lifestyle27 can modify high genetic risk and now, demonstrate that statin therapy may modify risk as well. Overall, these data may contribute to the conversation regarding statin eligibility in the primary prevention setting. Currently, statin eligibility is determined based on an estimation of absolute 10-year risk from demographic and clinical parameters. Age remains the key determinant of cardiovascular risk estimation.36 High genetic risk may identify statin candidates to prevent a first myocardial infarction event who otherwise would not have been considered by clinical criteria, a hypothesis that can be tested in more contemporary cohorts with sizeable proportions of statin-ineligible patients. Furthermore, disclosure of genetic risk may motivate greater adherence with statin therapy.37
Our analyses have potential limitations. First, the entry criteria in the three randomized controlled trials were different with varying follow-up times. However, there was no significant heterogeneity in effect estimates in the genetic risk groups across clinical trials. Second, our analyses were performed on individuals of European ancestry. The genetic determinants of CHD and their effects on statin benefit in other ancestries may be different.38, 39. Third, in the WOSCOPS trial, we included events beyond trial cessation. However, likely crossover occurring after the termination of the trial would bias results to the null. Finally, the polygenic risk score captures common genetic variation, but about 1 in 200 individuals are affected by a monogenic disease, namely familial hypercholesterolemia, that markedly increases risk for CHD.40, 41 The clinical utility of a polygenic risk score in those with familial hypercholesterolemia is uncertain.
A key goal of precision medicine is to identify subsets of individuals more likely to have clinical benefit from preventive strategies. We show that a 57-SNP polygenic risk score for CHD can identify individuals 1) at higher risk for developing a coronary event, 2) more likely to experience clinical benefit from preventive statin therapy, and 3) with a greater burden of subclinical atherosclerosis.
Supplementary Material
CLINICAL PERSPECTIVE.
What is New?
A recent analysis of primary prevention statin trials surprisingly suggested those at high genetic risk for coronary heart disease (CHD) derive greater relative benefit from statin therapy.
We now developed an expanded genetic risk score for CHD with 57 SNPs to identify individuals at high genetic risk.
We now show in an independent study (WOSCOPS) statin therapy was associated with a relative risk reduction of 44% for CHD among those at high genetic risk versus 24% among all others.
Additionally, we observe that those at high genetic risk have an increased burden of atherosclerosis in both coronary and carotid arteries.
What Are the Clinical Implications?
Stratifying by genetic risk may identify a subset of adults who have a greater burden of subclinical atherosclerosis and derive the greatest benefit from statin therapy to prevent a first CHD event.
Acknowledgments
We thank the investigators and participants in the WOSCOPS, CARDIA, and BioImage studies for their contributions to this study.
Funding Sources
Dr. Natarajan is supported by the John S. LaDue Memorial Fellowship from Harvard Medical School. Dr. Kathiresan is supported by an Ofer and Shelly Nemirovsky Research Scholar Award from Massachusetts General Hospital and grants from the NIH (HL127564 and UM1HG008895)
Footnotes
Twitter handles: Pradeep Natarajan, MD MMSc: @pnatarajanmd, Sekar Kathiresan, MD: @skathire
Disclosures
Dr. Natarajan reports consulting fees from Amarin Corporation. Dr. Reilly is an employee of Merck & Co. Dr. Kathiresan reports grant support from Regeneron, grant support and personal fees from Bayer Healthcare, scientific advisory board of Regeneron Genetics Center, Merck, Celera, Genomics PLC, Novartis, Sanofi, AstraZeneca, Alnylam, Eli Lilly, Leerink Partners, Noble Insights, and Ionis, and Catabasis, and equity in Catabasis and San Therapeutics.
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA, 3rd, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cholesterol Treatment Trialists C. Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC, Jr, Watson K, Wilson PW. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S1–S45. doi: 10.1161/01.cir.0000437738.63853.7a. [DOI] [PubMed] [Google Scholar]
- 4.Cholesterol Treatment Trialists C. Fulcher J, O’Connell R, Voysey M, Emberson J, Blackwell L, Mihaylova B, Simes J, Collins R, Kirby A, Colhoun H, Braunwald E, La Rosa J, Pedersen TR, Tonkin A, Davis B, Sleight P, Franzosi MG, Baigent C, Keech A. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385:1397–1405. doi: 10.1016/S0140-6736(14)61368-4. [DOI] [PubMed] [Google Scholar]
- 5.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ, Nordio F, Hyde CL, Cannon CP, Sacks FM, Poulter NR, Sever PS, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271. doi: 10.1016/S0140-6736(14)61730-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sever PS, Dahlof B, Poulter NR, Wedel H, Beevers G, Caulfield M, Collins R, Kjeldsen SE, Kristinsson A, McInnes GT, Mehlsen J, Nieminen M, O’Brien E, Ostergren J, investigators A Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–1158. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- 7.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ, Group JS Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 8.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 9.A coronary primary prevention study of Scottish men aged 45–64 years: trial design. The West of Scotland Coronary Prevention Study Group. J Clin Epidemiol. 1992;45:849–860. doi: 10.1016/0895-4356(92)90068-x. [DOI] [PubMed] [Google Scholar]
- 10.Hughes GH, Cutter G, Donahue R, Friedman GD, Hulley S, Hunkeler E, Jacobs DR, Jr, Liu K, Orden S, Pirie P. Recruitment in the Coronary Artery Disease Risk Development in Young Adults (Cardia) Study. Control Clin Trials. 1987;8:68S–73S. doi: 10.1016/0197-2456(87)90008-0. [DOI] [PubMed] [Google Scholar]
- 11.Yan LL, Liu K, Daviglus ML, Colangelo LA, Kiefe CI, Sidney S, Matthews KA, Greenland P. Education, 15-year risk factor progression, and coronary artery calcium in young adulthood and early middle age: the Coronary Artery Risk Development in Young Adults study. JAMA. 2006;295:1793–1800. doi: 10.1001/jama.295.15.1793. [DOI] [PubMed] [Google Scholar]
- 12.Baber U, Mehran R, Sartori S, Schoos MM, Sillesen H, Muntendam P, Garcia MJ, Gregson J, Pocock S, Falk E, Fuster V. Prevalence, Impact, and Predictive Value of Detecting Subclinical Coronary and Carotid Atherosclerosis in Asymptomatic Adults: The BioImage Study. J Am Coll Cardiol. 2015;65:1065–1074. doi: 10.1016/j.jacc.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Natarajan P, Kohli P, Baber U, Nguyen KD, Sartori S, Reilly DF, Mehran R, Muntendam P, Fuster V, Rader DJ, Kathiresan S. Association of APOC3 Loss-of-Function Mutations With Plasma Lipids and Subclinical Atherosclerosis: The Multi-Ethnic BioImage Study. J Am Coll Cardiol. 2015;66:2053–2055. doi: 10.1016/j.jacc.2015.08.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.CARDIoGRAMplusC4D. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CARDIoGRAMplusC4D. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, Konig IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikainen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, Consortium D, Consortium C. Doney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Muller-Nurasyid M, Mu TC, Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schafer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, van der Schoot CE, Wagner PJ, Wellcome Trust Case Control C. Wells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrieres J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kahonen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee JY, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Tregouet DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen T, Syvanen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimaki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O’Donnell C, Reilly MP, Marz W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stitziel NO, Stirrups K, Masca NGD, Erdmann J, Ferrario PG, Konig IR, Weeke PE, Webb TR, Auer PL, Schick UM, Lu Y, Zhang H, Dube MP, Goel A, Farrall M, Peloso GM, Won HH, Do R, Van Iperen E, Kanoni S, Kruppa J, Mahajan A, Scott RA, Willenborg C, Braund PS, JC VC, Doney AS, Donnelly LA, Asselta R, Merlini PA, Duga S, Marziliano N, Denny JC, Shaffer CM, El Mokhtari NE, Franke A, Gottesman O, Heilmann S, Hengstenberg C, Hoffmann P, Holmen OL, Hveem K, Jansson JH, Jockel KH, Kessler T, Kriebel J, Laugwitz KL, Marouli E, Martinelli N, McCarthy MI, Van Zuydam NR, Meisinger C, Esko T, Mihailov E, Escher SA, Alver M, Moebus S, Morris AD, Muller-Nurasyid M, Nikpay M, Olivieri O, Perreault LPL, AlQarawi A, Robertson NR, Akinsaya KO, Reilly DF, Vogt TF, Yin W, Asselbergs FW, Kooperberg C, Jackson RD, Stahl E, Strauch K, Varga TV, Waldenberger M, Zeng L, Kraja AT, Liu C, Ehret GB, Newton-Cheh C, Chasman DI, Chowdhury R, Ferrario M, Ford I, Jukema JW, Kee F, Kuulasmaa K, Nordestgaard BG, Perola M, Saleheen D, Sattar N, Surendran P, Tregouet DA, Young R, Howson JMM, Butterworth AS, Danesh J, Ardissino D, Bottinger EP, Erbel R, Franks PW, Girelli D, Hall AS, Hovingh GK, Kastrati A, Lieb W, Meitinger T, Kraus WE, Shah SH, McPherson R, Orho-Melander M, Melander O, Metspalu A, Palmer CNA, Peters A, Rader DJ, Reilly MP, Loos RJ, Reiner AP, Roden DM, Tardif JC, Thompson JR, Wareham NJ, Watkins H, Willer CJ, Kathiresan S, Deloukas P, Samani NJ, Schunkert H. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med. 2016;374:1134–1144. doi: 10.1056/NEJMoa1507652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, Devlin JJ, Kathiresan S, Shiffman D. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J. 2016;37:561–567. doi: 10.1093/eurheartj/ehv462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, Burtt NP, Fuchsberger C, Li Y, Erdmann J, Frayling TM, Heid IM, Jackson AU, Johnson T, Kilpelainen TO, Lindgren CM, Morris AP, Prokopenko I, Randall JC, Saxena R, Soranzo N, Speliotes EK, Teslovich TM, Wheeler E, Maguire J, Parkin M, Potter S, Rayner NW, Robertson N, Stirrups K, Winckler W, Sanna S, Mulas A, Nagaraja R, Cucca F, Barroso I, Deloukas P, Loos RJ, Kathiresan S, Munroe PB, Newton-Cheh C, Pfeufer A, Samani NJ, Schunkert H, Hirschhorn JN, Altshuler D, McCarthy MI, Abecasis GR, Boehnke M. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD, Hansen M, Borecki IB, Cupples LA, Fornage M, Gudnason V, Harris TB, Kathiresan S, Kraaij R, Launer LJ, Levy D, Liu Y, Mosley T, Peloso GM, Psaty BM, Rich SS, Rivadeneira F, Siscovick DS, Smith AV, Uitterlinden A, van Duijn CM, Wilson JG, O’Donnell CJ, Rotter JI, Boerwinkle E. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS One. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M, Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990;15:827–832. doi: 10.1016/0735-1097(90)90282-t. [DOI] [PubMed] [Google Scholar]
- 21.Sillesen H, Muntendam P, Adourian A, Entrekin R, Garcia M, Falk E, Fuster V. Carotid plaque burden as a measure of subclinical atherosclerosis: comparison with other tests for subclinical arterial disease in the High Risk Plaque BioImage study. JACC Cardiovasc Imaging. 2012;5:681–689. doi: 10.1016/j.jcmg.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 22.Cholesterol Treatment Trialists C. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM, Investigators I-I Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372:2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 24.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton-Cheh C, Orho-Melander M. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 25.Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376:1393–1400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, Yetukuri L, Tikkanen E, Perola M, Schunkert H, Sijbrands EJ, Palotie A, Samani NJ, Salomaa V, Ripatti S, Inouye M. Genomic prediction of coronary heart disease. Eur Heart J. 2016;37:3267–3278. doi: 10.1093/eurheartj/ehw450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, Chasman DI, Baber U, Mehran R, Rader DJ, Fuster V, Boerwinkle E, Melander O, Orho-Melander M, Ridker PM, Kathiresan S. Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med. 2016;375:2349–2358. doi: 10.1056/NEJMoa1605086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, O’Leary DH, Tracy R, Watson K, Wong ND, Kronmal RA. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336–1345. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 29.McClelland RL, Jorgensen NW, Budoff M, Blaha MJ, Post WS, Kronmal RA, Bild DE, Shea S, Liu K, Watson KE, Folsom AR, Khera A, Ayers C, Mahabadi AA, Lehmann N, Jockel KH, Moebus S, Carr JJ, Erbel R, Burke GL. 10-Year Coronary Heart Disease Risk Prediction Using Coronary Artery Calcium and Traditional Risk Factors: Derivation in the MESA (Multi-Ethnic Study of Atherosclerosis) With Validation in the HNR (Heinz Nixdorf Recall) Study and the DHS (Dallas Heart Study) J Am Coll Cardiol. 2015;66:1643–1653. doi: 10.1016/j.jacc.2015.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bis JC, Kavousi M, Franceschini N, Isaacs A, Abecasis GR, Schminke U, Post WS, Smith AV, Cupples LA, Markus HS, Schmidt R, Huffman JE, Lehtimaki T, Baumert J, Munzel T, Heckbert SR, Dehghan A, North K, Oostra B, Bevan S, Stoegerer EM, Hayward C, Raitakari O, Meisinger C, Schillert A, Sanna S, Volzke H, Cheng YC, Thorsson B, Fox CS, Rice K, Rivadeneira F, Nambi V, Halperin E, Petrovic KE, Peltonen L, Wichmann HE, Schnabel RB, Dorr M, Parsa A, Aspelund T, Demissie S, Kathiresan S, Reilly MP, Taylor K, Uitterlinden A, Couper DJ, Sitzer M, Kahonen M, Illig T, Wild PS, Orru M, Ludemann J, Shuldiner AR, Eiriksdottir G, White CC, Rotter JI, Hofman A, Seissler J, Zeller T, Usala G, Ernst F, Launer LJ, D’Agostino RB, Sr, O’Leary DH, Ballantyne C, Thiery J, Ziegler A, Lakatta EG, Chilukoti RK, Harris TB, Wolf PA, Psaty BM, Polak JF, Li X, Rathmann W, Uda M, Boerwinkle E, Klopp N, Schmidt H, Wilson JF, Viikari J, Koenig W, Blankenberg S, Newman AB, Witteman J, Heiss G, Duijn C, Scuteri A, Homuth G, Mitchell BD, Gudnason V, O’Donnell CJ. Meta-analysis of genome-wide association studies from the CHARGE consortium. Nat Genet. 2011;43:940–947. doi: 10.1038/ng.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peyser PA, Bielak LF, Chu JS, Turner ST, Ellsworth DL, Boerwinkle E, Sheedy PF., 2nd Heritability of coronary artery calcium quantity measured by electron beam computed tomography in asymptomatic adults. Circulation. 2002;106:304–308. doi: 10.1161/01.cir.0000022664.21832.5d. [DOI] [PubMed] [Google Scholar]
- 32.Natarajan P, Bis JC, Bielak LF, Cox AJ, Dorr M, Feitosa MF, Franceschini N, Guo X, Hwang SJ, Isaacs A, Jhun MA, Kavousi M, Li-Gao R, Lyytikainen LP, Marioni RE, Schminke U, Stitziel NO, Tada H, van Setten J, Smith AV, Vojinovic D, Yanek LR, Yao J, Yerges-Armstrong LM, Amin N, Baber U, Borecki IB, Carr JJ, Chen YI, Cupples LA, de Jong PA, de Koning H, de Vos BD, Demirkan A, Fuster V, Franco OH, Goodarzi MO, Harris TB, Heckbert SR, Heiss G, Hoffmann U, Hofman A, Isgum I, Jukema JW, Kahonen M, Kardia SL, Kral BG, Launer LJ, Massaro J, Mehran R, Mitchell BD, Mosley TH, Jr, de Mutsert R, Newman AB, Nguyen KD, North KE, O’Connell JR, Oudkerk M, Pankow JS, Peloso GM, Post W, Province MA, Raffield LM, Raitakari OT, Reilly DF, Rivadeneira F, Rosendaal F, Sartori S, Taylor KD, Teumer A, Trompet S, Turner ST, Uitterlinden AG, Vaidya D, van der Lugt A, Volker U, Wardlaw JM, Wassel CL, Weiss S, Wojczynski MK, Becker DM, Becker LC, Boerwinkle E, Bowden DW, Deary IJ, Dehghan A, Felix SB, Gudnason V, Lehtimaki T, Mathias R, Mook-Kanamori DO, Psaty BM, Rader DJ, Rotter JI, Wilson JG, van Duijn CM, Volzke H, Kathiresan S, Peyser PA, O’Donnell CJ, Consortium C Multiethnic Exome-Wide Association Study of Subclinical Atherosclerosis. Circ Cardiovasc Genet. 2016;9:511–520. doi: 10.1161/CIRCGENETICS.116.001572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA, Levy D, D’Agostino RB, Hwang SJ, O’Donnell CJ. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet. 2012;5:113–121. doi: 10.1161/CIRCGENETICS.111.961342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Donnell CJ, Cupples LA, D’Agostino RB, Fox CS, Hoffmann U, Hwang SJ, Ingellson E, Liu C, Murabito JM, Polak JF, Wolf PA, Demissie S. Genome-wide association study for subclinical atherosclerosis in major arterial. BMC Med Genet. 2007;8(Suppl 1):S4. doi: 10.1186/1471-2350-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Donnell CJ, Kavousi M, Smith AV, Kardia SL, Feitosa MF, Hwang SJ, Sun YV, Province MA, Aspelund T, Dehghan A, Hoffmann U, Bielak LF, Zhang Q, Eiriksdottir G, van Duijn CM, Fox CS, de Andrade M, Kraja AT, Sigurdsson S, Elias-Smale SE, Murabito JM, Launer LJ, van der Lugt A, Kathiresan S, Krestin GP, Herrington DM, Howard TD, Liu Y, Post W, Mitchell BD, O’Connell JR, Shen H, Shuldiner AR, Altshuler D, Elosua R, Salomaa V, Schwartz SM, Siscovick DS, Voight BF, Bis JC, Glazer NL, Psaty BM, Boerwinkle E, Heiss G, Blankenberg S, Zeller T, Wild PS, Schnabel RB, Schillert A, Ziegler A, Munzel TF, White CC, Rotter JI, Nalls M, Oudkerk M, Johnson AD, Newman AB, Uitterlinden AG, Massaro JM, Cunningham J, Harris TB, Hofman A, Peyser PA, Borecki IB, Cupples LA, Gudnason V, Witteman JC. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124:2855–2864. doi: 10.1161/CIRCULATIONAHA.110.974899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karmali KN, Goff DC, Jr, Ning H, Lloyd-Jones DM. A systematic examination of the 2013 ACC/AHA pooled cohort risk assessment tool for atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64:959–968. doi: 10.1016/j.jacc.2014.06.1186. [DOI] [PubMed] [Google Scholar]
- 37.Kullo IJ, Jouni H, Austin EE, Brown SA, Kruisselbrink TM, Isseh IN, Haddad RA, Marroush TS, Shameer K, Olson JE, Broeckel U, Green RC, Schaid DJ, Montori VM, Bailey KR. Incorporating a Genetic Risk Score Into Coronary Heart Disease Risk Estimates: Effect on Low-Density Lipoprotein Cholesterol Levels (the MI-GENES Clinical Trial) Circulation. 2016;133:1181–1188. doi: 10.1161/CIRCULATIONAHA.115.020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lettre G, Palmer CD, Young T, Ejebe KG, Allayee H, Benjamin EJ, Bennett F, Bowden DW, Chakravarti A, Dreisbach A, Farlow DN, Folsom AR, Fornage M, Forrester T, Fox E, Haiman CA, Hartiala J, Harris TB, Hazen SL, Heckbert SR, Henderson BE, Hirschhorn JN, Keating BJ, Kritchevsky SB, Larkin E, Li M, Rudock ME, McKenzie CA, Meigs JB, Meng YA, Mosley TH, Newman AB, Newton-Cheh CH, Paltoo DN, Papanicolaou GJ, Patterson N, Post WS, Psaty BM, Qasim AN, Qu L, Rader DJ, Redline S, Reilly MP, Reiner AP, Rich SS, Rotter JI, Liu Y, Shrader P, Siscovick DS, Tang WH, Taylor HA, Tracy RP, Vasan RS, Waters KM, Wilks R, Wilson JG, Fabsitz RR, Gabriel SB, Kathiresan S, Boerwinkle E. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. 2011;7:e1001300. doi: 10.1371/journal.pgen.1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wojczynski MK, Li M, Bielak LF, Kerr KF, Reiner AP, Wong ND, Yanek LR, Qu L, White CC, Lange LA, Ferguson JF, He J, Young T, Mosley TH, Smith JA, Kral BG, Guo X, Wong Q, Ganesh SK, Heckbert SR, Griswold ME, O’Leary DH, Budoff M, Carr JJ, Taylor HA, Jr, Bluemke DA, Demissie S, Hwang SJ, Paltoo DN, Polak JF, Psaty BM, Becker DM, Province MA, Post WS, O’Donnell CJ, Wilson JG, Harris TB, Kavousi M, Cupples LA, Rotter JI, Fornage M, Becker LC, Peyser PA, Borecki IB, Reilly MP. Genetics of coronary artery calcification among African Americans, a meta-analysis. BMC Med Genet. 2013;14:75. doi: 10.1186/1471-2350-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, Guella I, Asselta R, Lange LA, Peloso GM, Auer PL, Project NES, Girelli D, Martinelli N, Farlow DN, DePristo MA, Roberts R, Stewart AF, Saleheen D, Danesh J, Epstein SE, Sivapalaratnam S, Hovingh GK, Kastelein JJ, Samani NJ, Schunkert H, Erdmann J, Shah SH, Kraus WE, Davies R, Nikpay M, Johansen CT, Wang J, Hegele RA, Hechter E, Marz W, Kleber ME, Huang J, Johnson AD, Li M, Burke GL, Gross M, Liu Y, Assimes TL, Heiss G, Lange EM, Folsom AR, Taylor HA, Olivieri O, Hamsten A, Clarke R, Reilly DF, Yin W, Rivas MA, Donnelly P, Rossouw JE, Psaty BM, Herrington DM, Wilson JG, Rich SS, Bamshad MJ, Tracy RP, Cupples LA, Rader DJ, Reilly MP, Spertus JA, Cresci S, Hartiala J, Tang WH, Hazen SL, Allayee H, Reiner AP, Carlson CS, Kooperberg C, Jackson RD, Boerwinkle E, Lander ES, Schwartz SM, Siscovick DS, McPherson R, Tybjaerg-Hansen A, Abecasis GR, Watkins H, Nickerson DA, Ardissino D, Sunyaev SR, O’Donnell CJ, Altshuler D, Gabriel S, Kathiresan S. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–106. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S, Kathiresan S. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. doi: 10.1016/j.jacc.2016.03.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.