

Abstract

Preclinical evidence in support of the potential utility of mGlu5 NAMs for the treatment of a variety of psychiatric and neurodegenerative disorders is extensive, and multiple such molecules have entered clinical trials. Despite some promising results from clinical studies, no small molecule mGlu5 NAM has yet to reach market. Here we present the discovery and evaluation of N-(5-fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (27, VU0424238), a compound selected for clinical evaluation. Compound 27 is more than 900-fold selective for mGlu5 versus the other mGlu receptors, and binding studies established a Ki value of 4.4 nM at a known allosteric binding site. Compound 27 had a clearance of 19.3 and 15.5 mL/min/kg in rats and cynomolgus monkeys, respectively. Imaging studies using a known mGlu5 PET ligand demonstrated 50% receptor occupancy at an oral dose of 0.8 mg/kg in rats and an intravenous dose of 0.06 mg/kg in baboons.
Introduction
The metabotropic glutamate (mGlu) receptors are an eight-membered family of class C G protein-coupled receptors (GPCRs) that are activated by l-glutamic acid, the major excitatory neurotransmitter of the mammalian central nervous system (CNS). Further stratification of the mGlu receptors is made based on a variety of considerations, including structure, downstream signaling partners, and pharmacology. Whereas group I mGlu receptors (mGlu1 and mGlu5) are found in post-synaptic locations and couple via Gq to the activation of phospholipase C (PLC), both group II (mGlu2–3) and group III (mGlu4 and mGlu6–8) receptors are found mainly in presynaptic locations and couple via Gi/Go to the inhibition of adenylyl cylase activity. Common to all mGlu receptors are a seven transmembrane (7TM) α-helical domain that connects to a large bilobed extracellular amino-terminal domain, sometimes termed the “venus fly trap (VFT)” domain. Located within this VFT domain is the binding site for the native ligand; however, allosteric binding sites for small molecules have thus far been confined to the transmembrane domain. The structural and functional properties of the mGlu receptors have been reviewed extensively.1,2
In the search for compounds that are sufficiently selective for individual mGlu receptor subtypes, we have found allosteric modulation to be a successful approach. In fact, in many cases, such a strategy can be employed for the design of both positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) of the same receptor.3 Among the most actively investigated and advanced areas within this field is that of mGlu5 NAMs.4,5 Potential therapeutic applications for small molecule mGlu5 NAMs are extensive and include addiction,6 Alzheimer’s disease (AD),7 anxiety,8 autism spectrum disorder (ASD),9 fragile X syndrome (FXS),10 the levodopa-induced dyskinesia (LID) experienced by many patients with Parkinson’s disease (PD),11,12 major depressive disorder (MDD),13 and obsessive-compulsive disorder (OCD).14 Much of the preclinical work that has served to establish mGlu5 antagonism as an exciting new mechanism for targeting these various conditions has been conducted with two related tool compounds, methyl-6-(phenylethynyl)-pyridine (1, MPEP)15 and 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (2, MTEP)16 (Figure 1). Both compounds are structurally similar disubstituted alkynes, a motif that has been exploited for the design of several other highly optimized mGlu5 NAM compounds.4,5 While results from clinical studies have been reported with nonalkyne based mGlu5 NAMs such as 3 (fenobam),17,184 (AZD9272),19,20 and 5 (AZD2066),19,20 the most highly studied and advanced clinical compounds are three alkyne mGlu5 NAMs: 6 (mavoglurant),14,21−257 (basimglurant),26−29 and 8 (dipraglurant)30,31 (Figure 1). Still, failures to reach primary clinical end points with 6(14,22,23) and 7(28) point to the continued need for highly optimized mGlu5 NAMs. It should be noted that a recent report from Pfizer has described cutaneous toxicities in cynomolgus monkey toxicology studies with three structurally related nonalkyne based mGlu5 NAMs.32 Given that other mGlu5 NAMs that are not structurally related to the Pfizer compounds have been used in long-term, repeat dose studies in nonhuman primates (NHPs)33,34 and humans14,22−24,28 without notes of a similar significant toxicity, it seems reasonable that such effects are not endemic to the mechanism. Still, these results make a compelling argument for using cynomolgus monkeys as the nonrodent species for toxicology studies in the event that our conclusion is incorrect.
Figure 1.

Prototypical mGlu5 NAM preclinical tools 1 and 2 and clinically investigated mGlu5 NAMs 3–8.
Alkynes are potentially reactive functional groups. Pfizer has reported an instance of biliary epithelial hyperplasia in a NHP toxicology study with an alkyne-based mGlu5 NAM developed by Wyeth.35 This toxicity was believed to be linked to the reactivity of the alkyne based on metabolic studies that revealed extensive glutathione conjugation.35 While this type of biotransformation does not always manifest in humans with alkyne-containing compounds,25,27 many research groups have sought to design novel mGlu5 NAMs that are outside of this chemotype.4,5 We have actively pursued a variety of approaches toward that end over the past decade.36,37 For example, we conducted a high-throughput screen (HTS) of a collection of 160,000 compounds that identified multiple distinct mGlu5 NAM hits. This same platform functions as our primary assay for lead optimization by measuring the ability of novel compounds to block the mobilization of calcium induced by an EC80 concentration of glutamate in HEK293A cells that express rat mGlu5.
Results and Discussion
Initial Analogs
Recently, we described the discovery of an in vivo tool compound 9 (VU0409106)37−40 that resulted from the optimization of one of these HTS hits (Table 1). That report focused exclusively on compounds with a benzamide core;37 however, we were also interested in exploring picolinamide analogs as well (10–13, Table 1). Moving from the 3-fluorophenyl benzamide core (9) to an unsubstituted picolinamide core (10) reduced potency by approximately 8-fold; however, substitution of the picolinamide core at the 6-position with a methyl group (11) improved potency. In addition, structure–activity relationships (SARs) established in the benzamide series37 translated to the picolinamides in the form of enhanced potency with the 5-fluoropyridin-3-yl ether (12). Once again, 6-methyl substitution of the picolinamide core (13) improved potency relative to the unsubstituted analog 12. Buoyed by the encouraging results from this limited set of analogs and in search of a compound that represented a step forward from 9 to a compound worthy of advancement to clinical development, we devised a plan to prepare additional 6-alkyl-picolinamide analogs. Herein, we report the successful execution of that strategy.
Table 1. Initial Picolinamide Core Analogs of 9.

| no. | mGlu5 pIC50 (±SEM)a | mGlu5 IC50 (nM)a | % Glu maxa,b |
|---|---|---|---|
| 9 | 7.62 ± 0.03 | 24 | 1.2 ± 0.1 |
| 10 | 6.70 ± 0.15 | 200 | 1.2 ± 0.1 |
| 11 | 7.01 ± 0.09 | 98 | 1.3 ± 0.1 |
| 12 | 7.22 ± 0.23 | 60 | 1.4 ± 0.1 |
| 13 | 8.01 ± 0.16 | 10 | 1.3 ± 0.1 |
Calcium mobilization mGlu5 assay; values are an average of n ≥ 3 independent experiments.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); an average of n ≥ 3 independent experiments.
Synthesis of Compounds
The synthetic routes to the picolinamide analogs largely mirror those described for the benzamide mGlu5 NAMs.37 Briefly, 4-nitropyridine-N-oxides 14 were refluxed in concentrated hydrochloric acid to afford 4-chloropyridine-N-oxides 16 (Scheme 1). Alternatively, 4-chloropyridines 15 were oxidized with a hydrogen peroxide-urea adduct in the presence of trifluoroacetic acid to provide 16. Treatment of 16 with trimethylsilyl cyanide and dimethylcarbamyl chloride generated 2-cyanopyridines 17. A nucleophilic aromatic substitution reaction between heteroaryl alcohols 18 and 17 was employed to generate ether products 19. Hydrolysis of the nitrile moieties of 19 was accomplished via thermal heating with aqueous sodium hydroxide solution to give the penultimate intermediate acids 20. Coupling with the appropriate primary heteroaryl amines (H2NR4) generated the target analogs.
Scheme 1. Synthesis of Picolinamide Analogs 10–13 and 21–37.

Reagents and conditions: R1 = H, Me, CHF2; R2 = H, Me, C3H5; A = N, CH, CF, CCl; R4 = 4-methylthiazol-2-yl or substituted pyridin-2-yl; (a) conc. HCl, reflux, 53–75%; (b) H2O2·urea, (F3CCO)2O, THF, 0 °C to rt, 44–99%; (c) Me3SiCN, CH2Cl2, Me2NCOCl, 75–86%; (d) 18, K2CO3, DMF, 80 °C, 45–77%; (e) 2 N aq. NaOH, dioxane, reflux, 82–99%; (f) H2NR4, POCl3, pyridine, −15 °C, 22–81%.
Initial SAR
Even though compound 9 is a useful tool for acute behavioral studies in mice and rats,37,39 it has several properties that prevent its further development. In particular, 9 has moderate to high clearance in rats (Clp 43 mL/min/kg) and cynomolgus monkeys (Clp 24 mL/min/kg).40 In addition, 9 has poor bioavailability following oral administration in rats (F < 5%) and is an inhibitor of CYP1A2 (IC50 < 100 nM). Evaluation of the picolinamide analogs of 9 indicated that the 4-methylthiazol-2-yl amide contributed substantially to the potent inhibition of CYP1A2 (e.g., 10 CYP1A2 IC50 < 100 nM), and SAR for mGlu5 activity within the benzamide series showed limited tolerance for even subtle modifications on the thiazol-2-yl moiety.37 Fortunately, analogs with a pyridin-2-yl replacement for the thiazol-2-yl ring appeared more amenable to substitution, and initial work in the picolinamide series was carried out in that context while taking advantage of SAR knowledge gained from the benzamide work (Table 2).
Table 2. Substituted 2-Pyridyl Amine Analogs.

| protein
binding (fu)c |
P450 inhibition
IC50 (μM)d |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| no. | series | R | mGlu5 pIC50 (±SEM)a | mGlu5 IC50 (nM)a | % Glu maxa,b | rat | human | 3A4 | 2D6 | 2C9 | 1A2 |
| 21 | A | 6-Me | 7.70 ± 0.04 | 20 | 1.4 ± 0.1 | 0.076 | 0.066 | >30 | >30 | 4.1 | 2.4 |
| 22 | B | 6-Me | 8.04 ± 0.12 | 9.1 | 1.4 ± 0.1 | 0.028 | 0.016 | >30 | >30 | 1.3 | 0.3 |
| 23 | A | 6-Et | 8.03 ± 0.07 | 9.3 | 1.5 ± 0.2 | 0.027 | 0.034 | >30 | >30 | 1.8 | 0.9 |
| 24 | B | 6-Et | 7.49 ± 0.31 | 32 | 1.2 ± 0.1 | 0.010 | 0.005 | ||||
| 25 | A | 6-F | 7.94 ± 0.21 | 11 | 1.3 ± 0.1 | NDe | 0.132 | >30 | >30 | 1.8 | 0.6 |
| 26 | B | 6-F | 8.45 ± 0.11 | 3.6 | 1.4 ± 0.1 | NDe | 0.023 | 8.0 | >30 | 1.8 | 0.1 |
| 27 | A | 5-F | 7.97 ± 0.05 | 11 | 1.6 ± 0.1 | 0.106 | 0.120 | >30 | >30 | 12.1 | 1.0 |
| 28 | B | 5-F | 8.51 ± 0.04 | 3.1 | 1.2 ± 0.1 | 0.015 | 0.011 | 4.3 | >30 | 5.4 | 0.2 |
| 29 | A | 5-Cl | 8.17 ± 0.14 | 6.8 | 1.3 ± 0.1 | 0.067 | 0.049 | 16.4 | >30 | 11.0 | 0.9 |
| 30 | B | 5-Cl | 8.02 ± 0.16 | 9.6 | 1.3 ± 0.1 | 2.0 | >30 | >30 | <0.1 | ||
| 31 | A | 4-F | 7.27 ± 0.22 | 54 | 1.6 ± 0.3 | >30 | >30 | >30 | 6.0 | ||
| 32 | B | 4-F | 7.72 ± 0.13 | 19 | 1.3 ± 0.2 | 27.4 | >30 | 5.7 | 0.4 | ||
Calcium mobilization mGlu5 assay; values are an average of n ≥ 3 independent experiments.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); an average of n ≥ 3 independent experiments.
fu = fraction unbound; equilibrium dialysis assay.
Assayed in pooled HLM in the presence of NADPH with CYP-specific probe substrates.
Compound was unstable in rat plasma.
As expected, the majority of these pyridin-2-yl analogs (21–32) proved to be potent antagonists of mGlu5 on par with their thiazol-2-yl analogs (11 and 13). In a majority of instances, analogs with the 5-fluoropyridin-3-yl ether exhibited slightly superior mGlu5 potency compared to the pyrimidin-5-yl ether analogs; however, the 6-ethylpyridin-2-yl analogs 23 and 24 and 5-chloropyridin-2-yl derivatives 29 and 30 were an exception to this trend. Assays that measured the degree of binding to rat and human plasma and a human liver microsomes (HLM) cocktail assay with probe substrates for four common P450s41 were used to profile several selected compounds. SAR with regard to the heteroaryl ether group was clear, as the pyrimidin-5-yl ether (Series A) analogs were consistently less bound to plasma than their 5-fluoropyridin-3-yl ether (Series B) analogs. No activity at 30 μM test compound was noted with regard to inhibition of CYP2D6, and inhibition of CYP3A4 and CYP2C9 ranged considerably. As anticipated, the most consistently potent P450 inhibition was observed with CYP1A2; however, there was another clear SAR with respect to the heteroaryl ether group. In this case, the pyrimidin-5-yl ether (Series A) analogs were less potent CYP1A2 inhibitors than the 5-fluoropyridin-3-yl ether (Series B) analogs.
Moving our attention to other portions of the chemotype, we prepared further analogs that revealed additional SAR. Modifications made in the context of the 5-fluoropyridin-2-yl amide found in analogs 27 and 28 are depicted here (Table 3). Installation of a methyl group at the 2-position of the pyrimidin-5-yl ether (33) resulted in a 3-fold loss in potency relative to 27. While this modification increased the extent of binding to plasma, it also reduced inhibition versus CYP1A2 and CYP2C9. Moving to a larger cyclopropyl group at the 2-position of the pyrimidin-5-yl ether (34) substantially eroded potency at mGlu5, indicating minimal tolerance for steric bulk at this position. Likewise, replacement of the 6-methyl group on the picolinamide core with a difluoromethyl group (35) led to an approximately 5-fold loss in mGlu5 potency, illustrating little tolerance for modification at that position. Replacement of the 5-fluoro group of the pyridin-3-yl of 28 with a 5-chloro group (36) resulted in only a slight drop in potency. Replacement of the meta-halogen atoms found in 28 and 36 with the para-methyl group in 6-methylpyridin-3-yl ether 37 reduced mGlu5 potency by 50- and 20-fold, respectively.
Table 3. Analogs of Compound 27 and 28.

| protein
binding (fu)c |
P450 inhibition
IC50 (μM)d |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| no. | mGlu5 pIC50 (±SEM)a | mGlu5 IC50 (nM)a | % Glu maxa,b | rat | human | 3A4 | 2D6 | 2C9 | 1A2 |
| 27 | 7.97 ± 0.05 | 11 | 1.6 ± 0.1 | 0.106 | 0.120 | >30 | >30 | 12.1 | 1.0 |
| 33 | 7.48 ± 0.04 | 33 | 1.5 ± 0.2 | 0.081 | 0.074 | >30 | >30 | >30 | >30 |
| 34 | 6.60 ± 0.25 | 251 | 1.5 ± 0.1 | ||||||
| 35 | 7.32 ± 0.02 | 48 | 1.3 ± 0.1 | ||||||
| 28 | 8.51 ± 0.04 | 3.1 | 1.2 ± 0.1 | 0.015 | 0.011 | 4.3 | >30 | 5.4 | 0.2 |
| 36 | 8.14 ± 0.27 | 7.2 | 1.0 ± 0.1 | 0.058 | 0.014 | 3.8 | >30 | 21.5 | 0.2 |
| 37 | 6.80 ± 0.11 | 158 | 1.2 ± 0.1 | ||||||
Calcium mobilization mGlu5 assay; values are an average of n ≥ 3 independent experiments.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); an average of n ≥ 3 independent experiments.
fu = fraction unbound; equilibrium dialysis assay.
Assayed in pooled HLM in the presence of NADPH with CYP-specific probe substrates
In Vivo Pharmacokinetics (PK) Studies
Previous studies in our laboratories with pyrimidin-5-yl ethers identified this ring as a substrate for aldehyde oxidase (AO)-mediated metabolism.38,40 Thus, given the likelihood that several of our interesting new compounds would be subject to non-P450-mediated metabolism, we decided to move directly into an in vivo setting to evaluate the clearance of a variety of select analogs (Table 4). In these studies, the three analogs with the 5-fluoropyridin-2-yl amide group (27, 28, and 33) exhibited superior profiles compared to the other compounds that contained pyridin-2-yl amides with 6-alkyl (21–23) or 6-fluoro (26) groups. The plasma clearance of 26 was greater than hepatic blood flow,42 which suggests that the instability previously observed in rat plasma for this compound (Table 2) is not merely an in vitro phenomenon. Blocking the 2-position of pyrimidin-5-yl ether (33) reduced clearance and lengthened mean residence time relative to unsubstituted analog 27. Such a result points toward this position as one potential site for metabolism, which is consistent with prior studies on related analog 9.38,40 Compounds 27, 28, and 33 were subsequently evaluated in tissue distribution studies (Table 5) using oral dosing. Acceptable CNS penetration was achieved with all three compounds, and unbound brain to unbound plasma ratios (Kp,uu) were near unity with 27 and 28, indicating distribution equilibrium between the compartments and a low probability of these two compounds being substrates for transporters in rats.43 The Kp,uu observed for 33 raises the possibility of active transport with this compound; however, additional studies would be required to conclusively determine if that was indeed the case. All compounds achieved unbound brain levels in excess of their respective mGlu5 functional potency at the dose used in these experiments (10 mg/kg). Compounds 27 and 28 were particularly attractive from this perspective, reaching unbound brain levels of 18- and 10-fold over their functional potency, respectively.
Table 4. Preliminary Rat PK Results Using Intravenous (IV) Dosinga.
| no. | mGlu5 IC50 (nM)b | t1/2 (min)c | MRT (min)c | Clp (mL/min/kg)c | VSS (L/kg)c |
|---|---|---|---|---|---|
| 21d | 20 | 105 | 39 | 34.2 | 1.6 |
| 22e | 9.1 | 268 | 120 | 32.5 | 4.5 |
| 23f | 9.3 | 404 | 80 | 43.7 | 6.6 |
| 26d | 3.6 | 82 | 157 | 96 | 11 |
| 27d | 11 | 390 | 93 | 19.3 | 1.8 |
| 28d | 3.1 | 281 | 208 | 9.2 | 1.9 |
| 33f | 33 | 229 | 191 | 6.2 | 1.2 |
Average n = 2, male Sprague–Dawley rats; dose = 1.0 mg/kg.
Calcium mobilization mGlu5 assay; values are an average of n ≥ 3 independent experiments.
t1/2 = Terminal phase plasma half-life; MRT = mean residence time; Clp = plasma clearance; VSS = volume of distribution at steady-state.
Vehicle = 10% EtOH, 70% PEG 400, 20% saline.
Vehicle = 10% EtOH, 60% PEG 400, 30% saline.
Vehicle = 10% EtOH, 50% PEG 400, 40% saline.
Table 5. Tissue Distribution Studies Using Oral Dosinga.
| protein
binding (fu)c |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no. | mGlu5 IC50 (nM)b | plasma | brain | total plasma (nM) | total brain (nM) | unbound plasma (nM) | unbound brain (nM) | Kpd | Kp,uue | unbound brain/mGlu5 IC50 |
| 27 | 11 | 0.106 | 0.078 | 3000 | 2580 | 318 | 201 | 0.86 | 0.63 | 18 |
| 28 | 3.1 | 0.015 | 0.011 | 2570 | 2870 | 38.6 | 31.6 | 1.1 | 0.82 | 10 |
| 33 | 33 | 0.081 | 0.044 | 4820 | 3220 | 390 | 142 | 0.67 | 0.36 | 4.3 |
Average n = 2, male Sprague–Dawley rats; dose = 10 mg/kg; vehicle = 10% polysorbate 80 in 0.5% methyl cellulose; t = 1 h post-dose.
Calcium mobilization mGlu5 assay; values are an average of n ≥ 3 independent experiments.
fu = Fraction unbound; equilibrium dialysis assay; brain = mouse brain homogenates.
Kp = total brain to total plasma ratio.
Kp,uu = unbound brain (brain fu × total brain) to unbound plasma (plasma fu × total plasma) ratio.
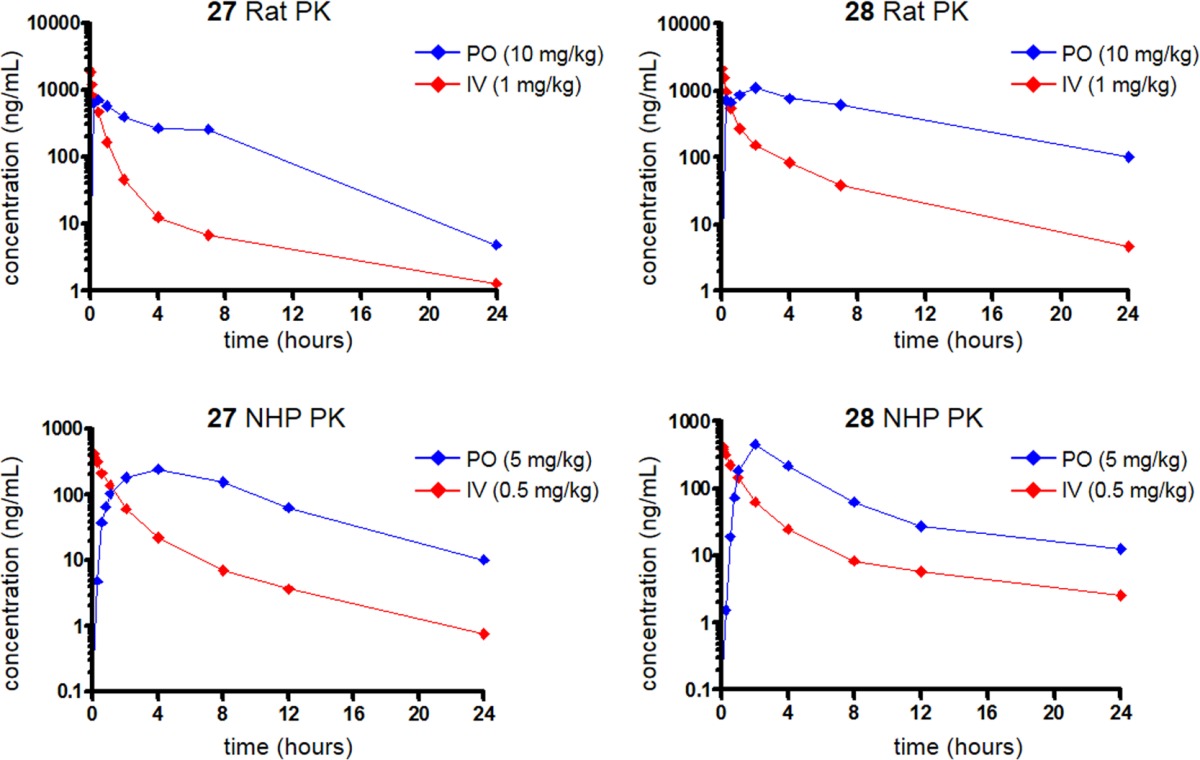
Considering their favorable profiles, compounds 27 and 28 were selected for oral pharmacokinetic and bioavailability studies in rats and cynomolgus monkeys (Table 6). Cynomolgus monkeys were chosen as a second preclinical species as it has been established that AO activity varies across species. Specifically, AO activity is highest in humans and NHPs, moderate in rats, and undetectable in dogs.44 Using in vitro methods described previously for 9,38,40 we confirmed that AO contributed significantly to the metabolism of 27 in rats, cynomolgus monkey, and human hepatic S9 fractions (Figure 2). Oral exposure was significant as bioavailability exceeded 50% with both compounds in rats. Compound 27 exhibited an earlier and slightly higher plasma Cmax than 28; however, the lower clearance of 28 resulted in prolonged exposure and larger area under the curve (AUC) than observed for 27. The PK results from analogous studies in cynomolgus monkeys showed these compounds to be similar in that both compounds showed a clearance of approximately one-third of hepatic blood flow.42 Bioavailability, while lower than that observed in rats, was still >30% in each case. With largely attractive in vivo PK profiles, both compounds were advanced into further studies to identify additional factors for differentiation.
Table 6. Rat and Cynomolgus Monkey Pharmacokinetics of Compounds 27 and 28a.
| rat IV
PKb |
rat oral
PKb |
|||||||
|---|---|---|---|---|---|---|---|---|
| no. | t1/2 (min)c | MRT (min)c | Clp (mL/min/kg)c | VSS (L/kg)c | Tmax (min)d | Cmax (μM)d | AUC (μM·h)d | % Fd |
| 27 | 390 | 93 | 19.3 | 1.8 | 30 | 4.34 | 14.6 | 53 |
| 28 | 281 | 208 | 9.2 | 1.9 | 90 | 3.33 | 38.0 | 71 |
| cynomolgus
monkey IV PKe |
cynomolgus monkey oral PKe |
|||||||
|---|---|---|---|---|---|---|---|---|
| no. | t1/2 (min)c | MRT (min)c | Clp (mL/min/kg)c | VSS (L/kg)c | Tmax (min)d | Cmax (μM)d | AUC (μM·h)d | % Fd |
| 27 | 302 | 163 | 15.5 | 2.5 | 180 | 1.72 | 7.32 | 44 |
| 28 | 563 | 303 | 13.3 | 4.0 | 120 | 1.36 | 6.43 | 35 |
Oral vehicle = 10% polysorbate 80 in 0.5% methyl cellulose; IV vehicle = 10% EtOH, 70% PEG 400, 20% saline.
Average n = 2 male Sprague–Dawley rats.
t1/2 = Terminal phase plasma half-life; MRT = mean residence time; Clp = plasma clearance; VSS = volume of distribution at steady-state.
Tmax = time at which maximum concentration occurs; Cmax = maximum concentration; AUC = area under the curve; F = bioavailability.
Average n = 2 male cynomolgus monkeys.
Figure 2.

Metabolite (38) of 27 identified in rat, cynomolgus monkey, and human hepatic S9 fractions that is produced predominately by AO.
Ancillary Pharmacology
To judge the selectivity of these analogs versus the other members of the mGlu receptor family, the effects of both 27 and 28 at 10 μM on the orthosteric agonist concentration response curve (CRC) were measured in fold-shift experiments.45,46 No significant effects on the agonist CRC were noted with either compound, indicating excellent selectivity for mGlu5. To assess potential ancillary pharmacology, both compounds were screened at 10 μM in a commercial radioligand binding assay panel of 68 diverse and clinically relevant targets (see Supporting Information).47 While 27 had no significant responses at any target, where significant responses are defined as inhibition of more than 50% of radioligand binding, one significant response of 86% inhibition was noted for 28 at the dopamine transporter (DAT). For comparison, 27 only inhibited DAT at a level of 34%. A subsequent CRC was obtained, and 28 was found to inhibit radioligand binding with an IC50 of 1.54 μM. Since inhibition of DAT is associated with abuse potential,48 we next evaluated 28 in a functional cell-based assay that measured inhibition of dopamine uptake.49 Indeed, consistent with results obtained from the binding assay, 28 exhibited an IC50 of 1.38 μM in this assay. While 28 is more than 400-fold selective for mGlu5 versus DAT, the lack of concern regarding this target in the case of 27 was viewed as a positive for that compound. With its preferable P450 inhibition profile (Table 2) and the higher unbound brain concentrations achieved in rats also in its favor, the decision was made to proceed with additional detailed characterization of 27.
Detailed Characterization of 27
Having narrowed our interest to analog 27, we carried out a more thorough characterization of its interaction with mGlu5 (Figure 3). First, to ensure that there was no appreciable species difference in the mGlu5 activity of 27, we carried out our calcium mobilization assay in HEK293A cells expressing the human receptor (Figure 3A). As anticipated, the potency difference between rat (IC50 = 11 nM) and human mGlu5 (IC50 = 14 nM) was negligible. To study the effects of 27 on mGlu5 activity in a native system, experiments in rat cortical astrocytes were conducted (Figure 3B). As expected, 27 induced a concentration-dependent rightward shift of the glutamate CRC and decreased the maximal response to glutamate. To investigate whether 27 interacted with the mGlu5 allosteric site that is common to many mGlu5 NAMs such as 1 and 2 (Figure 1), we also carried out a competition radioligand binding assay with [3H]-3-methoxy-5-(pyridin-2-ylethynyl)pyridine (39)50 (Figure 3C) in membranes prepared from HEK293A cells expressing rat mGlu5. This study confirmed the interaction of 27 with this known binding site (mGlu5Ki = 4.4 nM). We also performed saturation binding experiments with increasing concentrations of [3H]-38 in the presence and absence of multiple concentrations of 27 (Figure 3D,E). Increasing concentrations of 27 had no effect on the Bmax of the radioligand but increased the Kd as shown (Figure 3D), a finding that suggests a competitive interaction between 27 and [3H]-39. Scatchard analysis (Figure 3E) of the results plotting bound/free versus bound ligand demonstrated that addition of 27 induced a change in slope of the regression line (change in Kd) but had no effect on the x-intercept (no change in Bmax). These data are consistent with 27 competitively displacing [3H]-39 at the aforementioned known allosteric binding site. A final set of radioligand kinetic binding experiments was conducted that examined the effect of 27 on the dissociation of [3H]-38. In these studies, neither concentration (300 nM or 100 nM) of 27 had any effect on the dissociation rate of the radioligand (data not shown), a result consistent with prior studies and a competitive interaction. Lastly, we examined the effects of increasing concentrations of the mGlu5 neutral site ligand 5-methyl-6-(phenylethynyl)-pyridine (40)51 on the calcium mobilization CRC of 27 in HEK293A cells expressing rat mGlu5 (Figure 3F). In these experiments, 40 induced a parallel rightward shift in the 27 concentration response relationship, which is consistent with 40 competing for the known allosteric binding site with 27. Furthermore, a Schild analysis (data not shown) of the effects of 40 on the 27 concentration response yielded a linear regression with a slope of 0.96, indicating the relationship between the two compounds was competitive. Extrapolation of the line to the x-intercept established a Ki of approximately 240 nM, a result consistent with the Ki value for 40 at the [3H]-39 site as described in the literature (388 nM).51 Each of these studies combine to conclusively establish that 27 binds in a competitive manner to the aforementioned known mGlu5 allosteric binding site.
Figure 3.

(A) Ca2+ mobilization assay CRC for 27 in HEK293A cells expressing human mGlu5. (B) Effects of 27 on glutamate CRC in rat cortical astrocytes. (C) Competition radioligand binding CRC with 27 and [3H]-3-methoxy-5-(pyridin-2-ylethynyl)pyridine (39). (D) Saturation binding experiment: increasing concentrations of [3H]-39 in the presence and absence of multiple concentrations of 27. (E) Saturation binding experiment: Scatchard analysis of bound/free versus bound ligand. (F) Effects of the neutral mGlu5 site ligand 5-methyl-6-(phenylethynyl)-pyridine (40) on the functional inhibition of glutamate responses induced by 27.
With a detailed understanding of the pharmacology of 27 in hand, we moved the compound into a host of additional assays designed to identify potential risks for clinical development (Table 7). Permeability and potential for P-glycoprotein (P-gp)-mediated efflux was assessed in both Madin–Darby canine kidney (MDCK) cells transfected with the human MDR1 gene52 and human colon carcinoma derived Caco-2 cells.53 Permeability was moderate, and efflux ratios were near unity in both cell lines, indicating a low potential for transporter-mediated efflux. A definitive and expanded assessment of the ability of 27 to reversibly inhibit the metabolism of isoform-selective probe substrates of P450s was conducted. Results were generally consistent with the preliminary results (Table 2) with moderate inhibition of CYP1A2 observed. In parallel to these studies, the potential for 27 to time-dependently inhibit each P450 was assessed with no evidence of time-dependent inhibition noted in any case. To shed further light on the likely human PK profile of 27, the stability of the compound was assessed in human cryopreserved hepatocytes, a system which is potentially useful for evaluating compounds with non-P450-mediated routes of metabolism such as AO.54 Gratifyingly, the predicted hepatic clearance in humans using this method was low with a half-life of more than 5 h. To assess any potential cardiac liabilities, 27 was subjected to a commercial ion channel panel.55 Fortunately, no significant risks were identified in this panel. Finally, the potential mutagenic activity of 27 was investigated in the bacterial reverse mutation assay (Ames assay),56 and no mutagenic responses were noted in any of the five strains either in the presence or absence of S9 metabolic activation. Encouraged by the overall profile of 27, we advanced this compound to behavioral assays in the rat.
Table 7. Further in Vitro Characterization of 27.
| permeability assays | |||
|---|---|---|---|
| cell line | Papp (A-B) (cm/s) | Papp (B-A) (cm/s) | efflux ratio |
| MDCK-MDR1 | 6.2 × 10–6 | 7.2 × 10–6 | 1.2 |
| Caco-2 | 6.5 × 10–6 | 7.9 × 10–6 | 1.2 |
| definitive P450 inhibition assaysa | ||
|---|---|---|
| P450 | probe | IC50 (μM) |
| CYP3A4 | midazolam | 133 ± 23 |
| CYP3A4 | testosterone | >200 |
| CYP2D6 | (R)-bufuralol | >200 |
| CYP2C9 | diclofenac | 29.9 ± 7.3 |
| CYP2C19 | (S)-mephenytoin | 106 ± 10 |
| CYP2C8 | amodiaquine | >200 |
| CYP2B6 | efavirenz | >200 |
| CYP1A2 | phenacetin | 2.90 ± 0.72 |
| stability in human cryopreserved hepatocytes | |
|---|---|
| intrinsic clearance (CLINT) | 1.43 μL/min/million cells |
| half-life (t1/2) | 322 min |
| predicted hepatic clearance (CLHEP) | 3.59 mL/min/kg |
| cardiac
ion channel panelb | |||
|---|---|---|---|
| ion channel | IC50 (μM) | ion channel | IC50 (μM) |
| hERG | 73.6 | hCav1.2 | >100 |
| hNav1.5 (tonic) | >100 | hCav3.2 | >100 |
| hNav1.5 (phasic) | >100 | hKir2.1 | >100 |
| hKv4.3 | >100 | hHCN2 | >100 |
| hKv1.5 | >100 | hHCN4 | >100 |
| bacterial
reverse mutation assayc | ||||
|---|---|---|---|---|
| TA98 | TA100 | WP2 uvrA | TA135 | TA1537 |
| negative | negative | negative | negative | negative |
Assayed in pooled HLM (with and without NADPH) using CYP-specific probe substrates.
Evaluated using IonWorks Quattro system.
With and without S9 metabolic activation.
Behavioral Pharmacology
It is well-known that mice will bury foreign objects such as glass marbles in deep bedding. Likewise, pretreatment with low doses of benzodiazepines and multiple mGlu5 NAM tool compounds have been shown to inhibit this behavior.57 It has been argued recently that the marble burying assay models the repetitive and perseverative behavior associated with OCD as opposed to novelty-induced anxiety.58 We have also employed this rapid assay that utilizes naïve mice as a frontline behavioral screen for our discovery programs directed toward mGlu5 NAMs.36,37 Using intraperitoneal (IP) dosing as a convenient route for rapidly and accurately dosing large numbers of animals, we examined the effects of 27 in this test (Figure 4A). Dose-dependent effects on marble burying were observed, and all doses ≥0.3 mg/kg produced significant effects relative to the vehicle control. The 3.0 mg/kg dose produced an effect on marble burying on par with the positive control 2 (15 mg/kg) in this study. A satellite experiment was conducted using the 3.0 mg/kg dose, and brain samples were collected and analyzed for exposure of 27. At 15 min post-dose of 27, which correlates to the initiation time point of the marble burying study, the total brain concentration of 27 was 1.48 μM. Calculation of the unbound brain concentration using equilibrium dialysis data (Table 5) yields a level of 115 nM, which is approximately 10-fold more than the functional mGlu5 activity.
Figure 4.

(A) Dose-dependent inhibition of marble burying with 27; vehicle = 10% polysorbate 80 (IP); male CD-1 mice; n = 10 per treatment group; 15 min pretreatment; 30 min burying time; *, p < 0.05 versus vehicle control group, Dunnett’s test; bars denote marbles buried. (B) Dose-dependent inhibition of immobility in rats with 27; vehicle = 10% polysorbate 80 (27, intraperitoneal) and sterile saline (ketamine, subcutaneous); male Sprague–Dawley rats; n = 8–10 per treatment group; 30 min pretreatment; 6 min testing session; *, p < 0.05 versus vehicle control group, Dunnett’s test; bars denote duration of immobility in s.
Having observed efficacy in this model of anxiety/OCD, we next studied 27 in an animal model of depression as well. The forced swim test (FST) measures the duration of immobility observed in rats suspended in a tank of water from which they cannot escape.59 Many clinical antidepressants are able to reduce the time that the animals are immobile while increasing the time that they are active. Furthermore, certain mGlu5 NAMs have also shown efficacy in this model of depression.60 Examination of 27 in this assay showed a dose-dependent ability of the compound to reduce the duration of immobility indicative of antidepressant activity (Figure 4B). This result compares favorably to the literature report for 7 in the same assay, where that compound had a minimum efficacious dose of 10 mg/kg (PO).60 The high dose used in this study (30 mg/kg IP) produced an efficacy equivalent to that of the positive control, ketamine, a known antagonist of the N-methyl-d-aspartate (NMDA) receptor. Though ketamine is an older drug and produces undesirable psychotomimetic side effects, more recent clinical studies have shown that it can induce a rapid antidepressant effect in patients with treatment-resistant depression (TRD).61 A report from last year implicated the activation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor by a specific ketamine metabolite rather than direct antagonism of the NMDA receptor by ketamine as the potential source of these rapid antidepressant effects.62 Interestingly, prior studies have shown that the antidepressant activity in the FST with mGlu5 NAM 2 involved signaling through the NMDA but not the AMPA receptor.63 Thus, the mechanisms by which ketamine and mGlu5 NAMs illicit their antidepressant effects may well be distinct.
Imaging Studies
Recognizing that an ability to study receptor occupancy (RO) in the clinic would represent a potential advantage for advancing 27, we decided to investigate RO studies in preclinical species. [18F]-3-Fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile ([18F]-FPEB, [18F]-41)64 is a close structural analog of radioligand [3H]-38 that has been used both preclinically39 and in the clinic64 as a positron emission topography (PET) ligand for studying mGlu5 RO. Thus, we decided to carry out [18F]-41 μPET studies with 27 in rats (Figure 5). In these studies, 27 was dosed orally 30 min prior to IV dosing of [18F]-41. PET imaging was initiated and continued for 1 h. Representative images demonstrating dose-dependent displacement of [18F]-41 by 27 are depicted in Figure 5A. Maximum occupancy of 80% was achieved at 10 mg/kg 27 (Figure 5B). Plasma samples were collected throughout the study. Plotting the maximum unbound plasma concentration of 27 versus RO resulted in an unbound level Occ50 of 13 nM (Figure 5C). Plotting the dose of 27 versus the RO normalized to the maximum displacement of [18F]-41 gave an RO50 of 0.8 mg/kg (Figure 5D).
Figure 5.

Imaging studies in rodents; n = 4–8 Sprague–Dawley rats; 27 vehicle = 10% polysorbate 80 in 0.5% methyl cellulose; 27 dosed 30 min prior to [18F]-41 (IV). (A) Representative images of [18F]-41 by increasing oral doses of 27. (B) mGlu5 RO obtained with [18F]-41 and increasing oral doses of 27. (C) Unbound maximum plasma concentration of 27 versus mGlu5 RO. (D) Dose of 27 versus mGlu5 RO; *normalized to the maximum displacement of [18F]-41.
Based on the RO results in rats, we next investigated whether similar results would be obtained in NHPs. As such, we advanced 27 into studies in baboons using the same radiotracer [18F]-41 (Figure 6). The design for this experiment was very similar to the aforementioned study in rats; however, in this case, both 27 and the radiotracer were delivered via IV administration. There was a clear reduction in the uptake of [18F]-41 relative to baseline that followed a dose-dependent relationship with 27 (Figure 6A,D,E). The maximum dose resulted in 89% RO of mGlu5. Plasma samples were again taken throughout the study to analyze 27 levels, and RO was plotted against the plasma maximum to arrive at a total plasma Occ50 of 56 nM (Figure 6B). If one assumes that human and baboon plasma protein binding is similar, then this equates to a plasma unbound Occ50 of 6.7 nM, which is within 2-fold of that observed in the rodent study. The effective IV dose that produces 50% RO (RO50) was determined to be 0.06 mg/kg (Figure 6C).
Figure 6.

Imaging studies in female baboons: subject A received three doses (0.01, 0.1, and 0.18 mg/kg); subject B received two doses (0.03 and 0.56 mg/kg); 27 vehicle = 10% ethanol, 30% PEG 400, 20% hydroxypropyl-β-cyclodextrin in water; 27 dosed over 20 min, 30 min prior to [18F]-41 (IV, 3 min bolus) for all doses except the 0.18 mg/kg dose, which was dosed over 2 min, 50 min prior to [18F]-41. (A) mGlu5 RO obtained in two female baboons with [18F]-41 and increasing IV doses of 27. (B) Total maximum plasma concentration of 27 versus mGlu5 RO. (C) Dose of 27 versus mGlu5 RO. (D) Images for subject A averaged from 0 to 150 min. (E) Images for subject B averaged from 0 to 180 min.
Conclusion
Continuation of our mGlu5 NAM discovery program that began with a cell-based HTS has culminated in the identification of the highly optimized compound 27 (VU0424238).65 Compound 27 is a highly potent and selective antagonist of mGlu5 with a favorable preclinical profile. The molecule has low-moderate clearance and good oral bioavailability in two species. Compound 27 displays CNS penetration in rodents, and we believe that the compound is a low risk for transporter-mediated efflux and drug–drug interactions. While the compound is a moderate inhibitor of CYP1A2, this aspect is considered manageable. Importantly, the compound is active in preclinical models of anxiety/OCD and depression, both of which are assays where mGlu5 NAMs have shown activity in the past. Moreover, a clinically useful radiotracer has been used to demonstrate mGlu5 RO by 27 in both rodents and NHPs. Based on the totality of data, we have selected 27 for advancement to clinical development including studies designed to enable an investigational new drug (IND) application. Here is the public disclosure of the chemical structure of 27, our candidate for clinical studies. Future publications describing detailed metabolic profiling, chemical process development and formulation, and toxicology studies are under preparation and will be communicated in the near future.
Experimental Section
Synthetic Procedures and Characterization Data
General
All NMR spectra were recorded on a 400 MHz AMX Bruker NMR spectrometer. 1H and 13C chemical shifts are reported in δ values in ppm downfield with the deuterated solvent as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, b = broad, m = multiplet), integration, coupling constant (Hz). High-resolution mass spectra were obtained on an Agilent 6540 UHD Q-TOF with ESI source. MS parameters were as follows: fragmentor: 150, capillary voltage: 3500 V, nebulizer pressure: 60 psig, drying gas flow: 13 L/min, drying gas temperature: 275 °C. Samples were introduced via an Agilent 1200 UHPLC comprised of a G4220A binary pump, G4226A ALS, G1316C TCC, and G4212A DAD with ULD flow cell. UV absorption was observed at 215 and 254 nm with a 4 nm bandwidth. Column: Agilent Zorbax Extend C18, 1.8 μm, 2.1 × 50 mm. Gradient conditions: 5–95% CH3CN in H2O (0.1% formic acid) over 1 min, hold at 95% CH3CN for 0.1 min, 0.5 mL/min, 40 °C.
Purity
Low-resolution reverse phase LCMS analysis was used to assess compound purity at two wavelengths: 215 and 254 nm. All analogs were at least 95% pure according to this analysis. Low-resolution reverse phase LCMS analysis was performed using an Agilent 1200 system comprising a binary pump with degasser, high-performance autosampler, thermostated column compartment, diode-array detector (DAD), and a C18 column. Flow from the column was split to a 6130 SQ mass spectrometer and Polymer Laboratories ELSD. The MS detector was configured with an electrospray ionization source. Data acquisition was performed with Agilent Chemstation and Analytical Studio Reviewer software. Method 1: Samples were separated on a ThermoFisher Accucore C18 column (2.6 μm, 2.1 × 30 mm) at 1.5 mL/min, with column and solvent temperatures maintained at 45 °C. The gradient conditions were 7–95% acetonitrile in water (0.1% TFA) over 1.4 min. Low-resolution mass spectra were acquired by scanning from 135 to 700 atomic mass units (AMU) in 0.25 s with a step size of 0.1 AMU and peak width of 0.03 min. Drying gas flow was 11 L/min at a temperature of 350 °C and a nebulizer pressure of 40 psi. The capillary needle voltage was 3000 V, and the fragmentor voltage was 100 V. Method 2: Samples were separated on a ThermoFisher Accucore C18 column (2.6 μm, 2.1 × 30 mm) at 1.5 mL/min, with column and solvent temperatures maintained at 45 °C. The gradient conditions were 7–95% acetonitrile in water (0.1% TFA) over 1.1 min. Low-resolution mass spectra were acquired by scanning from 135 to 700 AMU in 0.25 s with a step size of 0.1 AMU and peak width of 0.03 min. Drying gas flow was 11 L/min at a temperature of 350 °C and a nebulizer pressure of 40 psi. The capillary needle voltage was 3000 V, and the fragmentor voltage was 100 V. Method 3: Samples were separated on a Restek Aqueous C18 column (3 μm, 3.2 × 30 mm) at 1.25 mL/min, with column and solvent temperatures maintained at 45 °C. The gradient conditions were 10–100% acetonitrile in water (0.1% TFA) over 2 min. Low-resolution mass spectra were acquired by scanning from 100 to 1500 AMU in 0.25 s with a step size of 0.1 AMU and peak width of 0.03 min. Drying gas flow was 11 L/min at a temperature of 350 °C and a nebulizer pressure of 40 psi. The capillary needle voltage was 3000 V, and the fragmentor voltage was 70 V.
Preparation of Compound 27
4-Chloro-2-methylpyridine-N-oxide (16a)
2-methyl-4-nitropyridine 1-oxide 14a (5.0 g, 32 mmol, 1.0 equiv) was dissolved in concentrated HCl (80 mL) and refluxed for 3 days. The reaction was cooled, and the excess concentrated HCl was removed in vacuo. The viscous oil was neutralized with 10% K2CO3 and extracted with CH2Cl2 (5×). The combined organics were dried (MgSO4), filtered, and concentrated in vacuo. Purification by flash chromatography on silica gel afforded 3.49 g (75%) of the title compound as a yellow solid.
Alternative Procedure: 4-Chloro-2-methylpyridine 15a (5.0 g, 39 mmol, 1.0 equiv) and hydrogen peroxide-urea adduct (7.37 g, 78.4 mmol, 2.0 equiv) were dissolved in THF (196 mL) and cooled to 0 °C. Trifluoroacetic anhydride (12 mL, 86.3 mmol, 2.2 equiv) was added dropwise over 15 min, and the reaction was allowed to warm to room temperature. After determination of the completion of the reaction by LCMS (approximately 45 min), the reaction was cooled to 0 °C and quenched with a 10% aqueous solution of sodium thiosulfate. The reaction was extracted with EtOAc (3×), dried (MgSO4), filtered, and concentrated in vacuo. Purification by flash chromatography on silica gel afforded 5.63 g (99%) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, J = 7.0 Hz, 1H), 7.66 (d, J = 3.0 Hz, 1H), 7.40 (dd, J = 7.0, 3.0 Hz, 1H), 2.32 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.53, 139.78, 128.73, 126.41, 124.17, 17.09 ppm; LCMS (Method 1): RT = 0.302 min, m/z = 144.2 [M + H]+; HRMS, calcd for C6H6ClNO [M], 143.0138; found 143.0139.
4-Chloro-6-methylpicolinonitrile (17a)
Compound 16a (8.97 g, 62.5 mmol, 1.0 equiv) was dissolved in CH2Cl2 and dried over MgSO4. The solution was added to a flame-dried round-bottom flask, and CH2Cl2 was added to give a total volume of 188 mL. Trimethylsilyl cyanide (10 mL, 75 mmol, 1.2 equiv) was added, and the reaction stirred for 15 min. Dimethylcarbamyl chloride (6.9 mL, 75 mmol, 1.2 equiv) was added dropwise over 20 min, and the reaction was stirred for 24 h. An additional 1 equiv each of trimethylsilylcyanide and dimethylcarbamyl chloride were added, and the reaction was stirred for another 72 h. The reaction was made basic with 10% K2CO3 and extracted with CH2Cl2 (3×). The combined organics were dried (MgSO4), filtered, and concentrated in vacuo. Purification by flash chromatography on silica gel afforded 7.2 g (76%) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 1.6 Hz, 1H), 7.82 (d, J = 1.7 Hz, 1H), 2.52 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ = 161.99, 144.13, 133.01, 127.65, 126.40, 116.60, 23.57 ppm; LCMS (Method 1): RT = 0.715 min, m/z = 153.2 [M + H]+; HRMS, calcd for C7H5ClN2 [M], 152.0141; found 152.0139.
6-Methyl-4-(pyrimidin-5-yloxy)picolinonitrile (19a)
Compound 17a (4.0 g, 26 mmol, 1.0 equiv), 5-hydroxypyrimidine 18a (5.56 g, 57.9 mmol, 2.2 equiv), K2CO3 (7.24 mg, 52.4 mmol, 2.0 equiv), and DMF (66 mL) were added to a reaction vessel and heated at 80 °C for 16 h. The reaction was filtered and concentrated on silica gel (25 g). The silica gel was loaded on top of a fresh bed of silica gel and washed with 50% EtOAc/hexane. The solvents were removed in vacuo, and the crude solid was purified by flash chromatography on silica gel to afford 4.31 g (77%) of the title compound as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.85 (s, 2H), 7.74 (d, J = 2.4 Hz, 1H), 7.32 (d, J = 2.3 Hz, 1H), 2.48 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ = 164.04, 162.28, 155.51, 150.02, 148.75, 133.54, 117.05, 115.59, 115.04, 23.82 ppm; LCMS (Method 1): RT = 0.535 min, m/z = 213.2 [M + H]+; HRMS, calcd for C11H8N4O [M], 212.0698; found 212.0697.
6-Methyl-4-(pyrimidin-5-yloxy)picolinic acid (20a)
Compound 19a (4.31 g, 20.3 mmol, 1.0 equiv) was dissolved in dioxane (90 mL), and 2 N NaOH (45 mL) was added. The mixture was refluxed for 18 h, and after cooling, the reaction was neutralized with 2 N HCl (45 mL). The water and solvent were removed in vacuo, and the crude reaction was dissolved in 10% MeOH/CH2Cl2. The undissolved salt was filtered off, and the solvents were removed in vacuo to afford 4.65 g (99%) of the title compound as a white solid which was used without further purification.
N-(5-Fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (27)
Compound 20a (16.84 g, 72.83 mmol, 1.0 equiv) and 5-fluoro-2-aminopyridine (16.33 g, 0.15 mol, 2.0 equiv) were dissolved in pyridine (485 mL) in a flame-dried round-bottom flask. The reaction was cooled to −15 °C, and phosphorus oxychloride (7.47 mL, 80.14 mmol, 1.1 equiv) was added dropwise while keeping the temperature below −15 °C. After stirring for 30 min at −15 °C, the reaction was quenched with ice–water and neutralized with 10% K2CO3. The mixture was extracted with EtOAc (3×), and the combined organics were dried (MgSO4), filtered, and concentrated in vacuo. Purification by flash chromatography on silica gel afforded 14.3 g (60%) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 9.18 (s, 1H), 8.88 (s, 2H), 8.40 (d, J = 3.0 Hz, 1H), 8.27 (dd, J = 9.2 Hz, 4.10 Hz, 1H), 7.86 (td, J = 8.6, 3.1 Hz, 1H), 7.55 (d, J = 2.3 Hz, 1H), 7.29 (d, J = 2.3 Hz, 1H), 2.58 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ = 165.15, 161.11, 160.57, 156.12 (d, J(C,F) = 249.0 Hz), 155.41, 150.29, 150.07, 149.01, 146.92 (d, J(C,F) = 2.1 Hz), 135.94 (d, J(C,F) = 25.5 Hz), 125.83 (d, J(C,F) = 19.9 Hz), 114.56, 114.27 (d, J(C,F) = 4.7 Hz), 108.01, 23.84 ppm; LCMS (Method 2): RT = 0.727 min, m/z = 326.2 [M + H]+; HRMS, calcd for C16H12FN5O2 [M], 325.0975; found 325.0979.
N-(4-Methylthiazol-2-yl)-4-(pyrimidin-5-yloxy)picolinamide (10)
Prepared analogous to compound 27. LCMS (Method 3): RT = 1.147 min, m/z = 314.2 [M + H]+.
6-Methyl-N-(4-methylthiazol-2-yl)-4-(pyrimidin-5-yloxy)picolinamide (11)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.670 min, m/z = 328.2 [M + H]+.
4-((5-Fluoropyridin-3-yl)oxy)-N-(4-methylthiazol-2-yl)picolinamide (12)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.732 min, m/z = 331.1 [M + H]+.
4-((5-Fluoropyridin-3-yl)oxy)-6-methyl-N-(4-methylthiazol-2-yl)picolinamide (13)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.740 min, m/z = 345.1 [M + H]+.
6-Methyl-N-(6-methylpyridin-2-yl)-4-(pyrimidin-5-yloxy)picolinamide (21)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.653 min, m/z = 322.2 [M + H]+.
4-((5-Fluoropyridin-3-yl)oxy)-6-methyl-N-(6-methylpyridin-2-yl)picolinamide (22)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.730 min, m/z = 339.2 [M + H]+.
N-(6-Ethylpyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (23)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.624 min, m/z = 336.1 [M + H]+.
N-(6-Ethylpyridin-2-yl)-4-((5-fluoropyridin-3-yl)oxy)-6-methylpicolinamide (24)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.777 min, m/z = 353.2 [M + H]+.
N-(6-Fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (25)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.780 min, m/z = 326.1 [M + H]+.
N-(6-Fluoropyridin-2-yl)-4-((5-fluoropyridin-3-yl)oxy)-6-methylpicolinamide (26)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.840 min, m/z = 343.2 [M + H]+.
N-(5-Fluoropyridin-2-yl)-4-((5-fluoropyridin-3-yl)oxy)-6-methylpicolinamide (28)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.822 min, m/z = 343.1 [M + H]+.
N-(5-Chloropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (29)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.803 min, m/z = 342.1 [M + H]+.
N-(5-Chloropyridin-2-yl)-4-((5-fluoropyridin-3-yl)oxy)-6-methylpicolinamide (30)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.874 min, m/z = 359.1 [M + H]+.
N-(4-Fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (31)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.744 min, m/z = 326.2 [M + H]+.
N-(4-Fluoropyridin-2-yl)-4-((5-fluoropyridin-3-yl)oxy)-6-methylpicolinamide (32)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.822 min, m/z = 343.2 [M + H]+.
N-(5-Fluoropyridin-2-yl)-6-methyl-4-((2-methylpyrimidin-5-yl)oxy)picolinamide (33)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.776 min, m/z = 340.2 [M + H]+.
4-((2-Cyclopropylpyrimidin-5-yl)oxy)-N-(5-fluoropyridin-2-yl)-6-methylpicolinamide (34)
Prepared analogous to compound 27. LCMS (Method 1): RT = 0.937 min, m/z = 366.2 [M + H]+.
6-(Difluoromethyl)-N-(5-fluoropyridin-2-yl)-4-(pyrimidin-5-yloxy)picolinamide (35)
Prepared analogous to compound 27. LCMS (Method 2): RT = 0.765 min, m/z = 362.1 [M + H]+.
4-((5-Chloropyridin-3-yl)oxy)-N-(5-fluoropyridin-2-yl)-6-methylpicolinamide (36)
Prepared analogous to compound 27. LCMS (Method 1): RT = 0.942 min, m/z = 359.1 [M + H]+.
N-(5-Fluoropyridin-2-yl)-6-methyl-4-((6-methylpyridin-3-yl)oxy)picolinamide (37)
Prepared analogous to compound 27. LCMS (Method 1): RT = 0.639 min, m/z = 339.1 [M + H]+.
Acknowledgments
We gratefully acknowledge the generous support of the National Institute of Mental Health (U19 MH097056-01) (P.J.C.), the National Institute on Drug Abuse (R01 DA023947-01) (C.W.L.), Seaside Therapeutics (VUMC33842), and The William K. Warren Foundation for their support of our programs focused on the development of mGlu5 NAMs.
Glossary
Abbreviations Used
- AD
Alzheimer’s disease
- AMU
atomic mass units
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AO
aldehyde oxidase
- ASD
autism spectrum disorder
- AUC
area under the curve
- Bmax
maximum amount of radioligand which can bind specifically to the receptors in a membrane preparation
- Cmax
maximum concentration
- CLHEP
predicted hepatic clearance
- CLINT
intrinsic clearance
- CLp
plasma clearance
- CNS
central nervous system
- CRC
concentration response curve
- DAT
dopamine transporter
- DMF
N,N-dimethylformamide
- DMPK
drug metabolism and pharmacokinetics
- EC50
the concentration of agonist that affords a half-maximal response
- Et
ethyl
- EtOAc
ethyl acetate
- FPEB
3-fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile
- FST
forced swim test
- F
bioavailability
- fu
fraction unbound
- FXS
fragile X syndrome
- Glu
glutamate
- GPCR
G-protein-coupled receptors
- h
hours
- HEK
human embryonic kidney
- HLM
human liver microsomes
- HTS
high-throughput screen
- IC50
the concentration of antagonist at which 50% of receptor function or radioligand binding is inhibited
- IND
investigational new drug
- IP
intraperitoneal
- IV
intravenous
- Ki
the concentration of compound that will bind half of the binding sites at equilibrium
- Kp
brain to plasma ratio
- Kp,uu
unbound brain to unbound plasma ratio
- L
liters
- LID
levodopa-induced dyskinesia
- MDCK
Madin–Darby canine kidney
- MDD
major depressive disorder
- Me
methyl
- mGlu
metabotropic glutamate receptor
- MPEP
methyl-6-(phenylethynyl)-pyridine
- min
minutes
- MRT
mean residence time
- MTEP
3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced form)
- NAM
negative allosteric modulator
- NMDA
N-methyl-d-aspartate
- NHP
nonhuman primate
- Occ50
concentration of drug that produces 50% receptor brain occupancy
- OCD
obsessive-compulsive disorder
- PAM
positive allosteric modulator
- PD
Parkinson’s disease
- PEG
polyethylene glycol
- PET
positron emission tomography
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- RO
receptor occupancy
- RO50
dose of compound that results in 50% receptor occupancy
- s
seconds
- SAR
structure–activity relationship
- SEM
standard error of the mean
- THF
tetrahydrofuran
- TRD
treatment-resistant depression
- Tmax
time at which maximum concentration of test compound occurs
- t1/2
half-life
- VSS
volume of distribution at steady-state
- VFT
venus fly trap
- 7TM
seven transmembrane
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00410.
Author Present Address
# Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, 3500 Camp Bowie Boulevard, Fort Worth, Texas 76107, United States.
Author Contributions
K.A.E. and C.W.L. directed and designed the chemistry. A.S.F. and B.S.B. performed the medicinal chemistry. P.J.C. and C.M.N. directed and designed the molecular pharmacology experiments. A.L.R. directed and performed molecular pharmacology experiments. D.F.V. and V.B.L. performed molecular pharmacology experiments. J.S.D. directed and designed the DMPK experiments. A.L.B. directed and designed DMPK experiments and performed bioanalytical work. R.D.M. performed bioanalytical work. S.C. performed in vitro DMPK work. F.W.B. performed in vivo DMPK work. C.K.J. directed, designed, and performed behavioral experiments. A.T.G. performed behavioral experiments. J.M.R. designed and performed the rat brain occupancy experiments. M.N.T. performed rat brain occupancy experiments. G.D.T. was the study director for the NHP brain occupancy study.
The authors declare no competing financial interest.
Supplementary Material
References
- Golubeva A. V.; Moloney R. D.; O’Connor R. M.; Dinan T. G.; Cryan J. F. Metabotropic glutamate receptors in central nervous system disease. Curr. Drug Targets 2016, 17, 538–616. 10.2174/1389450116666150316224011. [DOI] [PubMed] [Google Scholar]
- Gregory K. J.; Noetzel M. J.; Rook J. M.; Vinson P. N.; Stauffer S. R.; Rodriguez A. L.; Emmitte K. A.; Zhou Y.; Chun A. C.; Felts A. S.; Chauder B. A.; Lindsley C. W.; Niswender C. M.; Conn P. J. Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol. Pharmacol. 2012, 82, 860–875. 10.1124/mol.112.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A.; Hopkins C. R.; Bridges T. M.; Gregory K. J.; Niswender C. M.; Conn P. J. Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chem. Rev. 2016, 116, 6707–6741. 10.1021/acs.chemrev.5b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmitte K. A. mGlu5 negative allosteric modulators: a patent review (2013–2016). Expert Opin. Ther. Pat. 2017, 27, 691–706. 10.1080/13543776.2017.1280466. [DOI] [PubMed] [Google Scholar]
- Rocher J. P.; Bonnet B.; Boléa C.; Lütjens R.; Le Poul E.; Poli S.; Epping-Jordan M.; Bessis A. S.; Ludwig B.; Mutel V. mGluR5 negative allosteric modulators overview: a medicinal chemistry approach towards a series of novel therapeutics. Curr. Top. Med. Chem. 2011, 11, 680–695. 10.2174/1568026611109060680. [DOI] [PubMed] [Google Scholar]
- Mihov Y.; Hasler G. Negative allosteric modulators of metabotropic glutamate receptors subtype 5 in addiction: a therapeutic window. Int. J. Neuropsychopharmacol. 2016, 19, pyw002. 10.1093/ijnp/pyw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro F. M.; Vieira L. B.; Pires R. G.; Olmo R. P.; Ferguson S. S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191. 10.1016/j.phrs.2016.11.013. [DOI] [PubMed] [Google Scholar]
- Busse C. S.; Brodkin J.; Tattersall D.; Anderson J. J.; Warren N.; Tehrani L.; Bristow L. J.; Varney M. A.; Cosford N. D. P. The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 2004, 29, 1971–1979. 10.1038/sj.npp.1300540. [DOI] [PubMed] [Google Scholar]
- Silverman J. L.; Smith D. G.; Rizzo S. J.; Karras M. N.; Turner S. M.; Tolu S. S.; Bryce D. K.; Smith D. L.; Fonseca K.; Ring R. H.; Crawley J. N. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med. 2012, 4, 131ra51. 10.1126/scitranslmed.3003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop A. S.; Gomez-Mancilla B.; Neri G.; Willemsen R.; Gasparini F. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology (Berl) 2014, 231, 1217–1226. 10.1007/s00213-013-3330-3. [DOI] [PubMed] [Google Scholar]
- Litim N.; Morissette M.; Di Paolo T. Metabotropic glutamate receptors as therapeutic targets in Parkinson’s disease: an update from the last 5 years of research. Neuropharmacology 2017, 115, 166–179. 10.1016/j.neuropharm.2016.03.036. [DOI] [PubMed] [Google Scholar]
- Rascol O.; Fox S.; Gasparini F.; Kenney C.; Di Paolo T.; Gomez-Mancilla B. Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat. Disord. 2014, 20, 947–956. 10.1016/j.parkreldis.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Jaso B. A.; Niciu M. J.; Iadarola N. D.; Lally N.; Richards E. M.; Park M.; Ballard E. D.; Nugent A. C.; Machado-Vieira R.; Zarate C. A. Therapeutic modulation of glutamate receptors in major depressive disorder. Curr. Neuropharmacol. 2017, 15, 57–70. 10.2174/1570159X14666160321123221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutrick D.; Stein D. J.; Subramanian G.; Smith B.; Fava M.; Hasler G.; Cha J.; Gasparini F.; Donchev T.; Ocwieja M.; Johns D.; Gomez-Mancilla B. Mavoglurant augmentation in OCD patients resistant to selective serotonin reuptake inhibitors: a proof-of-concept, randomized, placebo-controlled, phase 2 study. Adv. Ther. 2017, 34, 524–541. 10.1007/s12325-016-0468-5. [DOI] [PubMed] [Google Scholar]
- Gasparini F.; Lingenhöhl K.; Stoehr N.; Flor P. J.; Heinrich M.; Vranesic I.; Biollaz M.; Allgeier H.; Heckendorn R.; Urwyler S.; Varney M. A.; Johnson E. C.; Hess S. D.; Rao S. P.; Sacaan A. I.; Santori E. M.; Veliocelebi G.; Kuhn R. Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systematically active mGlu5 receptor antagonist. Neuropharmacology 1999, 38, 1493–1503. 10.1016/S0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- Cosford N. D.; Tehrani L.; Roppe J.; Schweiger E.; Smith N. D.; Anderson J.; Bristow L.; Brodkin J.; Jiang X.; McDonald I.; Rao S.; Washburn M.; Varney M. A. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem. 2003, 46, 204–206. 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- Porter R. H. P.; Jaeschke G.; Spooren W.; Ballard T. M.; Büttelmann B.; Kolczewski S.; Peters J. U.; Prinssen E.; Wichmann J.; Vieira E.; Mühlemann A.; Gatti S.; Mutel V.; Malherbe P. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J. Pharmacol. Exp. Ther. 2005, 315, 711–721. 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E. M.; Hessl D.; Coffey S.; Hervey C.; Schneider A.; Yuhas J.; Hutchinson J.; Snape M.; Tranfaglia M.; Nguyen D. V.; Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 2009, 46, 266–271. 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg M. D. B.; Raboisson P. AZD9272 and AZD2066: selective and highly central nervous system penetrant mGluR5 antagonists characterized by their discriminative effects. J. Pharmacol. Exp. Ther. 2014, 350, 212–222. 10.1124/jpet.114.215137. [DOI] [PubMed] [Google Scholar]
- Ståhle L.; Karlsten R.; Jonzon B.; Eriksson B.; Kågedal M.; Dominicus A. Safety evaluation of the mGluR5 antagonists AZD9272, AZD2066, and AZD2516 in healthy volunteers and patients with neuropathic pain or major depressive disorders. Abstract PT444. Proceedings of the 14th World Congress on Pain, Milan, Italy, August 28, 2012; International Association for the Study of Pain: Washington, DC, 2012.
- Vranesic I.; Ofner S.; Flor P. J.; Bilbe G.; Bouhelal R.; Enz A.; Desrayaud S.; McAllister K.; Kuhn R.; Gasparini F. AFQ056/mavoglurant, a novel clinically effective mGluR5 antagonist: identification, SAR and pharmacological characterization. Bioorg. Med. Chem. 2014, 22, 5790–5803. 10.1016/j.bmc.2014.09.033. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E.; Des Portes V.; Hagerman R.; Jacquemont S.; Charles P.; Visootsak J.; Brinkman M.; Rerat K.; Koumaras B.; Zhu L.; Barth G. M.; Jaecklin T.; Apostol G.; von Raison F. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 2016, 8, 321ra5. 10.1126/scitranslmed.aab4109. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C.; Stocchi F.; Poewe W.; Dronamraju N.; Kenney C.; Shah A.; von Raison F.; Graf A. Mavoglurant in Parkinson’s patients with l-Dopa-induced dyskinesias: two randomized phase 2 studies. Mov. Disord. 2016, 31, 1054–1058. 10.1002/mds.26585. [DOI] [PubMed] [Google Scholar]
- Stocchi F.; Rascol O.; Destee A.; Hattori N.; Hauser R. A.; Lang A. E.; Poewe W.; Stacy M.; Tolosa E.; Gao H.; Nagel J.; Merschhemke M.; Graf A.; Kenney C.; Trenkwalder C. AFQ056 in Parkinson patients with levodopa-induced dyskinesia: 13-week, randomized, dose-finding study. Mov. Disord. 2013, 28, 1838–1846. 10.1002/mds.25561. [DOI] [PubMed] [Google Scholar]
- Walles M.; Wolf T.; Jin Y.; Ritzau M.; Leuthold L. A.; Krauser J.; Gschwind H. P.; Carcache D.; Kittelmann M.; Ocwieja M.; Ufer M.; Woessner R.; Chakraborty A.; Swart P. Metabolism and disposition of the metabotropic glutamate receptor 5 antagonist (mGluR5) mavoglurant (AFQ056) in healthy subjects. Drug Metab. Dispos. 2013, 41, 1626–1641. 10.1124/dmd.112.050716. [DOI] [PubMed] [Google Scholar]
- Jaeschke G.; Kolczewski S.; Spooren W.; Vieira E.; Bitter-Stoll N.; Boissin P.; Borroni E.; Büttelmann B.; Ceccarelli S.; Clemann N.; David B.; Funk C.; Guba W.; Harrison A.; Hartung T.; Honer M.; Huwyler J.; Kuratli M.; Niederhauser U.; Pähler A.; Peters J. U.; Petersen A.; Prinssen E.; Ricci A.; Rueher D.; Rueher M.; Schneider M.; Spurr P.; Stoll T.; Tännler D.; Wichmann J.; Porter R. H.; Wettstein J. G.; Lindemann L. Metabotropic glutamate receptor 5 negative allosteric modulators: discovery of 2-chloro-4-[1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-ylethynyl]pyridine (basimglurant, RO4917523), a promising novel medicine for psychiatric diseases. J. Med. Chem. 2015, 58, 1358–1371. 10.1021/jm501642c. [DOI] [PubMed] [Google Scholar]
- Guerini E.; Schadt S.; Greig G.; Haas R.; Husser C.; Zell M.; Funk C.; Hartung T.; Gloge A.; Mallalieu N. L. A double-tracer technique to characterize absorption, distribution, metabolism and excretion (ADME) of [14C]-basimglurant and absolute bioavailability after oral administration and concomitant intravenous microdose administration of [13C6]-labeled basimglurant in humans. Xenobiotica 2017, 47, 144–153. 10.3109/00498254.2016.1169334. [DOI] [PubMed] [Google Scholar]
- Quiroz J. A.; Tamburri P.; Deptula D.; Banken L.; Beyer U.; Rabbia M.; Parkar N.; Fontoura P.; Santarelli L. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry 2016, 73, 675–684. 10.1001/jamapsychiatry.2016.0838. [DOI] [PubMed] [Google Scholar]
- Fuxe K.; Borroto-Escuela D. O. Basimglurant for treatment of major depressive disorder: a novel negative allosteric modulator of metabotropic glutamate receptor 5. Expert Opin. Invest. Drugs 2015, 24, 1247–1260. 10.1517/13543784.2015.1074175. [DOI] [PubMed] [Google Scholar]
- Bezard E.; Pioli E. Y.; Li Q.; Girard F.; Mutel V.; Keywood C.; Tison F.; Rascol O.; Poli S. M. The mGluR5 negative allosteric modulator dipraglurant reduces dyskinesia in the MPTP macaque model. Mov. Disord. 2014, 29, 1074–1079. 10.1002/mds.25920. [DOI] [PubMed] [Google Scholar]
- Tison F.; Keywood C.; Wakefield M.; Durif F.; Corvol J. C.; Eggert K.; Lew M.; Isaacson S.; Bezard E.; Poli S. M.; Goetz C. G.; Trenkwalder C.; Rascol O. A phase 2A trial of the novel mGluR5-negative allosteric modulator dipraglurant for levodopa-induced dyskinesia in Parkinson’s disease. Mov. Disord. 2016, 31, 1373–1380. 10.1002/mds.26659. [DOI] [PubMed] [Google Scholar]
- Palanisamy G. S.; Marcek J. M.; Cappon G. D.; Whritenour J.; Shaffer C. L.; Brady J. T.; Houle C. Drug-induced skin lesions in cynomolgus macaques treated with metabotropic glutamate receptor 5 (mGluR5) negative allosteric modulators. Toxicol. Pathol. 2015, 43, 995–1003. 10.1177/0192623315588114. [DOI] [PubMed] [Google Scholar]
- Morin N.; Morissette M.; Grégoire L.; Gomez-Mancilla B.; Gasparini F.; Di Paolo T. Chronic treatment with MPEP, an mGlu5 receptor antagonist, normalizes basal ganglia glutamate neurotransmission in L-DOPA-treated parkinsonian monkeys. Neuropharmacology 2013, 73, 216–231. 10.1016/j.neuropharm.2013.05.028. [DOI] [PubMed] [Google Scholar]
- Masilamoni G. J.; Bogenpohl J. W.; Alagille D.; Delevich K.; Tamagnan G.; Votaw J. R.; Wichmann T.; Smith Y. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 2011, 134, 2057–2073. 10.1093/brain/awr137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See page 862 of:Zhang L.; Balan G.; Barreiro G.; Boscoe B. P.; Chenard L. K.; Cianfrogna J.; Claffey M. M.; Chen L.; Coffman K. J.; Drozda S. E.; Dunetz J. R.; Fonseca K. R.; Galatsis P.; Grimwood S.; Lazzaro J. T.; Mancuso J. Y.; Miller E. L.; Reese M. R.; Rogers B. N.; Sakurada I.; Skaddan M.; Smith D. L.; Stepan A. F.; Trapa P.; Tuttle J. B.; Verhoest P. R.; Walker D. P.; Wright A. S.; Zaleska M. M.; Zasadny K.; Shaffer C. L. Discovery and preclinical characterization of 1-methyl-3-(4-methylpyridin-3-yl)-6-(pyridin-2-ylmethoxy)-1H-pyrazolo-[3,4-b]pyrazine (PF470): a highly potent, selective, and efficacious metabotropic glutamate receptor 5 (mGluR5) negative allosteric modulator. J. Med. Chem. 2014, 57, 861–877. 10.1021/jm401622k. [DOI] [PubMed] [Google Scholar]
- Bates B. S.; Rodriguez A. L.; Felts A. S.; Morrison R. D.; Venable D. F.; Blobaum A. L.; Byers F. W.; Lawson K. P.; Daniels J. S.; Niswender C. M.; Jones C. K.; Conn P. J.; Lindsley C. W.; Emmitte K. A. Discovery of VU0431316: a negative allosteric modulator of mGlu5 with activity in a mouse model of anxiety. Bioorg. Med. Chem. Lett. 2014, 24, 3307–3314. 10.1016/j.bmcl.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts A. S.; Rodriguez A. L.; Morrison R. D.; Venable D. F.; Manka J. T.; Bates B. S.; Blobaum A. L.; Byers F. W.; Daniels J. S.; Niswender C. M.; Jones C. K.; Conn P. J.; Lindsley C. W.; Emmitte K. A. Discovery of VU0409106: a negative allosteric modulator of mGlu5 with activity in a mouse model of anxiety. Bioorg. Med. Chem. Lett. 2013, 23, 5779–5785. 10.1016/j.bmcl.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch R. D.; Morrison R. D.; Byers F. W.; Lindsley C. W.; Emmitte K. A.; Daniels J. S. Evaluating the disposition of a mixed aldehyde oxidase/cytochrome P450 substrate in rats with attenuated P450 activity. Drug Metab. Dispos. 2016, 44, 1296–1303. 10.1124/dmd.115.068338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook J. M.; Tantawy M. N.; Ansari M. S.; Felts A. S.; Stauffer S. R.; Emmitte K. A.; Kessler R. M.; Niswender C. M.; Daniels J. S.; Jones C. K.; Lindsley C. W.; Conn P. J. Relationship between in vivo receptor occupancy and efficacy of metabotropic glutamate receptor subtype 5 allosteric modulators with different in vitro binding profiles. Neuropsychopharmacology 2015, 40, 755–765. 10.1038/npp.2014.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison R. D.; Blobaum A. L.; Byers F. W.; Santomango T. S.; Bridges T. M.; Stec D.; Brewer K. A.; Sanchez-Ponce R.; Corlew M. M.; Rush R.; Felts A. S.; Manka J.; Bates B. S.; Venable D. F.; Rodriguez A. L.; Jones C. K.; Niswender C. M.; Conn P. J.; Lindsley C. W.; Emmitte K. A.; Daniels J. S. The role of aldehyde oxidase and xanthine oxidase in the biotransformation of a novel negative allosteric modulator of metabotropic glutamate receptor subtype 5. Drug Metab. Dispos. 2012, 40, 1834–1845. 10.1124/dmd.112.046136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zientek M.; Miller H.; Smith D.; Dunklee M. B.; Heinle L.; Thurston A.; Lee C.; Hyland R.; Fahmi O.; Burdette D. Development of an in vitro drug-drug interaction assay to simultaneously monitor five cytochrome P450 isoforms and performance assessment using drug library compounds. J. Pharmacol. Toxicol. Methods 2008, 58, 206–214. 10.1016/j.vascn.2008.05.131. [DOI] [PubMed] [Google Scholar]
- Davies B.; Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- Di L.; Rong H.; Feng B. Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 2013, 56, 2–12. 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
- Kitamura S.; Sugihara K.; Ohta S. Drug-metabolizing ability of molybdenum hydroxylases. Drug Metab. Pharmacokinet. 2006, 21, 83–98. 10.2133/dmpk.21.83. [DOI] [PubMed] [Google Scholar]
- Noetzel M. J.; Rook J. M.; Vinson P. N.; Cho H.; Days E.; Zhou Y.; Rodriguez A. L.; Lavreysen H.; Stauffer S. R.; Niswender C. M.; Xiang Z.; Daniels J. S.; Lindsley C. W.; Weaver C. D.; Conn P. J. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol. Pharmacol. 2012, 81, 120–133. 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Johnson K. A.; Luo Q.; Ayala J. E.; Kim C.; Conn P. J.; Weaver C. D. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008, 73, 1213–1224. 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- LeadProfilingScreen (catalog #68); Eurofins Panlabs, Inc.: St. Charles, MO. https://www.eurofinspanlabs.com/Catalog/Products/ProductDetails.aspx?prodId=0aCrd3Mu4RA%3d&path=128&leaf=128&track=Add%2f2%2fPackages%2fMolecular+Packages (accessed April 26, 2017).
- Zahniser N. R.; Sorkin A. Rapid regulation of the dopamine transporter: role in stimulant addiction?. Neuropharmacology 2004, 47 (S1), 80–91. 10.1016/j.neuropharm.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Dopamine uptake (catalog #316010); Eurofins Panlabs, Inc.: St. Charles, MO. https://www.eurofinspanlabs.com/Catalog/Products/ProductDetails.aspx?prodId=0WVKOLUf1Bw%3d (accessed April 26, 2017).
- Cosford N. D. P.; Roppe J.; Tehrani L.; Schweiger E. J.; Seiders T. J.; Chaudary A.; Rao S.; Varney M. A. [3H]-Methoxymethyl-MTEP and [3H]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg. Med. Chem. Lett. 2003, 13, 351–354. 10.1016/S0960-894X(02)00997-6. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. L.; Nong Y.; Sekaran N. K.; Alagille D.; Tamagnan G. D.; Conn P. J. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol. 2005, 68, 1793–1802. 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Rager J. D.; Weinstein K.; Kardos P. S.; Dobson G. L.; Li J.; Hidalgo I. J. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood–brain barrier. Int. J. Pharm. 2005, 288, 349–359. 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. 10.1016/0006-291X(91)91647-U. [DOI] [PubMed] [Google Scholar]
- Hutzler J. M.; Yang Y.-S.; Albaugh D.; Fullenwider C. L.; Schmenk J.; Fisher M. B. Characterization of aldehyde oxidase enzyme activity in cryopreserved human hepaotcytes. Drug Metab. Dispos. 2012, 40, 267–275. 10.1124/dmd.111.042861. [DOI] [PubMed] [Google Scholar]
- ChanTest Cardiac Channel Panel; Charles River Laboratories International, Inc.: Wilmington, MA. http://www.criver.com/products-services/drug-discovery/therapeutic-areas/oncology/safety-assessment/cardiac-risk/cardiac-ion-channel-panel (accessed April 26, 2017).
- McCann J.; Choi E.; Yamasaki E.; Ames B. N. Detection of carcinogens as mutagens in the Salmonella/microsome test: assay of 300 chemicals. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 5135–5139. 10.1073/pnas.72.12.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas L. B.; Kolb Y.; Prinssen E. P. M. A combined marble burying-locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur. J. Pharmacol. 2006, 547, 106–115. 10.1016/j.ejphar.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Thomas A.; Burant A.; Bui N.; Graham D.; Yuva-Paylor L. A.; Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology 2009, 204, 361–373. 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery D. A.; Cryan J. F. Using the rat forced swim test to assess antidepressant-like activity in rodents. Nat. Protoc. 2012, 7, 1009–1014. 10.1038/nprot.2012.044. [DOI] [PubMed] [Google Scholar]
- Lindemann L.; Porter R. H.; Scharf S. H.; Kuennecke B.; Bruns A.; von Kienlin M.; Harrison A. C.; Paehler A.; Funk C.; Gloge A.; Schneider M.; Parrott N. J.; Polonchuk L.; Niederhauser U.; Morairty S. R.; Kilduff T. S.; Vieira E.; Kolczewski S.; Wichmann J.; Hartung T.; Honer M.; Borroni E.; Moreau J. L.; Prinssen E.; Spooren W.; Wettstein J. G.; Jaeschke G. Pharmacology of basimglurant (RO4917523, RG7090), a unique metabotropic glutamate receptor 5 negative allosteric modulator in clinical development for depression. J. Pharmacol. Exp. Ther. 2015, 353, 213–233. 10.1124/jpet.114.222463. [DOI] [PubMed] [Google Scholar]
- Browne C. A.; Lucki I. Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. Front. Pharmacol. 2013, 4, 161. 10.3389/fphar.2013.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P.; Moaddel R.; Morris P. J.; Georgiou P.; Fischell J.; Elmer G. I.; Alkondon M.; Yuan P.; Pribut H. J.; Singh N. S.; Dossou K. S.; Fang Y.; Huang X. P.; Mayo C. L.; Wainer I. W.; Albuquerque E. X.; Thompson S. M.; Thomas C. J.; Zarate C. A. Jr.; Gould T. D. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomierny-Chamioło L.; Poleszak E.; Pilc A.; Nowak G. NMDA but not AMPA glutamatergic receptors are involved in the antidepressant-like activity of MTEP during the forced swim test in mice. Pharmacol. Rep. 2010, 62, 1186–1190. 10.1016/S1734-1140(10)70381-9. [DOI] [PubMed] [Google Scholar]
- Wong D. F.; Waterhouse R.; Kuwabara H.; Kim J.; Brašić J. R.; Chamroonrat W.; Stabins M.; Holt D. P.; Dannals R. F.; Hamill T. G.; Mozley P. D. 18F-FPEB, a PET radiopharmaceutical for quantifying metabotropic glutamate 5 receptors: a first-in-human study of radiochemical safety, biokinetics, and radiation dosimetry. J. Nucl. Med. 2013, 54, 388–396. 10.2967/jnumed.112.107995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Lindsley C. W.; Emmitte, Kyle A.; Rodriguez A. L.; Felts A. S.; Jones C. K.; Bates B. S.; Chauder B. A.. 6-Alkyl-N-(pyridin-2-yl)-4-aryloxypicolinamide analogs as mGluR5 negative allosteric modulators and methods of making and using the same. U.S. Patent 9085562, July 21, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.