

Abstract
More than 80% of all cancers arise from epithelial cells referred to as carcinomas. Adenocarcinomas are the most common type of carcinomas arising from the specialized epithelial cells that line the ducts of our major organs. Despite many advances in cancer therapies, metastatic and treatment-refractory cancers remain the 2nd leading cause of death. Immunotherapy has offered potential opportunities with specific targeting of tumor cells and inducing remission in many cancer patients. Numerous therapies using antibodies as antagonists or checkpoint inhibitors/immune modulators, peptide or cell vaccines, cytokines, and adoptive T cell therapies have been developed. The most innovative immunotherapy approach so far has been the use of engineered T cell, also referred to as chimeric antigen receptor T cells (CAR-T cells). CAR-T cells are genetically modified naïve T cells that express a chimeric molecule which comprises of the antigen-recognition domains (scFv) of an anti-tumor antibody and one, two, or three intracellular signaling domains of the T cell receptor (TCR). When these engineered T cells recognize and bind to the tumor antigen target via the scFv fragment, a signal is sent to the intracellular TCR domains of the CAR, leading to activation of the T cells to become cytolytic against the tumor cells. CAR-T cell therapy has shown tremendous success for certain hematopoietic malignancies, but this success has not been extrapolated to adenocarcinomas. This is due to multiple factors associated with adenocarcinoma that are different from hematopoietic tumors. Although many advances have been made in targeting multiple cancers by CAR-T cells, clinical trials have shown adverse effects and toxicity related to this treatment. New strategies are yet to be devised to manage side effects associated with CAR-T cell therapies. In this review, we report some of the promising immunotherapeutic strategies being developed for treatment of most common adenocarcinomas with particular emphasis on the future generation of CAR-T cell therapy.
Keywords: immunotherapy, cancer, tumor, chimeric antigen receptor T cell (CAR-T cell), engineered T cell, adoptive T cell therapy, solid tumor, adenocarcinoma, lung cancer, breast cancer, gastrointestinal cancer, prostate cancer, hepatocellular adenocarcinoma, pancreatic cancer, ovarian cancer
1. Introduction
14.1 million incidental cancer cases with 8.2 million deaths were estimated around the world in 2012. This number is anticipated to increase to 24 million by 2035 [1]. Among all types of cancer, carcinomas are the most common type that develop from epithelial cells. They are generally originated from endodermal or ectodermal germ layer during embryogenesis [2]. Carcinomas are further subtyped into squamous cell carcinomas (epithelial cell that form a protective lining such as the epithelial cell lining of the skin and the esophagus) and adenocarcinomas (specialized epithelial cells lining the ducts of all major organs). This review will be restricted to adenocarcinomas.
Chemotherapy and radiation have long been the mainstay non-surgical treatment option for cancer patients. However, many cancers exhibit or develop resistance to these treatment modalities within a short period of time. Despite recent significant advances in therapies such as the use of monoclonal antibodies (mAb) and small-molecule inhibitors, the responses in patients vary considerably and are usually accompanied by high recurrence rate and poor prognosis. Since there are few or no treatment strategies for eradicating resistant cells, more efficient therapies are necessary. The essential role of the immune system in controlling and eradicating tumors has been supported with compelling evidence. Many studies have been focused on harnessing the immune system to attain clinical efficacy [3] and developing novel targeted therapies for improving patient survival.
Genetically modified cytotoxic T lymphocytes to kill tumor cells were first demonstrated by Gross and colleagues nearly two decades ago [4]. This work paved the way for the development of a series of first generation chimeric antigen receptors (CARs) in which single chain variable fragment (scFv, which is made of variable parts of heavy and light chains connected by a linker sequence) of a tumor targeting antibody is fused directly to a signaling domain of the TCR complex member, CD3ζ [3, 5] (Figure 1a). The extracellular scFv domain of CAR recognizes the specific surface antigen on tumor cell as a mAb independently of MHC molecules, and delivers the signal to T cells via CD3ζ, which leads to T cell activation similarly to that of TCR signaling. Since then, in order to increase T-cell proliferation and persistence, other costimulatory genes (e.g. CD28, 4-1BB, and OX40) were added to the intracellular domain, creating second- and third-generation CARs [6–8]. Recently, modern CAR structures containing suicide or cytokine gene have been designed that may be dubbed as fourth generation CAR [9, 10] (Figure 1a). Although CAR-T cells have numerous designs and utilize various tumor-specific scFvs, their manufacturing procedure remains unchanged. In brief, this procedure encompasses harvesting T cells from patient via apheresis, ex vivo enrichment, genetic modification of T cells with CAR cDNA (using electroporation, lipofectamine or viral vectors) followed by large-scale expansion, final formulation, and lastly infusing back to the patients. CD4+ or CD8+ T cells may further be sorted depending on the application [11] (Figure 1b).
Figure 1. CAR architecture and manufacturing.
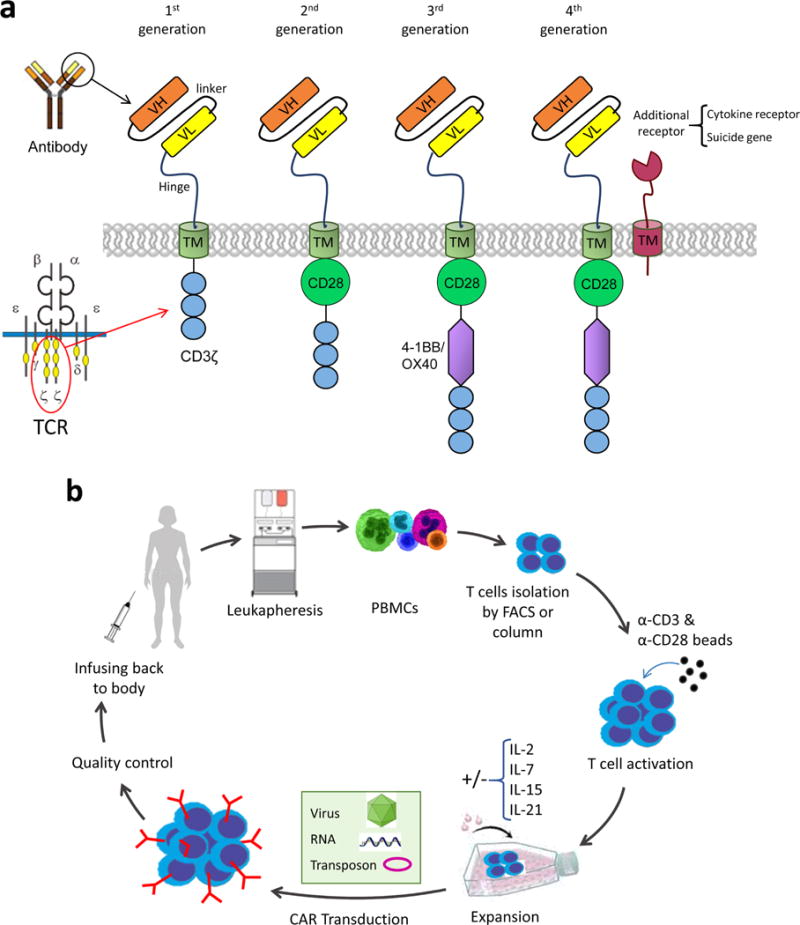
a. CAR molecule structure in four different designs. The simplest design of CAR consists of a scFv, transmembrane and CD3ζ domain. The 2nd, 3rd and 4th generation CARs have additional costimulatory genes incorporated in the C-terminal end of the molecule. A cytokine or inhibitory receptor is coexpressed with the CAR in 4th generation design. b. Manufacturing workflow of gene engineered T cells. T cells are harvested from the patients’ blood via leukaphresis and further activated by beads. The CAR gene is delivered to T cells by retro/lenti viruses, electroporation (RNA) or transposons. Then, transduced T cells get expanded and undergo quality control before injecting back to patient.
Enormous success has been generated in early phase clinical trials of hematologic malignancies, particularly, CD19-targeted CAR-T cells in leukemia [12, 13] and related CARs in lymphoma and myeloma. Successful result has been reported for metastatic melanoma as well [14]. Unfortunately, these successes, despite many attempts, have not yet been extended to adenocarcinomas. Many clinical trials have focused on solid tumors by targeting various proteins such as carcinoembryonic antigen (CEA), the diganglioside GD2, human epidermal growth factor receptor 2 (HER2), mesothelin (MSLN), fibroblast activation protein (FAP), interleukin 13 receptor α (IL13Rα), and L1 cell adhesion molecule (L1CAM) [12, 15]. Among these, GD2-specific CAR-T cells for neuroblastoma [16] and HER2 CARs for sarcoma [17] have shown the most encouraging results thus far.
A disadvantage of CAR-T cell therapy is that CAR-T cells are “living drugs”, therefore, failure in treatment may not be easily managed. Over activation or cross reactivity with antigens on healthy tissue may result in fatal outcome. Thus, effective strategies must be devised toward managing the safety issues [18]. As said by Robert Tepper, Chief Medical Officer at Jounce Therapeutics, “The good news - and the bad news - is that the immune system is incredibly powerful”.
2. Immunotherapy strategies with emphasis on CAR-T cell therapies for the most common adenocarcinomas
Our immune system can be a powerful weapon against cancer, but researchers are still struggling with how to control it [19]. Immunotherapy of cancers usually comprises of monoclonal antibodies, immune checkpoint inhibitors, therapeutic tumor vaccines, and adoptive T cell therapies. Here, we will discuss the main studies done in each adenocarcinoma ordered based on their mortality and incidence rate.
2.1. Lung cancer
Lung cancer is the most common cancer in the world, both in term of new cases (1.8 million cases in 2012) and deaths (1.6 million deaths) because of the high case fatality [1]. Non-small cell lung cancer (NSCLC) is the most common form of lung cancer accounting for 80–85% of all cases. The most commonly diagnosed type of NSCLC is adenocarcinoma that begins in the ductal epithelial cells of the lung [20]. NSCLC was conventionally considered non-immunogenic tumors. However, advancements in tumor immunology and recent immunotherapeutic successes have challenged this view [21]. Most common approaches in lung cancer immunotherapy are the use of immune checkpoint inhibitors, such as blockade of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (via ipilimumab and tremelimumab), programmed cell death-1 (PD-1) (via nivolumab and pembrolizumab), and PD-1 ligand (PDL1) (via atezolizumab). Other approaches include tumor vaccines against relevant tumor-associated antigens [22]. Even though progress has been made in the treatment of advanced NSCLC, more effective treatments are needed for progressive lung cancer. Immunotherapy seems the most promising approach to the future of lung cancer treatment. Yet, to fully exploit the effect of these new treatments, new biomarkers/tumor-specific antigens must be identified. Furthermore, to determine the patients who will benefit from this therapy, knowing the mechanism of these treatments seems critical. Besides, to certify the role of immunotherapy for lung cancer, immunotherapy should be tested in early-stage disease setting and in combination with other agents such as chemotherapy, radiotherapy and molecular targeted therapy [23].
Adoptive T cell therapy
Studies have confirmed that MSLN is overexpressed at metastatic sites of most of lung adenocarcinomas [24], and its expression is correlating with tumor aggressiveness and KRAS mutation [25]. These data provide the rationale to design CARs for targeting MSLN-expressing tumors. MSLN CART cells have been tested by several groups in subcutaneous or orthotopic mouse models of lung cancer and other malignancies. They showed that 30 times fewer T cells were needed when T cells were administered intra-pleurally compared to systemically and that these T cells were more effective in inducing long-term remission of tumors [26]. A phase I clinical trial investigating MSLN CAR-T cells administered regionally in patients with lung cancer, and breast cancer with pleural metastases started in 2015 and is ongoing (NCT02414269) [27]. There are numerous overexpressed antigens to target via CAR-T cells, but since CAR-T cells are very sensitive to even low levels of antigens (higher sensitivity than mAb), we need to be concerned with their safety and “on-target/off-tumor” side effects. One of the worst examples which will be discussed later is, high dose administration of CAR-T cells against HER2, causing a fatal adverse reaction. An increase in IFN-γ, GM-CSF, TNF-α, IL-6 and IL-10 was confirmed in the serum of the patient post treatment which was consistent with a cytokine storm (severe systemic inflammation caused by overwhelming release of cytokines). It was speculated that immediate localization of large number of infused cells to the lung after infusion which may be due to detection of low levels of HER2 on lung epithelial cells probably triggered the massive cytokine release leading to fatality [28]. To address this issue, researchers have suggested using CARs with lower affinity to HER2, or CARs only recognizing the tumor associated form of HER2 [29].
Glypican-3 (GPC3) which is a member of heparan sulfate proteoglycans family plays an essential role in cellular growth, differentiation, and migration [30]. This protein is overexpressed in tumor tissues, including lung cancer, while it is nearly absent in normal tissues [31]. In a recent study, the anti-tumor potential of 3rd generation anti-GPC3-CAR-T cells was explored in tumor models of lung squamous cell carcinoma (LSCC). GPC3-CAR-T cells mediated efficient eradication of GPC3+ tumor cells but not normal cells in two established LSCC xenograft models. Moreover, GPC3-CAR-T cells were persistent in vivo and could successfully infiltrate into the tumor tissues. These findings suggest that GPC3-CAR-T cells could be a novel potential treatment for lung cancer patients [32].
In a different approach, cancer-associated fibroblasts (CAFs) were targeted by CAR-T cells. In an A549 established lung cancer model, adoptive transfer of FAP-redirected CAR-T cells remarkably reduced FAP+ stromal cells and stunted tumor growth. In this study, the authors showed that combining this FAP-specific T cells with T cells targeting hepatocellular carcinoma A2 (EphA2) antigen on the A549 cancer cells significantly improved anti-tumor response and conferred a survival benefit compared to single treatment [33].
2.2. Breast cancer
Breast cancer is the second most common cancer overall (1.7 million cases in 2012) but by far is the most common cancer in women worldwide [1]. The most common type of breast cancer is invasive ductal carcinoma (IDC) [34]. Clinical trials utilizing vaccines and immune checkpoints in breast cancer have produced promising results which encourages for more immunotherapy approaches [35].
Trastuzumab, a mAb against HER2 antigen, has proved to be the most successful immunotherapeutic agent in the treatment of HER2+ breast cancers [36]. Studies have shown that trastuzumab can stimulate cytotoxicity by natural killer cells [37], and elicit HER2-specific CD4+ T cell and antibody response [38]. Different trials such as N9831 [39], FinHER [40] and Bowel Project B-31 [41] have recently shown promising results with trastuzumab in combination with chemotherapy in HER2+ breast cancer patients. PDL-1 has been targeted in a phase Ib study (KEYNOTE-012) using mAb called pembrolizumab in patients with metastatic triple-negative breast cancer (mTNBC). Only five out of 32 patients responded and 56% of patients showed adverse effect (AE) with one patient dying due to disseminated intravascular coagulation [42]. Another PD-L1 antibody, atezolizumab, was tested in a phase Ia study on 12 PD-L1+ TNBC patients. Only 8% of patients showed AE and no patient died due to treatment. Atezolizumab in combination with chemotherapy (nab-paclitaxel) was tested in a phase 1b trial in mTNBC patients and showed tolerability and positive impact on mTNBC patients [43]. A phase III trial assessing this regimen for first-line treatment of (previously untreated) mTNBC patients started in 2015 and is ongoing (NCT01633970) [44].
A phase III clinical trial is assessing HER2 peptide nelipepimut-S (E75) vaccine (NeuVax™) in breast cancer patients with low to intermediate HER2 expression. Phase I/II of this trial evaluated nelipepimut-S combined with GM-CSF administered in patients in an adjuvant setting in order to prevent recurrence. Most patients with various levels of HER2/neu expression showed immune response and the vaccine was efficacious [45]. Several other vaccines are being investigated in patients with breast cancer, including additional HER2-derived peptide in combination with Mucin 1 (MUC1) peptide vaccine, a HER2 peptide-pulsed dendritic cell vaccine, an allogeneic GM-CSF-secreting vaccine [46], and PANVAC, which is a genetically engineered vaccine incorporating vaccinia and fowlpox viruses expressing tumor-associated CEA and MUC1 [47]. All of these vaccine tactics have shown evidence of clinical benefit in specific disease setting.
Adoptive T cell therapy
Tumor-associated MUC1 (tMUC1) which is found in many tumors, has been the target of various therapies including CAR-T cells treatment. Wilkie and et al. showed that tMUC1 could be targeted by CAR-T cells and it could significantly delay the tumor growth in xenograft mouse model of breast cancer [48]. In order to increase specificity and safety of CAR-T cells treatment, they have developed a bispecific CAR-T targeting HER2 and MUC1 and showed that “dual-targeted” T-cells are capable of efficient killing of the HER2+ tumor cells and their proliferation requires co-expression of both MUC1 and HER2 on target cells. However only a modest increase in IL-2 production was seen compared to control CAR-T cells with a single administration [49]. Other trials designed to target HER2 with CAR-T cells in breast cancer [50, 51] showed severe toxicity and death (due to infused T cells attacking the patient’s lungs). Thus, targeting HER2 must be with ultimate caution [28].
A phase I clinical trial with CAR-T cells targeting MSLN plus cyclophosphamide in 14 metastatic breast cancer patients revealed partial remission (NCT02414269) [27]. A CEA 2nd generation CAR-T phase Ia trial in metastatic breast cancers patients (26 patients) was started by Roger Williams Medical Center and is still ongoing (NCT00673829). TARP which is a nuclear protein derived from alternative reading frame of TCR gamma chain locus is expressed on normal prostate epithelium and in adenocarcinomas of the prostate and breast [52]. Hillerdal and et al. have engineered T cells that recognize HLA-A2-restricted TARP epitope (TARP-TCR-T). These T cells could kill HLA-A2+ prostate and breast cancer cells expressing TARP [53].
2.3. Gastrointestinal cancer (colorectal, colon, esophagus and stomach cancer)
In this category, colorectal cancer is the 3rd most common cancer in men and 2nd in women in the world (1.4 million overall new cases in 2012). Stomach cancer is the fifth most common malignancy worldwide (1 million new cases in 2012) [1]. The number of T cells present within colorectal cancer metastases is correlated to response to chemotherapy, and high expression of PDL-1 is associated with poor prognosis in advanced gastric cancer patients [54]. Immune checkpoint blockade with antibodies targeting CTLA-4, PD-1 and PDL-1 have shown promising results in gastrointestinal (GI) cancer. However, tumors might get resistant to these treatments and ultimately relapse. Hence, new strategies are emerging and being evaluated in the clinic in order to defeat this resistance. Other checkpoints targeted in early clinical trials are OX40, LAG3 and TIM3. Blocking OX40, which is a member of TNFR family, has produced encouraging results in preclinical studies. Therefore, OX40 appears to be a great candidate to target, especially in combination with other agents [55]. A clinical trial with an anti-OX40 antibody (MEDI6469) is currently ongoing in patients with metastatic colorectal cancer (NCT02559024).
LAG3 is another promising target, since it is primarily expressed on regulatory T cells (Tregs). Dual targeting of LAG3 and PD-1 by antibodies remarkably improved the survival in a MC38 colorectal cancer mouse model and out-performed the single antibody treatment [56]. Moreover, safety and efficacy of LAG3 antibody as a single agent and in combination with PDR001 (an anti-PD-1 mAb) are being tested in patients with advanced malignancies (NCT02460224 and NCT01968109).
TIM3, which is exclusively expressed on IFN-γ producing Th1 and not on Th2 cells, negatively regulates IFN-γ secretion by stimulating cell death upon binding to its ligand galectin-9 [57]. Hence, TIM3 has been explored as a potential target [58]. Safety and efficacy of MBG453 (anti-human TIM-3 mAb) are being investigated in a clinical trial (NCT02608268) as a single arm study and in combination with PDR001 in patients with advanced malignancies. Combinational therapies using immune checkpoint inhibitors with other agents, including chemo/radiotherapy or angiogenesis inhibitors, are alternative strategies currently in early development [59, 60].
Adoptive T cell therapy
There are a few clinical trials utilizing CAR-T cells in GI cancers. Results of a study evaluating CEA-redirected-CAR-T cells in CEA transgenic mice showed eradication of CEA+ tumors in a primary response and an efficient recall response upon second challenge with CEA+ tumor cells. This approach showed no toxicity towards normal tissues [61]. A CEA-TCR T cell therapy was tested in a small clinical trial and provided the first example of an objective regression of metastatic colorectal cancer in three patients with slight toxicity [62].
In GI cancers, trafficking of T cells to the tumor site remains a challenge. To improve trafficking, Kobold et al. have devised a strategy combining TCR-engineered T cells with a bispecific antibody that can simultaneously recognize T cells and tumor cells resulting in bringing T cells and tumor cells in close vicinity. In their study, SV40 T antigen-specific T cells from TCR-I-transgenic mice were transduced to express a truncated form of epithelial growth factor receptor (EGFR) and were combined with a bispecific antibody against EGFR and EpCAM. This therapy was tested in a C57Bl/6 mice bearing subcutaneous tumors of murine gastric cancer cell line GC8 (SV40+ and EpCAM+) and resulted in increased T cell infiltration of tumors, delayed tumor growth, and extended survival in vivo [63]. Another study tested the efficacy of human anti-FITC CAR-T cells plus FITC-labeled cetuximab (FITC-Ctx) in an immunocompromised mouse model. A delay in the growth of colon cancer was observed; however, this treatment resulted in an unexpected outgrowth of EGFR-negative tumor cells [64]. An ongoing phase I/II clinical trial is evaluating anti-MUC1 CAR-T cell therapy in patients with a MUC1+ solid tumor, including colorectal and gastric cancer (NCT02617134).
2.4. Prostate cancer
Prostate cancer (PCa) is the fourth most common cancer in both sexes combined and the second most common cancer in men in the world. An estimated 1.1 million people were diagnosed with PCa in 2012 worldwide [1]. Localized PCa is curable by surgical treatment; however, metastatic tumors recur in lymph nodes and bones of majority of patients. Current available treatments for metastatic PCa are not effective and comprises of alleviating androgen withdrawal that results in hormone-resistant disease usually within months. However, immunotherapy remains an optimistic option for treatment of metastatic PCa, considering a phase III study of provenge (sipuleucel-T, an antigen-presenting dendritic cell vaccine) has reported increased overall survival in hormone refractory PCa patients [65]. Among different immunotherapies, therapeutic vaccines and immune checkpoint inhibitors have been the most evaluated in clinical trials. Combining immunotherapy with established treatments for PCa, such as androgen deprivation therapy (ADT) and chemotherapy or radiotherapy, are being investigated in several ongoing trials [66]. Immune check point inhibitors used in PCa have been fully reviewed by Modena et al. [67]. There are many trials ongoing in PCa, testing ipilimumab, tremelimumab, nivolumab, sipuleucel-T, prostvac-VF, DCVAC/PCa, GVAX and tasquinimod as a single agent or in combination with other agents. These therapies have been reviewed extensively by Silvestri et al. [18].
Adoptive T cell therapy
Prostate stem cell antigen (PSCA) and prostate-specific membrane antigen (PSMA) have been the most attractive antigens targeted by CAR-T cells in PCa. Preclinical studies showed that PSMA-CAR-T cells are able to proliferate and detect PSMA+ cells both in vitro and in vivo [68, 69]. A first generation CAR containing scFv of 7F5, an antibody with high affinity to PSCA, was tested in mice and showed activation of anti-tumor response by CAR-T cells [70]. Other researchers have confirmed delayed tumor growth mediated by PSCA-CAR-T cells in mice utilizing 1G8 and Ha1-4.117 antibodies [71]. However, the mice were not cured, suggesting that the high cytotoxicity of CAR-T cells may not be enough for in vivo treatment. New strategies have been developed to increase the efficiency of CAR-T cell therapy. For example, a low affinity CAR-T against PSCA in combination with a high affinity CAR-T against PSMA has been tried by Kloss et al. This treatment showed efficient destruction of double positive cells, proposing a new strategy for attaining persistent anti-tumor response [72].
An in vivo study evaluated the ability of PSMA-CAR-T cells in metastatic PCa. Besides depicting the effectiveness of CAR-T in eradicating disseminated PCa, they demonstrated that human CAR-T cells can survive better in non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice than in SCID or Rag2−/−/γc−/− mice [73].
Another strategy is using bispecific antibodies (diabodies) or bispecific T cell engagers (BITEs), which bind to both CD3 on T cells and a specific antigen on tumor cells, potentially resulting in T cells coming in contact with tumor cells. Diabodies targeting PSCA and PSMA have been developed and tested in vitro [74–76]. However, using diabodies in animal studies only delayed tumor growth and did not stop tumor progression. Hence, diabodies as a single regimen may not be effective, since they are unable to provide the cellular memory response that CAR-T therapy can potentially elicit [77].
PCa metastasis commonly occurs in lymph nodes and bone. Bone microenvironment creates a serious challenge for the infusing CAR-T cells. Aberrant angiogenesis commonly occurs in bone metastases and vascular endothelial growth factor (VEGF) plays a key role in establishing the bone metastasis [78]. Therapies that normalize angiogenesis rather than demolishing them, have shown to improve responses to immunotherapy [79]. Therefore, vascular normalization seems to be required for increasing CAR-T cell efficacy targeting bone metastasis.
Unfortunately, there are not many clinical trials exploiting CAR-T cells in PCa patients and more research is required. An anti-PSMA-CAR has been evaluated in a clinical trial by Slovin et al. They have tested the safety of different doses and generated a protocol for ex vivo transduction, expansion and clinical administration of PSMA-CAR-T cells in clinical trials (NCT01140373). This study is estimated to be completed by June 2017 [80].
2.5. Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is the third leading cause of cancer death in the world and more than 600,000 patients die from this disease annually [81]. HCC usually arises in the clinical setting of chronic liver disease, defined as the hypercarcinogenic state. To date, available options for HCC ranges from loco-regional treatment (tumor ablation via different techniques such as radiofrequency, ethanol injection, etc.) to chemotherapies, including sorafenib. These therapies are effective on the disease with smaller and limited nodules, but they are not adequate for the HCC with large and abundant nodules, and recurrences. Recent studies have focused on developing immunotherapies targeting specific markers in HCC and numerous molecular targets have been explored. Immunological approaches in HCC have mainly concentrated on target discovery, adoptive T cells, dendritic cell-based vaccines, natural killer (NK) cells, NKT cells, and peptide vaccines [82]. GPC3 is an attractive target for HCC immunotherapy. GPC3 peptide vaccine was tested in an early trial in 33 patients with HCC, and vaccine tolerability and anti-tumor response was recorded. One patient showed partial response, and 19 patients showed stable disease after two months. GPC3-specific CTL response was observed in 30 out of 33 patients, and its frequency was correlated with patient’s overall survival [83].
Adoptive T cell therapy
To date, there are only a few clinical trials evaluating the efficacy of engineered T cells in HCC. Adoptive T cell therapy using GPC3-redirected CD8+ CART cell was carried out in a HCC mouse model. Results confirmed the ability of GPC3-CAR-T in elimination of GPC3+cancer cells, which offers a potential therapeutic strategy to treat HCC [84]. Chronic infection with the hepatitis B virus (HBV) has been linked to the development of HCC for more than 30 years [85]. In one study, it was proposed that genetically modified T cells could be used to potentially reactivate the virus-specific T cells in chronic HBV patients and initiate a response against tumors in HBV-mediated HCC. They showed that T cells engineered to express HBV-specific TCR were able to recognize and lyse HBV infected hepatocyte-like cell lines. Also, HCC cell lines with natural HBV-DNA integration could be detected by HBV-specific TCR-redirected T cells [86].
2.6. Pancreatic cancer
Pancreatic cancer is the twelfth most common cancer worldwide. However, it is almost always fatal and is the seventh leading cause of cancer related deaths in both sexes combined worldwide [1] and the 4th leading cause of cancer-related deaths in USA [87]. It is predicted to become the 2nd leading cause by 2030 [88] and five-year survival remains at a dismal 5% [87]. Pancreatic ductal adenocarcinoma (PDA) which accounts for over 90% of pancreatic cancers, remains treatment refractory [89]. Immunotherapy of PDA can be categorized into four main avenues: monoclonal antibodies/checkpoint inhibitors/immune modulators, therapeutic vaccines, cytokines, and adoptive T cell therapy [90].
Several mAbs have been tested in PDA such as PAM4 or clivatuzumab (a MUC1-specific 90Yttrium-labled mAb) [91], trastuzumab (anti-HER2) [92], cetuximab and matuzumab (anti-EGFR) [93], bevacizumab (anti-VEGF receptor or VEGFR) [94], CD40 agonist mAb [95], and NPC-1C or NEO-102 (anti-MUC5A) [96]. Our lab has developed a mAb named TAB004 (patent US20140010759 A1) which precisely detects tMUC1 in breast cancer mouse model [97], as well as shed tMUC1 and the MUC1 on cancer stem cells (CSC) in the blood and tissue of PDA patients [98, 99]. TAB004 antibody, as a whole molecule and its fragments, are being exploited as a targeting agent in various immunotherapies, including generating CAR-T cells (unpublished study).
PD-1, PDL-1, and CTLA-4 are the key immune regulators and have been immune targets. Ipilimumab (anti-CTLA-4), which had previously shown remarkable results in metastatic melanoma patients [100], was tested in a phase II clinical trial on 27 PDA patients, and the outcome was not encouraging [101]. However, a phase Ib study with ipilimumab, in combination with gemcitabine on 13 advanced or metastatic PDA patients, revealed more promising results [102]. An anti-PDL-1 mAb (MEDI4736) has been tested and has shown efficacy in xenograft mouse model [103, 104]. This antibody is being further evaluated in phase I and II clinical trial of metastatic PDA and in combination with other chemotherapeutic agents (NCT02558894) [105]. Two other PD-1 inhibitors, nivolumab and pembrolizumab, have been tried in PDA patients. Using these mAbs as single agent treatments has not proved very effective [106], and thus new trials combining these antibodies with other agents such as vaccines or chemotherapy have emerged.
Thus far, the most clinically progressed immunotherapy for PDA has been tumor vaccines. Vaccines targeting different antigens include mesothelin [107], MUC-1 [108–110], CEA & MUC1 [111], heat-shock proteins [112], Kras [113], personalized peptide vaccination (using multiple peptides based on the pre-existing immunity) [114, 115], Kinesin Family Member 20A (KIF20A) [116, 117], Wilms tumor-1 [118], and VEGFR-2 [119, 120]. The most promising results arise from allogenic whole tumor cell expressing GM-CSF, dendritic cells expressing the antigens, and telomerase peptide vaccines in combination with chemotherapy drugs. Among the antigens, MUC1 has been one of the most popular antigens to target PDA. We have previously shown that combination of a MUC1-based vaccine with a cyclooxygenase-2 inhibitor (celecoxib), and low-dose chemotherapy (gemcitabine) prevents PDA progression in a preclinical PDA model [121].
Cytokines, such as IL-10 which has been known to promote anti-tumor response, have been administered to patients with limited efficacy [122, 123]. mAb against IL-17B receptor signaling showed improved survival in an orthotropic xenograft model of PDA [124].
Adoptive T cell therapy
MSLN is a cell surface antigen present on normal mesothelial cells and overexpressed in several human tumors such as mesothelioma, lung, pancreas, breast, ovarian, and other solid cancers [27]. MSLN-CAR-T cell therapy is in a phase I study and has demonstrated anti-tumor response for the first time in PDA and has shown the development of new antibodies [125]. Ongoing trials are testing genetically engineered T cells to target the immunogenic peptide derived from the cancer-testis antigen (NY-ESO-1), an antigen usually found in normal testis and in a variety of tumors (NCT01967823). Other researchers have shown MUC1-CAR-T cells as a potential candidate for PDA therapy [126, 127], including our lab (manuscript in preparation). A clinical trial has started recently using anti-MUC1 CAR-T cells for patients with MUC1+ advanced refractory solid tumors (NCT02587689). The orphan tyrosine kinase receptor, ROR1, is expressed by many tissues during embryogenesis but is absent in many organs after maturation, except for B cell precursor and in low levels in the pancreas, lung, and adipose tissue. ROR1 targeting CAR-T cells have also been evaluated in preclinical trials and it showed effectiveness in leukemia and some solid tumors, including pancreatic cancer [128]. ROR1-CAR-T safety was confirmed in non-human primates [129].
2.7. Ovarian cancer
Ovarian cancer is the seventh most common cancer and the eighth leading cause of cancer related deaths in women in the world (239,000 cases and 152,000 deaths) [1]. Augmented number of tumor infiltrating lymphocytes (TILs) in ovarian tumor has been associated with better prognosis. Tregs play a key role in immune tolerance to cancer cells by suppressing TILs in tumor microenvironment. This fact has led to emergence of numerous immunotherapies in ovarian cancer. Despite many studies supporting the evidence of successful induction of anti-tumor T cells, these results have not been significantly translated to clinical. As already shown for other tumors, immune checkpoint blockade, primarily antibodies targeting PD-1 (nivolumab, pembrolizumab) and PD-L1 (BMS-936559, avelumab), could hold the key to the future of ovarian cancer treatment. Among them avelumab and nivolumab have been more effective in controlling the disease [130].
Adoptive T cell therapy
Folate receptor α (FRα) is glycosylphosphatidyl-inositol (GPI)-anchored cell surface protein [131] overexpressed in epithelial ovarian cancer and is an excellent target for new promising therapies [132]. Although FRα-specific CAR-T cells showed specific killing of cancer cells in vitro and in preclinical mouse models, the clinical responses to this treatment was rather disappointing in patients with metastatic ovarian cancer. No tumor reduction was observed in 14 patients studied. Inefficacy of treatment was attributed to lack of specific T cells trafficking to tumor areas, short persistence of the transferred T cells, and presence of inhibitory factors in serum of patients [133]. A study showed that anti-tumor T cell function could be improved by blocking LAG-3 and PD-1. Circulating NY-ESO-1-specific CD8+ T cells derived from ovarian cancer patients exhibited higher proliferation and cytokine production in vitro after dual blockade of LAG-3 and PD-1 [134]. MUC-16ecto is another target for ovarian cancer, which is the retained portion of MUC-16 (CA125) after cleavage. MUC-16ecto is overexpressed on a majority of ovarian tumors, and shed CA125 is used as a diagnostic/prognostic marker for ovarian cancer patients. It was previously shown that MUC-16ecto-CAR-T cells eliminated orthotopic human ovarian cancer xenografts in SCID-Beige mice [135]. To improve function of T cells in the tumor microenvironment, MUC-16ecto-CAR-T cells were engineered to express IL-12. These IL-12-secreting CAR-T cells exhibited enhanced anti-tumor efficacy as determined by increased survival, prolonged persistence of T cells, and higher systemic IFN-γ in SCID-Beige mice with human ovarian cancer xenografts [136]. Thus, IL-12-secreting MUC-16ecto-CAR-T cells are being evaluated in a phase I clinical trial and have shown encouraging results [137]. Recently, CE7-epitope of L1-CAM, which is a cell adhesion molecule aberrantly expressed in multiple cancers, was targeted by CAR-T cells as a treatment for advanced ovarian cancer. Adoptive transfer of these CE7 receptor-CAR-T cells demonstrated a significant regression in SKOV3 xenograft tumors in mouse model [138].
3. Limitations of CAR-T cell therapy in adenocarcinomas
3.1. Hurdles in epithelial cancers: Trafficking & tumor microenvironment
Despite enormous success of CAR-T cell therapy for hematopoietic cancers, unfortunately, less encouraging results have been shown for epithelial tumors. This is due to several factors. The mechanical barrier in epithelial tumors, which is absent in liquid tumors, impede successful infiltrating of T cells to tumor sites. Unlike the “liquid tumor” setting of hematologic malignancies, CAR-T cells must pass multiple barriers in order to reach the tumor site. Potential mismatches of T cell chemokine receptors and their ligands on tumor cells and the presence of the dense stroma in most epithelial tumors makes T cell trafficking truly cumbersome. Numerous pre-clinical studies have shown that combining CAR-T cells with chemokine receptors improves migration and infiltration of T cells, once they are inside the tumor bed [139]. Moreover, antigen loss and heterogeneity of epithelial tumors contribute to the ineffectiveness of CAR-T cell [140]. Even after reaching the tumor sites, T cells must overcome various challenges in order to exert their anti-tumor effect. These challenges include a hostile tumor microenvironment enriched by oxidative stress, nutritional depletion, acidic pH, hypoxia, existence of numerous suppressive soluble factors and cytokines, suppressor immune cells such as Tregs, myeloid derived suppressor cells (MDSC), tumor-associated macrophages (TAM) or neutrophils (TAN), intrinsic negative regulatory mechanisms of T cells (expression of inhibitory receptors), and over-expression of inhibitory molecules and immune checkpoints [140]. To reduce these inhibitory effects, coupling pro-inflammatory cytokines, such as IL-12, with CAR-T cells, and also combinations of CAR-T cells with immune-checkpoint inhibitors, are currently being tested [141].
A possible down side of CAR-T cell therapy is the fact that these T cells recognize antigen independently of MHC molecules. Although this independency is considered a strength of CAR-T cells, it also limits the pool of antigens targetable by CAR, as a majority of tumor-associated antigens (TAAs) are intracellular neoantigens being solely expressed in the context of MHC [142]. This hurdle is not exclusive to epithelial solid tumors.
3.2. Toxicity
Lastly, the review cannot be completed without providing information on the potential toxicities attributed to CAR-T cells. Much of the toxicity data exists for the CAR-T cells against hematopoietic tumors, but not as much in epithelial solid tumors. The toxicities include cytokine release syndrome (CRS, systemic inflammatory response following CAR-T cells activation), B cell aplasia (specific for CD19 CAR-T cells), neurological toxicity, “on-target, off-tumor” toxicity (reactivity to target antigen on healthy tissues), anaphylaxis or allergy (host immune reaction to foreign antigen on CART cell), and insertional oncogenesis (mutagenesis by viral genes) [143].
The most prominent cytotoxicity seen in CD19 CAR-T cell trials is CRS which is associated with rapid T cell proliferation [144]. The main reason for this effect is not clearly explained yet; however, it is likely due to infused CAR-T cells secreting products that can trigger a toxic release of pro-inflammatory cytokines such as IL-6, IFN-γ, and TNFα [145]. CRS symptoms range from high fever and myalgia to unstable hypotension and respiratory failure. This effect was not observed in pre-clinical animal models, and hence it was unexpected [146].
Other types of toxicity which have been profoundly described in recent CD19 CAR-T cell trials are B cell aplasia and a diverse array of neurological toxicities [147]. B cell aplasia, which is an on-target/off-tumor consequence, is associated with hypogammaglobulinemia which is easily managed by γ-globulin replacement therapy [148]. Neurologic toxicities seem to be exclusive to CD19-targeted therapies and have been reported in almost all CD19 CAR constructs as well as the CD19 BiTE® blinatumomab [141].
It is said that CRS has not yet been reported for solid tumor trials. This could be due to low level of T cell engraftment and proliferation in solid tumors compared to leukemia. However, with development of enhanced CAR and utilizing stronger lymphodepletion regimens, this kind of toxicity may become a problem [140]. In one case, CRS in solid tumors has occurred due to “on-target, off-tumor” toxicity. To avoid T cells reacting to normal tissues, the most suitable tumor antigens need to be targeted. In addition, we need to develop tumor-specific antibody that does not recognize and bind to normal tissue. Unlike CD19 which is exclusively expressed on B lymphocytes, majority of solid tumor antigens are also present at low levels on normal cells [141]. In contrast to the ease of management of B cell aplasia, off-tumor toxicities that may occur in solid tumors are not easily controlled. A prominent example is targeting ERBB2 (HER2/neo), a marker overexpressed on both breast and colon cancers. A patient with metastatic colon cancer was treated with 3rd generation ERBB2-CD28-41BBζ CAR (based on Trastuzumab mAb), in 2010, and experienced a severe respiratory distress associated with the development of new lung infiltrates. Infiltrates were also found at low levels in several normal organs including heart and the pulmonary vasculature. The patient died 5 days after from CRS, despite intensive medical management. CRS was attributed to CAR-T cell targeting ERBB2 on lung epithelium [28]. On the contrary, this unexpected toxicity has not been reported repeatedly in solid tumors. It may have been stimulated by high dose of infused CART cells (10 million cells) and preconditioning therapy in the patient, since subsequent studies using a different ERBB2-specific CAR with considerably lower doses of CAR-T cells have proven safe in treating patients without preconditioning chemotherapy [17].
Anaphylaxis, allergy, or graft versus host disease (GVHD) has not been a major concern with CART cell therapy, since autologous T cells are used, and infused cells are the donor’s own cells. Also, no reaction to exogenous viral genes has been recorded following CAR-T treatment [149, 150]. Despite this proven safety to reduce the time and cost of treatment, establishment of an “off-the-shelf” or “third-party” cell bank, wherein universal CAR-T cells are available to anyone, seems like an attractive solution. In this case, immune reactivity to graft is of higher concern. To manage this, two strategies have been suggested so far, including using specific cells for CAR expression and silencing the immunogenic molecule, TCR, on T cells [151]. These approaches are in process and can broaden the application of CAR-T cells in the future. Lastly, insertional oncogenesis may occur via engineered T cells. Viral vectors or plasmid DNAs used to transfer gene into T cells might carry the risk of malignant transformation in clinical setting. Despite copious studies demonstrating safety of viral vectors, it is too early to decide if this approach is safe for larger patient populations [3].
3.3. Mechanisms to combat toxicity
Despite impressive clinical efficacy of CAR-T cells, severe treatment-related toxicities have restricted the universal use of CAR-T cells [152]. To address CAR-T cell toxicity, numerous strategies prominently involving CAR design have recently been devised:
Dual targeting strategy to increase specificity and safety. For example, a modified T cell expresses two CARs, wherein T cell activation signal 1 (via CD3ζ) is physically separated from the costimulatory signal 2 (via CD28) and each CAR recognizes a separate tumor antigen. These CAR-T cells exhibit full activation and function only if both CARs are engaged [153, 49, 72]. This approach provides a path for controlling CAR-T cell selectivity and activity in a way that retains both effectiveness and safety.
Suicide genes. These specific genes have been introduced into the CAR vector [154] and CAR-T cells get permanently eradicated upon activation of these suicide genes. So far, use of herpes simplex virus thymidine kinase (HSV-TK) and an inducible caspase 9 (iCasp9) genes have shown success in clinical trials [153].
Co-expression of a depletable receptor. A polypeptide on cell surface can be targeted by depleting antibodies in order to eliminate engineered T cells. This strategy is already in clinical use, for example, EGFR and CD20 [155].
Inhibitory CARs (iCAR). When target antigen is shared between tumor and normal cells, off-tumor toxicity may occur. If normal tissues express another antigen which is not expressed by tumor cells, it could be targeted by CAR-T via a receptor which triggers an inhibitory response. This receptor which comprises intracellular domain of T cell inhibitory receptors (PD-1 and CTLA-4) is co-expressed by CAR-T cells. Fedorov et al. have shown that iCAR protects normal tissues in preclinical mouse model [156]. Significantly, primary regulatory effect of iCARs was selective and transitory, allowing for future activation of T cells upon later encounter with target cells that exclusively express the antigen [157].
-
Switchable CARs (sCAR) and multi-chain CARs (mcCARs). These CAR-T cells need an intermediate switch molecule to get fully activated [158, 159]. In sCAR design, an antibody-based switch molecule is co-infused, and it bridges the target cell and the sCAR expressing T cell, while mcCARs are only activated in the existence of the small-molecules such as rapamycin. Switchable CARs have been designed to reversibly control CAR-T cell activity and specificity in immunocompetent mouse model of CD19 and CD22 expressing cancers.
Moreover, multiple antigens could be targeted using the same CAR simply by infusing various or bispecific switch molecules. For instance, two or more switch molecules such as anti-CD19 and -CD22 fused to FITC could be targeted by the same anti-FITC CAR-T cells (“universal CAR”) [152].
Masked CARs. In this design, the antigen-recognition domain of CAR is sterically blocked by a substrate peptide which is cleaved in the presence of matrix metalloproteinases. When T cells enter the tumor microenvironment, which is enriched by these enzymes, substrate will be cleaved, and binding capacity of T cells will be unmasked. Once the receptor is unblocked, T cells mainly localize to the tumor area. Use of masked CARs provides a way to focus CAR-T activity toward targets shared with healthy tissues [160].
Self-limited CAR. This strategy uses mRNA rather than retro/lenti-virus to transiently express CAR receptor [161, 125].
Pharmacologic therapy. IL-6 receptor blockade with tocilizumab is used to control CRS. Corticosteroids are used for neurologic toxicities and for CRS not responsive to tocilizumab. However, pharmacologic management of these therapies remains challenging, since it entails the risk of suppressing anti-tumor immune response by CAR-T cells [162]. Hurdles in CAR-T cell therapy of epithelial tumors and the ways to combat them are illustrated in figure 2.
Figure 2. Hurdles in CAR-T cell therapy of epithelial tumors.
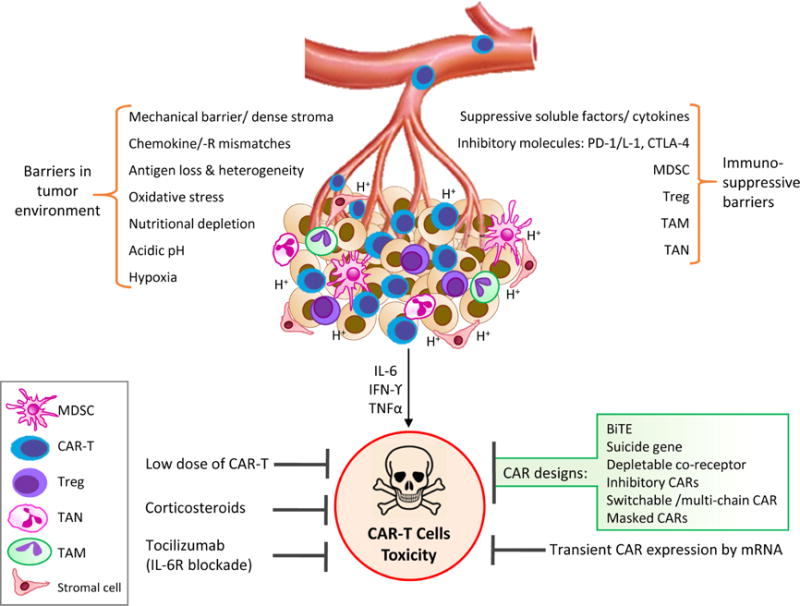
T cells face multiple barriers in solid epithelial tumors which negatively affect T cell trafficking and function within the tumor microenvironment. CAR-T cell therapy may result in toxicity which could be managed via different suggested mechanisms.
4. Conclusion and perspective
Despite challenges associated with CAR-T cell treatment, immunotherapy remains the most promising therapy that is revolutionizing how we treat cancer. This is exemplified with FDA’s approval of anti-tumor antibodies, immune checkpoint inhibitors, cellular and peptide tumor vaccines, antibody drug conjugates (ADCs), and adoptive T cell therapies for various epithelial malignancies. No cancer treatment is devoid of toxicity, and we have learnt over the years how to manage it. Therefore, much still needs to be learnt about how to mitigate the risks associated with immunotherapy. For targeted CAR-T cell therapy, most critical is the selection of the tumor antigen, and even more critical, is the generation of antibodies that bind to tumor cells but spares normal tissue. One such antigen that is recognized as the 2nd most targetable antigen by NCI is the tumor associated form of MUC1 [163]. The recent MUC1-CAR-T cell therapeutic effect in a preclinical model for breast and pancreatic cancer shows promise [127] but needs further validations. The generation of the novel antibody, TAB004, that is highly specific against tMUC1 [97–99, 164, 165] is clinically relevant for future development of targeted therapy, including generation of ADCs/nano-ADCs and CAR-T cells for various epithelial solid tumors including those of breast, pancreatic, lung, colon, stomach, and others that express tMUC1 in high percentage of patient tumors.
Cancer rates throughout the world are increasing at an alarming rate. Yet, we have not seen a significant change in the standard of care treatment options. It is time to bring cancer research to the 21st century through the exploration of novel therapies including immunotherapy. We have already witnessed the potential benefits of immunotherapy, with the use of antibodies and T cell therapies, in treating multiple cancers. We are at the forefront of an era in cancer research and treatment where we no longer have to rely solely on surgery, radiotherapy, and chemotherapy. The time for immunotherapy is here, and the potential is bright.
Acknowledgments
We are grateful to Priyanka Grover for editing the manuscript.
Footnotes
Search strategy
Authors scanned the PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and Google Scholar website using key words (aforementioned) and the website https://clinicaltrials.gov/ was scanned for existing, running and eligible studies.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to disclose.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Int J Cancer. 2015;136:E359. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Kirkham N, R Lemoine N. Progress in Pathology. Cambridge University Press; Cambridge: 2001. [Google Scholar]
- 3.Almåsbak H, Aarvak T, Vemuri MC. J Immunol Res. 2016;2016:5474602. doi: 10.1155/2016/5474602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross G, Waks T, Eshhar Z. PNAS. 1989;86:10024. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruella M, June CH. Curr Hematol Malig Rep. 2016;11:368. doi: 10.1007/s11899-016-0336-z. [DOI] [PubMed] [Google Scholar]
- 6.Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. Gene Ther. 2001;8:1601. doi: 10.1038/sj.gt.3301570. [DOI] [PubMed] [Google Scholar]
- 7.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH. PNAS. 2009;106:3360. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Mol Ther. 2009;18:413. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chmielewski M, Abken H. Expert Opin Biol Ther. 2015;15:1145. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 10.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. N Engl J Med. 2011;365:1673. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Rivière I. Mol Ther Oncolytics. 2016;3:16015. doi: 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gill S, Maus MV, Porter DL. Blood Rev. 2016;30:157. doi: 10.1016/j.blre.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. J Natl Cancer Inst. 1994;86:1159. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 15.Fousek K, Ahmed N. Clin Cancer Res. 2015;21:3384. doi: 10.1158/1078-0432.CCR-14-2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Blood. 2011;118:6050. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, Gray T, Wu MF, Liu H, Hicks J, Rainusso N, Dotti G, Mei Z, Grilley B, Gee A, Rooney CM, Brenner MK, Heslop HE, Wels WS, Wang LL, Anderson P, Gottschalk S. J Clin Oncol. 2015;33:1688. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silvestri I, Cattarino S, Giantulli S, Nazzari C, Collalti G, Sciarra A. Cancers (Basel) 2016;8:E64. doi: 10.3390/cancers8070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ledford H. Nature. 2014;508:24. doi: 10.1038/508024a. [DOI] [PubMed] [Google Scholar]
- 20.PDQ Adult Treatment Editorial Board. Non-Small Cell Lung Cancer Treatment (PDQ): Patient Version. National Cancer Institute; Bethesda (MD), USA: 2002. (Book Chapter NBK65917). [PubMed] [Google Scholar]
- 21.Rice SJ, Miller B, Wagman M, Jamorabo DS, Liu X, Belani C. Curr Mol Pharmacol. :2015. doi: 10.2174/1874467208666150716120108. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 22.Herzberg B, Campo MJ, Gainor JF. The Oncologist. 2016;0189 doi: 10.1634/theoncologist.2016-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khanna P, Blais N, Gaudreau PO, Corrales-Rodriguez L. Clin Lung Cancer. 2016:S1525. doi: 10.1016/j.cllc.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Kachala SS, Bograd AJ, Villena-Vargas J, Suzuki K, Servais EL, Kadota K, Chou J, Sima CS, Vertes E, Rusch VW, Travis WD, Sadelain M, Adusumilli PS. Clin Cancer Res. 2014;20:1020. doi: 10.1158/1078-0432.CCR-13-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas A, Chen Y, Steinberg SM, Luo J, Pack S, Raffeld M, Abdullaev Z, Alewine C, Rajan A, Giaccone G, Pastan I, Miettinen M, Hassan R. Oncotarget. 2015;6:11694. doi: 10.18632/oncotarget.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Science Translational Medicine. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morello A, Sadelain M, Adusumilli PS. Cancer Discovery. 2016;6:133. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Mol Ther. 2010;18:843. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, Zhou L, Loew A, Young RM, June CH, Zhao Y. Cancer Res. 2015;75:3596. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBaun MR, Ess J, Saunders S. Mol Genet Metab. 2001;72:279. doi: 10.1006/mgme.2001.3150. [DOI] [PubMed] [Google Scholar]
- 31.Baumhoer D, Tornillo L, Stadlmann S, Roncalli M, Diamantis EK, Terracciano LM. Am J Clin Pathol. 2008;129:899. doi: 10.1309/HCQWPWD50XHD2DW6. [DOI] [PubMed] [Google Scholar]
- 32.Li K, Pan X, Bi Y, Xu W, Chen C, Gao H, Shi B, Jiang H, Yang S, Jiang L, Li Z. Oncotarget. 2016;7:2496. doi: 10.18632/oncotarget.6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kakarla S, Chow KKH, Mata M, Shaffer DR, Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K, Gottschalk S. Mol Ther. 2013;21:1611. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li CI, Uribe DJ, Daling JR. Br J Cancer. 2005;93:1046. doi: 10.1038/sj.bjc.6602787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mittendorf EA, Hunt KK. Am J Hematol Oncol. 2015;11:6. [Google Scholar]
- 36.Hudis CA. N Engl J Med. 2007;357:39. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 37.Barok M, Isola J, Palyi-Krekk Z, Nagy P, Juhasz I, Vereb G, Kauraniemi P, Kapanen A, Tanner M, Vereb G, Szollosi J. Mol Cancer Ther. 2007;6:2065. doi: 10.1158/1535-7163.MCT-06-0766. [DOI] [PubMed] [Google Scholar]
- 38.Taylor C, Hershman D, Shah N, Suciu-Foca N, Petrylak DP, Taub R, Vahdat L, Cheng B, Pegram M, Knutson KL, Clynes R. Clin Cancer Res. 2007;13:5133. doi: 10.1158/1078-0432.CCR-07-0507. [DOI] [PubMed] [Google Scholar]
- 39.Perez EA, Thompson EA, Ballman KV, Anderson SK, Asmann YW, Kalari KR, Eckel-Passow JE, Dueck AC, Tenner KS, Jen J, Fan JB, Geiger XJ, McCullough AE, Chen B, Jenkins RB, Sledge GW, Winer EP, Gralow JR, Reinholz MM. J Clin Oncol. 2015;33:701. doi: 10.1200/JCO.2014.57.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Desmedt C, Piccart MJ, Loibl S, Denkert C, Smyth MJ, Joensuu H, Sotiriou C. Ann Oncol. 2014;25:1544. doi: 10.1093/annonc/mdu112. [DOI] [PubMed] [Google Scholar]
- 41.Loi S, Sirtaine N, Piette F, Salgado R, Viale G, van Eenoo F, Rouas G, Francis P, Crown JP, Hitre E, de Azambuja E, Quinaux E, Di Leo A, Michiels S, Piccart MJ, Sotiriou C. J Clin Oncol. 2013;31:860. doi: 10.1200/JCO.2011.41.0902. [DOI] [PubMed] [Google Scholar]
- 42.Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Dolled-Filhart M, Emancipator K, Gonzalez EJ, Houp J, Pathiraja K, Karantza V, Iannone R, Gause CK, Cheng JD, Buisseret L. Cancer Res. 2015;75:S1. [Google Scholar]
- 43.Adams S. J Clin Oncol. 2016;34 Board 114. [Google Scholar]
- 44.Emens LA, Braiteh FS, Cassier P, DeLord JP, Eder JP, Shen X, Xiao Y, Wang Y, Hegde PS, Chen DS, Krop I. Clin Cancer Res. 2015;25 Abstract PD1. [Google Scholar]
- 45.Mittendorf EA, Clifton GT, Holmes JP, Schneble E, van Echo D, Ponniah S, Peoples GE. Ann Oncol. 2014;25:1735. doi: 10.1093/annonc/mdu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mittendorf EA, Peoples GE. Oncology (Williston Park) 2016;30:475. [PubMed] [Google Scholar]
- 47.Madan RA, Arlen PM, Gulley JL. Expert Opin Biol Ther. 2007;7:543. doi: 10.1517/14712598.7.4.543. [DOI] [PubMed] [Google Scholar]
- 48.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM, Maher J. J Immunol (Baltimore, Md: 1950) 2008;180:4901. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 49.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J. J Clin Immunol. 2012;32:1059. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 50.Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. J Immuno (Baltimore, Md: 1950) 1993;151:6577. [PubMed] [Google Scholar]
- 51.Sun M, Shi H, Liu C, Liu J, Liu X, Sun Y. Breast Cancer Res. 2014;16:1. doi: 10.1186/bcr3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolfgang CD, Essand M, Vincent JJ, Lee B, Pastan I. Proc Natl Acad Sci USA. 2000;97:9437. doi: 10.1073/pnas.160270597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hillerdal V, Nilsson B, Carlsson B, Eriksson F, Essand M. Proc Natl Acad Sci USA. 2012;109:15877. doi: 10.1073/pnas.1209042109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halama N, Michel S, Kloor M, Zoernig I, Benner A, Spille A, Pommerencke T, von Knebel DM, Folprecht G, Luber B, Feyen N, Martens UM, Beckhove P, Gnjatic S, Schirmacher P, Herpel E, Weitz J, Grabe N, Jaeger D. Cancer Res. 2011;71:5670. doi: 10.1158/0008-5472.CAN-11-0268. [DOI] [PubMed] [Google Scholar]
- 55.Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Eur J Cancer. 2016;52:50. doi: 10.1016/j.ejca.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 56.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DAA. Cancer Res. 2012;72:917. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. Nat Immunol. 2005;6:1245. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 58.Anderson AC. Cancer Immunol Res. 2014;2:393. doi: 10.1158/2326-6066.CIR-14-0039. [DOI] [PubMed] [Google Scholar]
- 59.Turnis ME, Andrews LP, Vignali DA. Eur J Immunol. 2015;45:1892. doi: 10.1002/eji.201344413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baksh K, Weber J. Semin Oncol. 2015;42:363. doi: 10.1053/j.seminoncol.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 61.Chmielewski M, Rappl G, Hombach AA, Abken H. Gene Ther. 2013;20:177. doi: 10.1038/gt.2012.21. [DOI] [PubMed] [Google Scholar]
- 62.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA. Mol Ther. 2011;19:620. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kobold S, Steffen J, Chaloupka M, Grassmann S, Henkel J, Castoldi R, Zeng Y, Chmielewski M, Schmollinger JC, Schnurr M, Rothenfusser S, Schendel DJ, Abken H, Sustmann C, Niederfellner G, Klein C, Bourquin C, Endres S. J Natl Cancer Inst. 2015;107:364. doi: 10.1093/jnci/dju364. [DOI] [PubMed] [Google Scholar]
- 64.Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. Clin Cancer Res. 2012;18:6436. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]
- 65.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF. N Engl J Med. 2010;363:411. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 66.Arlen PM, Mohebtash M, Madan RA, Gulley JL. Future Oncol (London, England) 2009;5:187. doi: 10.2217/14796694.5.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Modena A, Ciccarese C, Iacovelli R, Brunelli M, Montironi R, Fiorentino M, Tortora G, Massari F. Oncol Rev. 2016;10:293. doi: 10.4081/oncol.2016.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gade TP, Hassen W, Santos E, Gunset G, Saudemont A, Gong MC, Brentjens R, Zhong XS, Stephan M, Stefanski J, Lyddane C, Osborne JR, Buchanan IM, Hall SJ, Heston WD, Riviere I, Larson SM, Koutcher JA, Sadelain M. Cancer Res. 2005;65:9080. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 69.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Nature Biotechnology. 2002;20:70. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 70.Morgenroth A, Cartellieri M, Schmitz M, Gunes S, Weigle B, Bachmann M, Abken H, Rieber EP, Temme A. The Prostate. 2007;67:1121. doi: 10.1002/pros.20608. [DOI] [PubMed] [Google Scholar]
- 71.Hillerdal V, Ramachandran M, Leja J, Essand M. BMC Cancer. 2014;14:30. doi: 10.1186/1471-2407-14-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Nat Biotech. 2013;31:71. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuccolotto G, Fracasso G, Merlo A, Montagner IM, Rondina M, Bobisse S, Figini M, Cingarlini S, Colombatti M, Zanovello P, Rosato A. PLoS One. 2014;9:e109427. doi: 10.1371/journal.pone.0109427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arndt C, Feldmann A, Koristka S, Cartellieri M, Dimmel M, Ehninger A, Ehninger G, Bachmann M. The Prostate. 2014;74:1335. doi: 10.1002/pros.22850. [DOI] [PubMed] [Google Scholar]
- 75.Baum V, Buhler P, Gierschner D, Herchenbach D, Fiala GJ, Schamel WW, Wolf P, Elsasser-Beile U. Immunotherapy. 2013;5:27. doi: 10.2217/imt.12.136. [DOI] [PubMed] [Google Scholar]
- 76.Buhler P, Wolf P, Gierschner D, Schaber I, Katzenwadel A, Schultze-Seemann W, Wetterauer U, Tacke M, Swamy M, Schamel WW, Elsasser-Beile U. Cancer Immuno Immunother. 2008;57:43. doi: 10.1007/s00262-007-0348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hillerdal V, Essand M. BioDrugs. 2015;29:75. doi: 10.1007/s40259-015-0122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roberts E, Cossigny DA, Quan GM. Prostate Cancer. 2013;2013:418340. doi: 10.1155/2013/418340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi S, Chen L, Huang G. Med Oncol. 2013;30:1. doi: 10.1007/s12032-013-0698-1. [DOI] [PubMed] [Google Scholar]
- 80.Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, Schöder H, Zanzonico P, Scher HI, Sadelain M, Riviere I. J Clin Oncol. 2013;31 abstr 72. [Google Scholar]
- 81.Breous E, Thimme R. J Hepatol. 2011;54:830. doi: 10.1016/j.jhep.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 82.Nakamoto Y. Hepatol Res. 2016 doi: 10.1111/hepr.12795. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 83.Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, Mizuno S, Ishii H, Nakachi K, Konishi M, Nakagohri T, Takahashi S, Gotohda N, Takayama T, Yamao K, Uesaka K, Furuse J, Kinoshita T, Nakatsura T. Clin Cancer Res. 2012;18:3686. doi: 10.1158/1078-0432.CCR-11-3044. [DOI] [PubMed] [Google Scholar]
- 84.Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, Kong J, Wang H, Yang S, Gu J, Li Z. Clin Cancer Res. 2014;20:6418. doi: 10.1158/1078-0432.CCR-14-1170. [DOI] [PubMed] [Google Scholar]
- 85.Di Bisceglie AM. Hepatology (Baltimore, Md) 2009;49:S56. doi: 10.1002/hep.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gehring AJ, Xue SA, Ho ZZ, Teoh D, Ruedl C, Chia A, Koh S, Lim SG, Maini MK, Stauss H, Bertoletti A. J Hepatol. 2011;55:103. doi: 10.1016/j.jhep.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 87.Poruk KE, Firpo MA, Adler DG, Mulvihill SJ. Annals of Surgery. 2013;257:17. doi: 10.1097/SLA.0b013e31825ffbfb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Cancer Res. 2014;74:2913. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 89.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, Song S, Harliwong I, Idrisoglu S, Nourse C, Nourbakhsh E, Manning S, Wani S, Gongora M, Pajic M, Scarlett CJ, Gill AJ, Pinho AV, Rooman I, Anderson M, Holmes O, Leonard C, Taylor D, Wood S, Xu Q, Nones K, Fink JL, Christ A, Bruxner T, Cloonan N, Kolle G, Newell F, Pinese M, Mead RS, Humphris JL, Kaplan W, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chou A, Chin VT, Chantrill LA, Mawson A, Samra JS, Kench JG, Lovell JA, Daly RJ, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Australian Pancreatic Cancer Genome, I. Kakkar N, Zhao F, Wu YQ, Wang M, Muzny DM, Fisher WE, Brunicardi FC, Hodges SE, Reid JG, Drummond J, Chang K, Han Y, Lewis LR, Dinh H, Buhay CJ, Beck T, Timms L, Sam M, Begley K, Brown A, Pai D, Panchal A, Buchner N, De Borja R, Denroche RE, Yung CK, Serra S, Onetto N, Mukhopadhyay D, Tsao MS, Shaw PA, Petersen GM, Gallinger S, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Schulick RD, Wolfgang CL, Morgan RA, Lawlor RT, Capelli P, Corbo V, Scardoni M, Tortora G, Tempero MA, Mann KM, Jenkins NA, Perez-Mancera PA, Adams DJ, Largaespada DA, Wessels LFA, Rust AG, Stein LD, Tuveson DA, Copeland NG, Musgrove EA, Scarpa A, Eshleman JR, Hudson TJ, Sutherland RL, Wheeler DA, Pearson JV, McPherson JD, Gibbs RA, Grimmond SM. Nature. 2012;491:399. [Google Scholar]
- 90.Kotteas E, Saif MW, Syrigos K. J Cancer Res Clin Oncol. 2016;142:1795. doi: 10.1007/s00432-016-2119-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gulec SA, Cohen SJ, Pennington KL, Zuckier LS, Hauke RJ, Horne H, Wegener WA, Teoh N, Gold DV, Sharkey RM, Goldenberg DM. Clin Cancer Res. 2011;17:4091. doi: 10.1158/1078-0432.CCR-10-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pratesi G, Petrangolini G, Tortoreto M, Addis A, Zunino F, Calcaterra C, Merlo A, Tagliabue E, Menard S, Balsari A. J Immunother (Hagerstown, Md: 1997) 2008;31:537. doi: 10.1097/CJI.0b013e31817c37ff. [DOI] [PubMed] [Google Scholar]
- 93.Assenat E, Azria D, Mollevi C, Guimbaud R, Tubiana-Mathieu N, Smith D, Delord JP, Samalin E, Portales F, Larbouret C, Robert B, Bibeau F, Bleuse JP, Crapez E, Ychou M, Pelegrin A. Oncotarget. 2015;6:12796. doi: 10.18632/oncotarget.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O’Reilly E, Wozniak TF, Picus J, Bhargava P, Mayer RJ, Schilsky RL, Goldberg RM. J Clin Oncol. 2010;28:3617. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, O’Dwyer PJ. Clin Cancer Res. 2013;19:6286. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morse M, Diaz LA, Azad NS, Laheru D, Haley S, Sleer LS, Arlen PM. J Clin Oncol. 2012;30 abstr 233. [Google Scholar]
- 97.Moore LJ, Roy LD, Zhou R, Grover P, Wu ST, Curry JM, Dillon LM, Puri PM, Yazdanifar M, Puri R, Mukherjee P, Dreau D. Trans Oncol. 2016;9:295. doi: 10.1016/j.tranon.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Curry JM, Thompson KJ, Rao SG, Besmer DM, Murphy AM, Grdzelishvili VZ, Ahrens WA, McKillop IH, Sindram D, Iannitti DA, Martinie JB, Mukherjee P. J Surg Oncol. 2013;107:713. doi: 10.1002/jso.23316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou R, Curry JM, Roy LD, Grover P, Haider J, Moore LJ, Wu ST, Kamesh A, Yazdanifar M, Ahrens WA, Leung T, Mukherjee P. Oncogene. 2016 doi: 10.1038/onc.2015.516. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain J-F, Testori A, Grob J-J, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen T-T, Humphrey R, Hoos A, Wolchok JD. N Engl J Med. 2011;364:2517. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 101.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA. J Immunother (Hagerstown, Md: 1997) 2010;33:828. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mohindra NA, Kircher SM, Nimeiri HS, Benson AB, Rademaker A, Alonso E, Blatner N, Khazaie K, Mulcahy MF. J Clin Oncol. 2015;33 abstr. e15281. [Google Scholar]
- 103.Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, Watkins A, Mullins S, Chodorge M, Andrews J, Bannister D, Dick E, Crawford N, Parmentier J, Alimzhanov M, Babcook JS, Foltz IN, Buchanan A, Bedian V, Wilkinson RW, McCourt M. Cancer Immunol Res. 2015;3:1052. doi: 10.1158/2326-6066.CIR-14-0191. [DOI] [PubMed] [Google Scholar]
- 104.Ibrahim R, Stewart R, Shalabi A. Semin Oncol. 2015;42:474. doi: 10.1053/j.seminoncol.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 105.Li Y, Li F, Jiang F, Lv X, Zhang R, Lu A, Zhang G. Int J Mol Sci. 2016;17:1151. doi: 10.3390/ijms17071151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. N Engl J Med. 2012;366:2455. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL, Onners B, Uram JN, Laheru DA, Lutz ER, Solt S, Murphy AL, Skoble J, Lemmens E, Grous J, Dubensky T, Jr, Brockstedt DG, Jaffee EM. J Clin Oncol. 2015;33:1325. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H, Day R, Troetschel M, Finn OJ. Cancer Immunol Immunother. 2005;54:254. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rong Y, Qin X, Jin D, Lou W, Wu L, Wang D, Wu W, Ni X, Mao Z, Kuang T, Zang YQ, Qin X. Clin Exp Med. 2012;12:173. doi: 10.1007/s10238-011-0159-0. [DOI] [PubMed] [Google Scholar]
- 110.Torres MP, Chakraborty S, Souchek J, Batra SK. Curr Pharm Des. 2012;18:2472. doi: 10.2174/13816128112092472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaufman HL, Kim-Schulze S, Manson K, DeRaffele G, Mitcham J, Seo KS, Kim DW, Marshall J. J Transl Med. 2007;5:60. doi: 10.1186/1479-5876-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maki RG, Livingston PO, Lewis JJ, Janetzki S, Klimstra D, Desantis D, Srivastava PK, Brennan MF. Dig Dis Sci. 2007;52:1964. doi: 10.1007/s10620-006-9205-2. [DOI] [PubMed] [Google Scholar]
- 113.Abou-Alfa GK, Chapman PB, Feilchenfeldt J, Brennan MF, Capanu M, Gansukh B, Jacobs G, Levin A, Neville D, Kelsen DP, O’Reilly EM. Am J Clin Oncol. 2011;34:321. doi: 10.1097/COC.0b013e3181e84b1f. [DOI] [PubMed] [Google Scholar]
- 114.Yutani S, Komatsu N, Yoshitomi M, Matsueda S, Yonemoto K, Mine T, Noguchi M, Ishihara Y, Yamada A, Itoh K, Sasada T. Oncol Rep. 2013;30:1094. doi: 10.3892/or.2013.2556. [DOI] [PubMed] [Google Scholar]
- 115.Yanagimoto H, Shiomi H, Satoi S, Mine T, Toyokawa H, Yamamoto T, Tani T, Yamada A, Kwon AH, Komatsu N, Itoh K, Noguchi M. Oncol Rep. 2010;24:795. doi: 10.3892/or_00000923. [DOI] [PubMed] [Google Scholar]
- 116.Asahara S, Takeda K, Yamao K, Maguchi H, Yamaue H. J Transl Med. 2013;11:291. doi: 10.1186/1479-5876-11-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Suzuki N, Hazama S, Ueno T, Matsui H, Shindo Y, Iida M, Yoshimura K, Yoshino S, Takeda K, Oka M. J Immunother (Hagerstown, Md: 1997) 2014;37:36. doi: 10.1097/CJI.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mayanagi S, Kitago M, Sakurai T, Matsuda T, Fujita T, Higuchi H, Taguchi J, Takeuchi H, Itano O, Aiura K, Hamamoto Y, Takaishi H, Okamoto M, Sunamura M, Kawakami Y, Kitagawa Y. Cancer Sci. 2015;106:397. doi: 10.1111/cas.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schmitz-Winnenthal FH, Hohmann N, Niethammer AG, Friedrich T, Lubenau H, Springer M, Breiner KM, Mikus G, Weitz J, Ulrich A, Buechler MW, Pianka F, Klaiber U, Diener M, Leowardi C, Schimmack S, Sisic L, Keller AV, Koc R, Springfeld C, Knebel P, Schmidt T, Ge Y, Bucur M, Stamova S, Podola L, Haefeli WE, Grenacher L, Beckhove P. Oncoimmunology. 2015;4:e1001217. doi: 10.1080/2162402X.2014.1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miyazawa M, Ohsawa R, Tsunoda T, Hirono S, Kawai M, Tani M, Nakamura Y, Yamaue H. Cancer Sci. 2010;101:433. doi: 10.1111/j.1349-7006.2009.01416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mukherjee P, Basu GD, Tinder TL, Subramani DB, Bradley JM, Arefayene M, Skaar T, De Petris G. J Immunol (Baltimore, Md: 1950) 2009;182:216. [PMC free article] [PubMed] [Google Scholar]
- 122.Kaufman HL, Rao JB, Irvine KR, Bronte V, Rosenberg SA, Restifo NP. J Immunother (Hagerstown, Md: 1997) 1999;22:489. doi: 10.1097/00002371-199911000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chard LS, Lemoine NR, Wang Y. Oncoimmunology. 2015;4:e1038689. doi: 10.1080/2162402X.2015.1038689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu Heng-Hsiung, Hwang-Verslues Wendy W, Lee Wen-Hsin, Huang Chun-Kai, Wei Pei-Chi, Chen Chia-Lin, Shew Jin-Yuh, Lee Eva Y-HP, Jeng Yung-Ming, Tien Yu-Wen, Che Ma, Lee WH. JEM. 2015;212:333. doi: 10.1084/jem.20141702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, Kalos M, June CH. Cancer Immunol Res. 2014;2:112. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, Fisher WE, Heslop HE, Rooney CM, Brenner MK, Leen AM, Vera JF. Mol Ther. 2014;22:623. doi: 10.1038/mt.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Posey AD, Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, Cogdill AP, Chen TJ, Song D, Scholler J, Kranz DM, Feldman MD, Young R, Keith B, Schreiber H, Clausen H, Johnson LA, June CH. Immunity. 2016;44:1444. doi: 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Clin Cancer Res. 2013;19:3153. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ, Kosasih PL, Rader C, Riddell SR. Cancer Immunol Res. 2015;3:206. doi: 10.1158/2326-6066.CIR-14-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weiss L, Huemer F, Mlineritsch B, Greil R. Memo. 2016;9:82. doi: 10.1007/s12254-016-0267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elnakat H, Ratnam M. Adv Drug Deliv Rev. 2004;56:1067. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 132.Lutz RJ. Transl Cancer Res. 2015;4:118. [Google Scholar]
- 133.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P. Clin Cancer Res. 2006;12:6106. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, Old LJ, Odunsi K. Proc Natl Acad Sci USA. 2010;107:7875. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chekmasova AA, Rao TD, Nikhamin Y, Park KJ, Levine DA, Spriggs DR, Brentjens RJ. Clin Cancer Res. 2010;16:3594. doi: 10.1158/1078-0432.CCR-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. J Transl Med. 2015;13:102. doi: 10.1186/s12967-015-0460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hong H, Brown CE, Ostberg JR, Priceman SJ, Chang WC, Weng L, Lin P, Wakabayashi MT, Jensen MC, Forman SJ. PLoS One. 2016;11:e0146885. doi: 10.1371/journal.pone.0146885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. Blood. 2009;113:6392. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Newick K, Moon E, Albelda SM. Mol Ther Oncolytics. 2016;3:16006. doi: 10.1038/mto.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frigault MJ, Maus MV. Int Immunol. 2016;28:355. doi: 10.1093/intimm/dxw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ikeda H, Lethe B, Lehmann F, van Baren N, Baurain JF, de Smet C, Chambost H, Vitale M, Moretta A, Boon T, Coulie PG. Immunity. 1997;6:199. doi: 10.1016/s1074-7613(00)80426-4. [DOI] [PubMed] [Google Scholar]
- 143.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Mol Ther Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ramos CA, Savoldo B, Dotti G. Cancer J. 2014;20:112. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Blood. 2014;124:188. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Barrett DM, Grupp SA, June CH. J Immunol (Baltimore, Md: 1950) 2015;195:755. doi: 10.4049/jimmunol.1500751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sadelain M. J Clin Invest. 2015;125:3392. doi: 10.1172/JCI80010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. N Eng J Med. 2014;371:1507. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL. Lancet (London, England) 2015;385:517. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, Lebuhotel C, Eyquem J, Cheung GW, Duclert A, Gouble A, Arnould S, Peggs K, Pule M, Scharenberg AM, Smith J. Cancer Res. 2015;75:3853. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 152.Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, Fonslow BR, Kochenderfer JN, Wright TM, Schultz PG, Young TS, Kim CH, Cao Y. Proc Natl Acad Sci USA. 2016;113:E450. doi: 10.1073/pnas.1524193113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, Powell DJ., Jr Cancer Immunol Res. 2013;1:43. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jones BS, Lamb LS, Goldman F, Di Stasi A. Front Pharmacol. 2014;5:254. doi: 10.3389/fphar.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. Blood. 2011;118:1255. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fedorov VD, Themeli M, Sadelain M. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gross G, Eshhar Z. Annu Rev Pharmacol Toxicol. 2016;56:59. doi: 10.1146/annurev-pharmtox-010814-124844. [DOI] [PubMed] [Google Scholar]
- 158.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, Ma J, Nunez V, Shen J, Woods AK, Wright TM, Schultz PG, Kim CH, Young TS. Proc Natl Acad Sci USA. 2016;113:E459. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, Duchateau P. Sci Rep. 2016;6:18950. doi: 10.1038/srep18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Klebanoff CA, Rosenberg SA, Restifo NP. Nat Med. 2016;22:26. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Hum Gene Ther. 2011;22:1575. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Brudno JN, Kochenderfer JN. Blood. 2016;127:3321. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM. Clin Cancer Res. 2009;15:5323. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Roy LD, Zhou R, Moore LJ, Dillon LM, Puri R, Lyerly K, Jeffrey R Marks, Mukherjee P. J Clin Oncol. 2015;33 abstr e22153. [Google Scholar]
- 165.Dréau D, Moore LJ, Alvarez-Berrios MP, Tarannum M, Mukherjee P, Vivero-Escoto JL. J Biomed Nanotechnol. 2016;12:1. doi: 10.1166/jbn.2016.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]