

Abstract
We aim to screen the mutations of 3 hearing loss (HL) genes (GJB2, SLC26A4, and 12S rRNA) in 71 cases with nonsyndromic hearing loss (NSHL) using microarray and SNPscan, and identify the roles of nonhotspot mutation of these genes in the screening of NSHL. Seventy-one cases with moderate or severe neurosensory deafness confirmed in our department from July 2014 to December 2015 including 25 Uyghur minorities and 46 Han Chinese were included in this study. The type of mutations in GJB2, SLC26A4, and 12S rRNA genes were detected using microarray and SNPscan, respectively. Statistical difference was noticed in the detection rate of the HL genes in 71 cases. Using microassay, deafness genes were identified in 10 subjects (14.08%), while 22 cases (30.98%) were confirmed with the presence of deafness genes using the SNPscan. Compared with the microarray, remarkable difference was noticed in the detection rate of SNPscan (P < .05). Nonhotspot mutation in GJB2, SLC26A4, and 12S rRNA genes played a crucial role in the pathogenesis of NSHL. SNPscan contributed to elevation of detection rate of NSHL in clinical practice.
Keywords: deafness gene, detection rate, mutation sites, nonsyndromic hearing loss
1. Introduction
Hearing loss (HL) has been reported to be associated with various factors such as trauma, medication, as well as environmental or genetical factors.[1,2] In a global survey, the prevalence of HL in children was 1/1000, and more than half of the HL was triggered by genetic factors.[3] The majority (70%) of patients with genetic deafness were classified into nonsyndromic hearing loss (NSHL), while the others (30%) were syndromic HS with anomalies in the other organs or functions.[4]
Up to now, 177 genetic loci have been identified to be responsible for the pathogenesis of NSHL, involving more than 1000 mutation sites in 108 genes.[5,6] As too many genes involved in the NSLH, it is not possible to screen all the pathogenic mutations from these genes. Instead, gene locus with a high mutation frequency among a large population may serve as an alternative method to the screening of pathogenic mutations. Increasing evidence reveals the pathogenesis of NSHL is extremely associated with few genes such as GJB2, SLC26A4, and 12S rRNA despite a higher heterogeneity between the gene and locus of various deafness genes. This aspect contributes to the gene screening of genetic deafness in clinical practices.[7,8]
Currently, several methods have been developed for the screening of genes associated with HL, including real-time PCR, gene microarray, SNPscan, and matrix-assisted laser desorption ionization-time-of-flight mass spectrometry. However, up to now, rare studies have been carried out for the screening of HL genes in the population in China. In this study, gene microarray and SNP methods were used to analyze the mutation sites in GJB2, SLC26A4, and 12S rRNA genes in the population.
2. Materials and methods
2.1. Subjects
Seventy-one cases with moderate or severe neurosensory deafness confirmed in our department from July 2014 to December 2015 including 25 Uyghur minorities and 46 Han Chinese were included in this study. The medical information of each subject participated in this study was collected by a questionnaire including the case history, family history, medication, and personal history. Besides, the subjects were subject to ENT tests and pure-tone audiogram. The category of the deafness was carried out using the noise exposure criterion based on noise immission level (NIL) as follows: normal, <20 dB nIL, mild HL, 21 to 40 dB nIL, moderate HL, 41 to 70 dB nIL, severe HL, 71 to 95 dB nIL, and complete HL, >95 dB nIL. The average of the frequencies was 0.5, 1, 2, and 4 kHz. The inclusion criteria were as follows: patients with no relations, those with moderate or severe neurosensory deafness. Those with deafness caused by environmental and/or traumatic factors were excluded from the study. All the cases signed the informed consent. The study protocols were approved by the Ethical Committee of the First Affiliated Hospital of Xinjiang Medical University.
2.2. Microarray
Genomic DNA was extracted from peripheral blood leukocytes of each subject using a commercial available DNA isolation kit (Tiangen Biotech Corporation, Beijing, China). The concentration and purity of DNA were determined using an ultraviolet spectrometry (ActGene Inc, Taipei, China). The Tag labeled specific primers designed based on the sequences of GJB2, SLC26A4, and 12SrRNA downloaded from the GeneBank were used for the amplification of genes. Subsequently, general gene chips containing the corresponding sequences of the Tag were used for the hybridization. Chip scan was conducted as recommended by the manufacturer of the microarray kit (CapitalBio Corporation, Beijing, China) for the simultaneous detection of 8 hotspot mutations in 3 most prevalent genes including GJB2, SLC26A4, and mitochondrial 12S rRNA. The test results were determined based on the fluorescent hybridization signal and the distribution of microarray probe by HybSet system (Packard BioChip Technology, MA).
2.3. SNPscan
Venous blood (3–5 mL) was collected from each family member, followed by DNA extraction using the commercial kit (Qiagen, Germany). SNPscan method was used to screen against the 115 sites in the common deafness genes (ie, GJB2, 12S rRNA, and SLC26A4) as previously described.[9] Subsequently, subjects with SNPscan negativity were subject to Sanger DNA sequencing for the sequencing of genes known to be responsible for HS.
2.4. Statistical analysis
SPSS17.0 software was used for the data analysis. Chi square test was used for the intergroup comparison. P < .05 was considered to be statistically significant.
3. Results
Using microassay, deafness genes were identified in 10 subjects (14.08%). Whereas, 22 cases (30.98%) were confirmed with the presence of deafness genes using the SNPscan technique. Compared with the microarray, remarkable difference was noticed in the detection rate of SNPscan (P < .05, Table 1).
Table 1.
Comparison of detection rate of gene mutation in 71 patients with nonsyndromic hearing loss (SNHL) deafness.

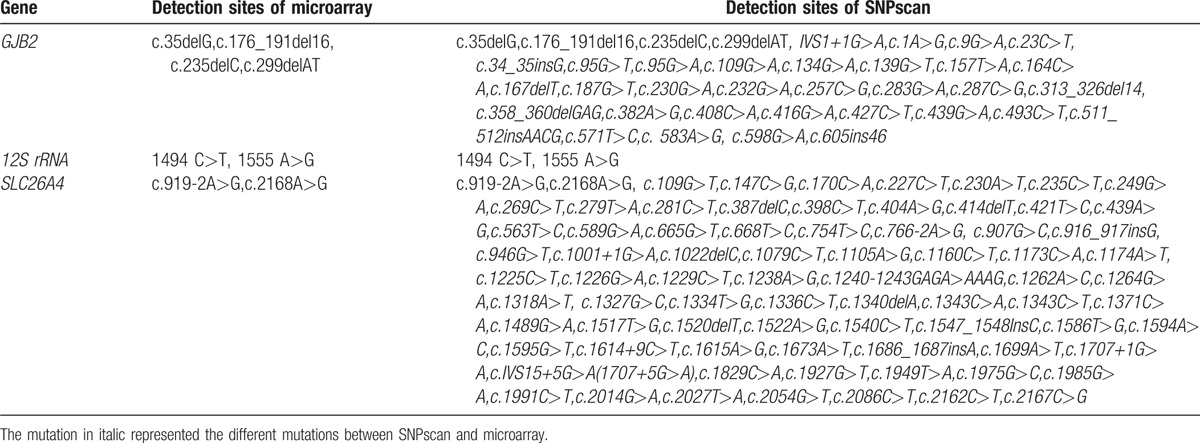
In the microarray, 8 hotspot mutation sites of the GJB2, SLC26A4, and 12S rRNA, while in the SNPscan, 115 mutation sites were analyzed including 8 hotspot mutation sites and 107 nonhotspot mutation sites. Compared with the microarray, another 75 mutation sites were detected using the SNPscan for the SLC26A4 gene with high mutation variants. Compared with the microarray, 32 new mutation sites were added for GJB2 gene using the SNPscan (Table 2).
Table 2.
Specific detection sites of the 2 detection methods.
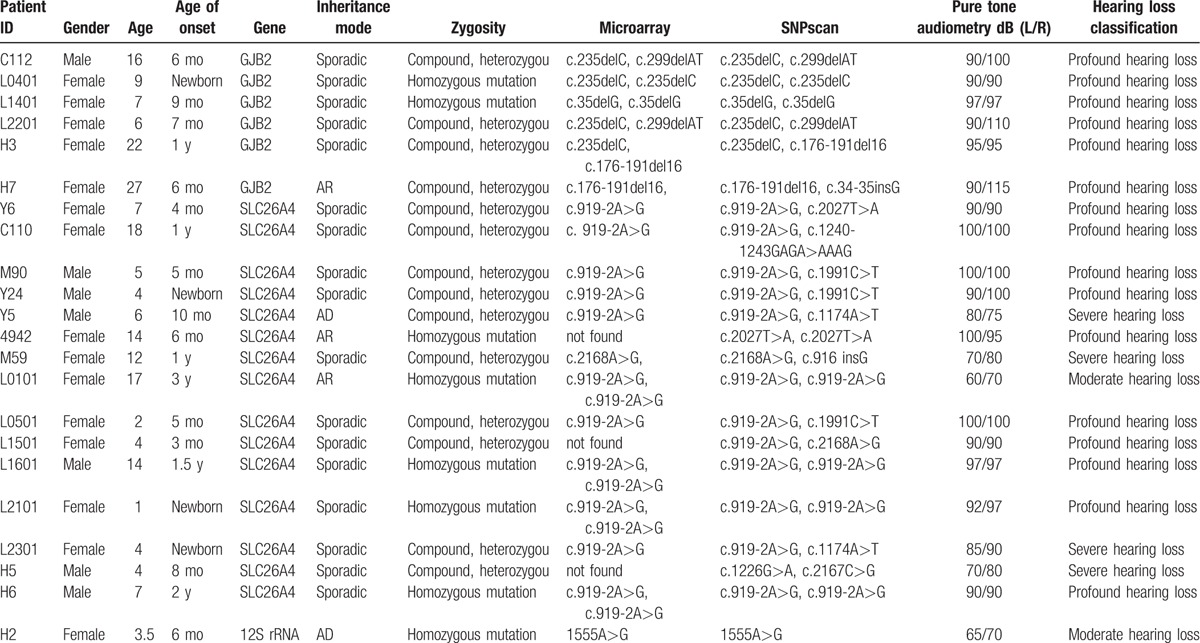
Among the 22 patients with mutation of deafness gene, 17 aged less than 14 years, 3 aged 14 to 18 years, and 2 aged >18 years. Two cases showed a family history of suspicious genetic deafness, 3 showed nonlineal relationship, while 17 cases showed no family history of deafness. Compared with the microassay, c.34-35insG was identified in GJB2 gene in H7 sample using the SNPscan method. As revealed by the SNPscan, several nontypical pathogenic mutations were identified in the SLC26A4 genes in many samples including 2027T>A (n = 2), 1240-1243GAGA>AAAG (n = 1), 1991C>T (n = 3), 1174A>T (n = 2), 916 insG (n = 1), 1226G>A (n = 1), and 2167C>G (n = 1). For the pure-tone audiometry, 2 cases were confirmed with moderate HL, 4 with severe HL, and 16 with extremely severe HL (Table 3).
Table 3.
Details of 22 diagnosed cases.
4. Discussion
The genetic etiology of HS may vary in different countries or races. Nowadays, rare studies have been carried out to investigate the genetic etiology of the genes associated with HS in Xinjiang Uyghur Autonomous region. In this study, gene microarray and SNP methods were used to analyze the mutation sites in GJB2, SLC26A4, and 12S rRNA genes in the population in the Xinjiang Uygur Autonomous region and the Han Chinese. Our study contributed to the understanding on the identification of pathogenic genes for the HL in China.
Genetic deafness shows higher genetic heterogeneity.[10] Up to now, 110 pathogenic genes of genetic deafness have been cloned including 34 DFNA-related genes, 69 DFNB-related genes, 5 DFN-related genes, and 2 mitochondrion-related genes, involving up to 2000 mutation types.[11] Identification of genetic deafness is helpful to the analysis of molecular pathways and functional structure of the internal ear. In addition to the genetic heterogeneity, the expression of a certain gene may be different due to the difference of mutation sites and genetic background. For instance, some genes may present in a form of dominant inheritance and/or recessive inheritance. All these lead to a challenge in the diagnosis of genetic deafness. In clinical practice, 3 major genes including GJB2, SLC26A4, and 12S rRNA have been commonly considered to play crucial roles in the pathogenesis of genetic deafness, and are preferentially used for the diagnosis of deafness. According to the previous description, GJB2 gene mutation was responsible for the half of the cases with moderate and even extremely severe HL.[12–14]SLC26A4 mutation was reported to involve in the 4.6% to 12.4% of the severe congenital deafness.[15] In studies performed in Asian regions, SLC26A4 mutation was observed in the majority of cases (80%) with enlargement of vestibular aqueduct and Mondini deformity in the cochlea.[16–18] Moreover, mutation of 12S rRNA was responsible for the deafness induced by aminoglycoside antibiotics.[19] Therefore, it is reasonable to improve the detection rate of mutations associated with deafness in these genes. In this study, SNPscan was used to screen the mutation sites in these genes, to increase the detection rate of NSRN.
In this study, microarray and SNPscan were used for the detection of mutation of deafness genes in 71 cases with bilateral neurosensory deafness. Statistical difference was noticed in the detection rates of these 2 methods (14.08% vs 30.98%, P < .05). Compared with the microarray method, 75 mutation sites of SLC26A4 with high mutation heterogeneity were added and analyzed, while 32 sites of GJB2 were added and analyzed, which contributed to the increase of detection rate of pathogenic mutations.
Among the 22 patients, 2 were confirmed with homozygous mutation of GJB2, 4 with heterozygous mutation of GJB2, 5 with homozygous mutation of SLC26A4, 10 with heterozygous mutation of SLC26A4, and 1 with homozygous mutation of 12S rRNA. In total, 15 cases showed SLC26A4 mutation, among which SLC26A4 c.919-2A>G(IVS7-2) was the most common type of pathogenic mutation in this study, which was in consistence with the previous report.[20] In addition, multiple nontypical pathogenic mutations were identified including 2027T>A (n = 2), 1240-1243GAGA>AAAG (n = 1), 1991C>T (n = 3), 1174A>T (n = 2), 916 insG (n = 1), 1226G>A (n = 1), and 2167C>G (n = 1). These types of mutation such as missense mutation, frame-shifting mutation, or mutations in the slicing position of the exon and the proximal sites may hamper the normal function of the protein through modulating the translational process, which may trigger enlargement in the vestibular aqueduct and neurosensory deafness.[21] Meanwhile, as these mutations are not distributed in each exon in a regular manner, it is hard to identify whether enlargement of vestibular aqueduct is responsible for the deafness in these patients. This may lead to missed diagnosis of enlargement of vestibular aqueduct caused by SLC26A4 mutation. In this study, no pathogenic mutations were identified in 49 patients (49/71), but we cannot exclude the presence of deafness caused by other mutations. On this basis, for the patients with enlargement of vestibular aqueduct, upon identification of single heterozygous mutations after typical mutation screening, it is necessary to search for new mutation sites through sequencing of the SLC26A4 exons.
In conclusion, remarkable differences were noticed in the screening of mutations in the GJB2, SLC26A4, and 12S rRNA genes using microarray and SNP methods in the 71 NSHL patients. Screening of 8 hotspot mutations in 3 genes could be achieved using microarray technique, while screening of 8 hotspot mutation sites and 107 non-hotspot mutation sites could be achieved using the SNPscan, which may increase the detection rate of pathogenetic mutations of HS. This indicated that analysis of nonhotspot mutation is necessary. Thus, SNPscan contributed to elevation of detection rate of NSHL in clinical practice, which may provide helpful information for the clinical screening of HS gene mutations.
Footnotes
Abbreviations: HL = hearing loss, NSHL = nonsyndromic hearing loss.
Funding/support: This project was supported by the National Natural Science Foundation (No. 81360158).
The authors have no conflicts of interest to disclose.
References
- [1].Fraser R. The Causes of Profound Deafness in Childhood. Baltimore: Johns Hopkins University Press; 1976. [Google Scholar]
- [2].Morton N. Genetic epidemiology of hearing impairment. Ann Acad Sci 1991;630:16–31. [DOI] [PubMed] [Google Scholar]
- [3].Marazita M, Ploughman L, Rawlings B, et al. Genetic epidemiological studies of early-onset deafness in the US school-age population. Am J Med Genet 1993;46:486–91. [DOI] [PubMed] [Google Scholar]
- [4].Van Camp G, Willems P, Smith R. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997;60:758–64. [PMC free article] [PubMed] [Google Scholar]
- [5].Gardner P, Oitmaa E, Messner A. Simultaneous multigene mutation detection in patients with sensorineural hearing loss through a novel diag-nostic microarray: a new approach for newborn screening follow-up. Pediatrics 2006;118:985–94. [DOI] [PubMed] [Google Scholar]
- [6].Siemering K, Manji S, Hutchison W, et al. Detection of mutations in genes associated with hearing loss using a microarray based approach. J Mol Diagn 2006;8:483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hilgert N, Smith RJ, Van Camp G. Function and expression pattern of nonsyndromic deafness genes. Curr Mol Med 2009;9:546–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ouyang XM, Yan D, Yuan HJ, et al. The genetic bases for non-syndromic hearing loss among Chinese. J Hum Genet 2009;54:131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mutation analysis of the common deafness genes in patients with nonsyndromic hearing loss in Linyi by SNPscan assay. Biomed Research Int 2016;3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lutman ME, Coles R. Asymmetric sensorineural hearing thresholds in the non-noise-exposed UK population: a retrospective analysis. Clin Otolaryngol 2009;34:316–21. [DOI] [PubMed] [Google Scholar]
- [11].Chen Y, Tudi M, Sun J, et al. Genetic mutations in non-syndromic deafoess patients of Uyghur and Han Chinese ethnicities in Xinjiang, China: a comparative study. J Transl Med 2011;9:154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cohn E, Kelley R. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. Am J Med Genet 1999;89:130–6. [PubMed] [Google Scholar]
- [13].Guilford P, Ben A, Blanchard S, et al. A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat Genet 1994;6:24–8. [DOI] [PubMed] [Google Scholar]
- [14].Trabelsi M, Bahri W, Habibi M, et al. GJB2 and GJB6 screening in Tunisian patients with autosomal recessive deafness. Int J Pediatr Otorhinolaryngol 2013;77:714–6. [DOI] [PubMed] [Google Scholar]
- [15].Du W, Guo Y, Wang C, et al. A systematic review and meta-analysis of common mutations of SLC26A4 gene in Asian populations. Int J Pediatr Otorhinolaiyngol 2013;77:1670–6. [DOI] [PubMed] [Google Scholar]
- [16].Yuan Y, You Y, Huang D, et al. Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med 2009;7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tsukamoto K, Suzuki H, Harada D, et al. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet 2003. 11916–22. [DOI] [PubMed] [Google Scholar]
- [18].Park H, Shaukat S, Liu X, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 2003;40:242–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu XZ, Angeli S, Ouyang XM, et al. Audiological and genetic features of the mtDNA mutations. Acta Otolaryngol 2008;128:732–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kelley P, Harris D, Comer B, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Q, Zhao Y, Rao S, et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet 2007;72:245–54. [DOI] [PubMed] [Google Scholar]