

Abstract
The chemical investigation of marine mollusks has led to the isolation of a wide variety of bioactive metabolites, which evolved in marine organisms as favorable adaptations to survive in different environments. Most of them are derived from food sources, but they can be also biosynthesized de novo by the mollusks themselves, or produced by symbionts. Consequently, the isolated compounds cannot be strictly considered as “chemotaxonomic markers” for the different molluscan species. However, the chemical investigation of this phylum has provided many compounds of interest as potential anticancer drugs that assume particular importance in the light of the growing literature on cancer biology and chemotherapy. The current review highlights the diversity of chemical structures, mechanisms of action, and, most importantly, the potential of mollusk‐derived metabolites as anticancer agents, including those biosynthesized by mollusks and those of dietary origin. After the discussion of dolastatins and kahalalides, compounds previously studied in clinical trials, the review covers potentially promising anticancer agents, which are grouped based on their structural type and include terpenes, steroids, peptides, polyketides and nitrogen‐containing compounds. The “promise” of a mollusk‐derived natural product as an anticancer agent is evaluated on the basis of its ability to target biological characteristics of cancer cells responsible for poor treatment outcomes. These characteristics include high antiproliferative potency against cancer cells in vitro, preferential inhibition of the proliferation of cancer cells over normal ones, mechanism of action via nonapoptotic signaling pathways, circumvention of multidrug resistance phenotype, and high activity in vivo, among others. The review also includes sections on the targeted delivery of mollusk‐derived anticancer agents and solutions to their procurement in quantity.
Keywords: mollusk; cancer, preclinical evaluation, treatment, innovative mechanism of action, targeted delivery
1. INTRODUCTION
A. Cancer Epidemiology and Treatment
As emphasized by Torre et al.1 the occurrence of cancer is increasing because of the growth and aging of the population, as well as an increasing prevalence of established risk factors such as use of tobacco products, obesity, physical inactivity, and changing reproductive patterns associated with urbanization and economic development. About 14 million new cancer cases and more than 8 million deaths occurred in 2012 worldwide.1
In contrast to benign tumors, which with few exceptions2 do not invade other sites in the body, cancer is a malignant tumor that is characterized by local tissue invasion and/or distant metastatic processes. At early stages of cancer development, surgery or surgery in combination with adjuvant radiotherapy are curative in most cases. Once the cancer is more developed and has invaded other tissues, systemic chemotherapy (oral or intravenous) is added to surgery and radiotherapy to eliminate isolated cancer cells and/or cancer cell subpopulations that have not been removed or destroyed. As a general rule (always with exceptions), the more advanced the cancer at the time of diagnosis, the more aggressive the adjuvant chemotherapy or polychemotherapy that will be applied. Apart from chemotherapy, a number of other treatments are often used, such as immunotherapy3, 4, 5 and photodynamic therapy.6, 7 Doroshow and Kumar8 state that more extensive molecular characterization of tumors and their supporting matrices are anticipated to become standard aspects of oncological practice, which will permit the design of the treatment type for each individual patient and its continuous reevaluation during the course of the treatment.
Paul Ehrlich, the founder of chemotherapy, who received the Nobel Prize for Physiology or Medicine a century ago, postulated the creation of “magic bullets” to be used as chemotherapeutic agents in the fight against human diseases.9 Unfortunately, such magic bullets are generally ineffective in oncology. One reason is related to the fact that, for the most part, cancers are heterogeneous and consist of multiple cell subpopulations.10, 11, 12 Combinations of various anticancer therapies (including polychemotherapies) involving drugs with different mechanisms of action are thus widely used.13 In addition, chemotherapy and hormone therapy have been combined with immunotherapy for the treatment of various solid cancers.14, 15 The US Food and Drug Administration (FDA) recently approved the first “immunotherapy combo,” the Bristol–Myers Squibb's combination of nivolumab and ipilumab, for the treatment of metastatic melanoma.16
B. Cytotoxic Versus Targeted Anticancer Therapies
Most chemotherapies consist of the use of cytotoxic drugs or molecularly targeted agents. Each type of therapy could be effective in the treatment of patients whose cancer has reached an advanced clinical stage and often these therapies are used in combination. However, for certain cancer types, current chemotherapeutic standards seem still to rely mainly on cytotoxic drugs (postsurgery and/or radiotherapy). This is the case, for example, for pancreatic cancer,17 advanced stage small cell lung cancer (SCLC),18 and glioblastoma (GBM).19 In their review entitled “An anticancer strategic dilemma: to kill or to contain,” Perret and Uzzan20 classify the use of cytotoxic compounds as “killing” tools and targeted therapies as containment tools, where the latter is aimed at the induction or extension of tumor dormancy.
Another difference between cytotoxic and targeted therapeutics consists of the way by which the compounds are discovered and developed. Many cytotoxic compounds are discovered from plants, fungi, or animals (especially from marine invertebrates) using bioactivity‐guided isolation, while targeted agents are typically designed and synthesized by researchers. Bioassay guided isolation of natural extracts, when successful, leads to the discovery of novel peptides, proteins, polysaccharides, lectins, small molecules, or other agents whose growth inhibitory activity is first assayed in vitro in normal and cancer cell lines and then in vivo in various murine syngeneic and/or human xenografted models. During these pharmacological (and early toxicological) evaluations, it is rarely possible to decipher the mechanism(s) of anticancer action. Targeted therapies, on the other hand, mainly rely on the screening of libraries of compounds against a specific target protein that is usually intracellular. Researchers have also developed biological agents (such as antibodies and nucleic acid aptamers) to target specific proteins that are usually presented extracellularly and are typically involved in cancer cell biology and/or characteristic of the tumor microenvironment.
C. Cancer Resistance to Chemotherapy
As will be seen later in the review, mollusk metabolites are evaluated based on the ability of these natural products to overcome cancer cell resistance to chemotherapy, a property which, in our view, makes a particular compound a promising anticancer agent. We thus summarize below some of the major mechanisms of cancer cell resistance to chemotherapy that generally lead to dismal prognoses. These discussed mechanisms are of most relevance to the compounds presented in the current review. It must however be emphasized that there exist many more types of cancer drug resistance, which are not mentioned herein. These, for example, include the involvement of noncoding RNAs and multiple repair mechanisms,21 such as DNA base excision22, 23 and DNA double‐strand break,24 among others.
1. The Multidrug Resistance (MDR) Phenotype
Chen et al.25 emphasize that one of the common mechanisms for cancer cells to resist cytotoxic insults is the overexpression of the ATP‐binding cassette (ABC) efflux transporters such as P‐glycoprotein (P‐gp/ABCB1), MDR‐associated protein 2 (MRP2/ABCC2), and breast cancer resistance protein (BCRP/ABCG2). These mechanisms belong to the so‐called MDR phenotype and limit the prolonged and effective use of chemotherapeutic drugs. For example, P‐gp overexpression in cancer cells leads to the decreased uptake of the drug and intracellular drug accumulation, minimizing drug–target interactions.26 As emphasized by Cui et al.,27 the superfamily of human ABC transporters comprises seven subfamilies with 48 members, which exclude structurally and/or functionally unrelated drugs.26 Dinic et al.26 report that there are two types of MDR: intrinsic and acquired. These authors26 further report that tumor microenvironment‐induced selection pressure leads to the development of intrinsic MDR, while acquired resistance is a consequence of chronic chemotherapy administrations. Cort and Ozben28 as well as Dinic et al.26 state that natural product‐based drugs are important in overcoming or reversing MDR in cancer therapy.
2. The Resistance to Targeted Therapies
Schmitt et al.29 recently reviewed the preexisting subclonal resistance mutations to various molecularly targeted agents that lead to clinical failures in the treatment of cancer patients with targeted therapies. In addition, as mentioned earlier in this review and also discussed Schmitt et al.,29 the problem of cancer heterogeneity leads to the inability of a single agent, whatever it may be, to kill all the subclones and the associated populations in a given cancer. Schmitt et al.29 accordingly state that early detection of preexisting or emerging drug resistance could enable more personalized use of targeted cancer therapy, as patients could be stratified to receive the therapies that are most likely to be effective. Further, Kim30 recently reviewed the mechanisms of resistance to targeted therapy, with a focus on acquired resistance involving mutations and amplification of genes in the same or parallel signaling pathways. This author also emphasizes that sequencing of primary tumors has revealed that therapy‐resistant clones already exist prior to targeted therapy, demonstrating once again that tumor heterogeneity in primary tumors confers a mechanism for inherent therapy resistance. Pazarentzos and Bivona31 also recently reviewed an important aspect of tumor resistance to targeted therapies involving the adaptive stress signaling process. They discussed the early adaptive changes by which tumor cells respond to the stress of a targeted therapy that may be crucial for tumor cell survival during treatment and the development of resistance.
An international task force of 180 scientists was recently assembled to explore the concept of a low‐toxicity “broad‐spectrum” therapeutic approach that could simultaneously target many key pathways and mechanisms.32 Block et al.32 report that using cancer hallmark phenotypes and the tumor microenvironment to account for the various aspects of relevant cancer biology, interdisciplinary teams reviewed each hallmark area and proposed a wide range of high‐priority targets (74 in total) that could be modified to improve patient outcomes. For these targets, corresponding low‐toxicity therapeutic approaches were then suggested, many of which were phytochemicals.32
3. Cancer Stem Cells (CSCs)
Several theories attempting to explain “the origin” of cancer(s) have been proposed. Some theories attribute cancer to problems in developmental biology and cell differentiation,33, 34, 35 while others point to a normal cell undergoing tumorigenic transformation as a result of genetic mutations.36, 37 In fact, some of these ideas were published in the early 1900s,38, 39 half a century before the solving of the DNA structure.40, 41 Regardless of its origin, as emphasized by Rycaj and Tang,37 an established clinical tumor is sustained by subpopulations of self‐renewing cancer cells, operationally called CSCs that can intraclonally generate both tumorigenic and nontumorigenic cells. Pattabiraman and Weinberg42 report that since their identification in 1994, CSCs have been demonstrated to be directly implicated in resistance to conventional anticancer therapeutics. Skrbo et al.11 have also recently revisited the scientific background of the CSC theory. In their recent review, Pattabiraman and Weinberg42 also propose ways to use the current knowledge of the complex biology of CSCs to design novel therapies to eliminate these cells. As detailed in Section 1.C.3, CSCs are implicated in both minimal residual disease (the major cause of cancer recurrence) and metastasis.43 Dragu et al.44 in their recent review discuss targeting CSC surface biomarkers, signaling pathways that regulate CSC self‐renewal and differentiation, drug‐efflux pumps involved in apoptosis resistance, and microenvironmental signals that sustain CSC growth, as well as manipulation of miRNA expression and induction of CSC apoptosis and differentiation as strategies for hampering CSC regeneration and cancer relapse. These authors44 also report that several anti‐CSC agents are under evaluation in preclinical and clinical studies, with most of them being designed to be used in combination with traditional therapies. Lisanti et al.45 are developing strategies to eradicate CSCs using mitochondria targeting46 or through inhibition of protein synthesis.47
4. Hypoxia
Span and Bussink48 recently discussed that: (i) hypoxia is a heterogeneous effect with oxygen tensions ranging from 0.01% (anoxia) to 5%, (ii) hypoxia can be chronic, acute, or cycling, all with differential effects on tumor cells, and (iii) low oxygen tension often occurs in tumor cells as a result of several processes, for example, poor angiogenesis and increased oxygen consumption. Dhani et al.49 accordingly report that hypoxia drives a complex compensatory response in cancer cells (and also in endothelial cells) leading to continued cell survival and induces genomic changes resulting in selection of hypoxia‐adapted cells with the propensity to invade locally, metastasize, and recur following surgery or radiotherapy. Along the same lines, Paolicchi et al.50 state that the hypoxic tumor microenvironment promotes metabolic changes, oncogene activation, epithelial to mesenchymal transition, and resistance to chemo‐ and radiotherapy, all of which are hallmarks of aggressive tumor behavior. All of these characteristics are orchestrated through the activation of the hypoxia‐inducible factor 1 alpha (HIF1A), which is an independent marker of poor prognosis.50, 51 Accumulating evidence in recent years suggests that hypoxia inducible factor 2 (HIF‐2) also contributes to chemo‐ and radio‐resistance in solid tumors.52
In a recent review, Parks et al.53 report that hypoxia promotes tumor growth by controlling nutrient import and acidic metabolite export. Eales et al.54 state that hypoxic tumor areas usually contain some of the most malignant cells in a given cancer. Liang et al.51 also state that the development of cancer therapies that target hypoxia is of vital importance and that one such targeting strategy is the design of hypoxia‐activated prodrugs, which release chemotherapeutic agents within hypoxic tumor regions. This targeting strategy is accomplished by attaching a hypoxia‐activated trigger to a chemotherapeutic agent such that, under oxygen‐poor conditions, the agent (effector) is released into the tumor, while remaining intact in normal tissue leaving nonhypoxic cells unaffected.
5. Resistance to Proapoptotic Stimuli
Apoptosis‐related signaling pathways have been extensively reviewed by Galluzzi et al.55, 56, 57 As stated by Simpson et al.,58 the ability of a cell to survive in an anchorage‐independent manner is a critical step in the development of metastatic potential. Such cells must be able to overcome anoikis, which is a type of cell death related to apoptosis and results from the loss of contact with neighboring cells or extracellular matrix.58, 59 Portt et al.59 reviewed the various molecular signaling pathways by which cancer cells evade apoptosis.59 These pathways involve, among others, activation or upregulation of mitogenic signaling pathways (Erk1/2, Akt, etc.), inactivation, or downregulation of certain proapoptotic pathways (Fas receptor, Bax, etc.), and the up‐regulation of a number of antiapoptotic genes (Bcl‐2, cFLIP, etc.). Portt et al.59 report that these signaling pathways leading to antiapoptotic phenotypes are activated in response to not only cytotoxic and proapoptotic insults, but also stressful environment such as hypoxia.
Speirs et al.60 state that an alternative approach to overcome the resistance of cancer cells to cytotoxic proapoptotic stimuli is to induce cell death pathways that are mechanistically distinct from apoptosis. These authors accordingly reviewed drugs that induce autophagic cell death or necrosis in cancer cells.60 However, whether inducing autophagy is beneficial or detrimental for cancer cells still remains an actual subject of debate that was recently reviewed by Belaid et al.61 Indeed, inhibiting (as opposed to inducing) autophagy in cancer cells also seems to be a promising approach to combat cancers associated with dismal prognoses.62, 63, 64 Furthermore, in addition to apoptosis, autophagy, and necrosis, there are multiple other cell death pathways that have been reviewed by Galluzzi et al.55, 56, 57 Some examples include necroptosis, mitotic catastrophe, senescence, lysosomal membrane permeabilization, oncosis, parthanatos, pyroptosis, ferroptosis, and autosis.55, 56, 57 We recently reviewed compounds that are able to induce these various death mechanisms in cancer cells.65 For example, lysosomal inhibition could emerge as a new therapeutic strategy to overcome drug resistance in cancer.66 Lysosomes are membrane‐bound intracellular organelles that receive macromolecules delivered by endocytosis, phagocytosis, and autophagy for degradation and recycling. Later in the review in Section 4.C, we describe a mollusk‐derived compound (belonging to the chemical family of kahalalides) that has reached clinical trials in oncology and that targets lysosomes in melanoma cells displaying marked resistance to apoptosis‐related cytotoxic insults.67
GBM is the deadliest type of cancer characterized by pronounced resistance to proapoptotic stimuli.68 The only chemotherapeutic drug that leads to real but still limited beneficial effects for GBM patients is the 30‐year‐old cytotoxic drug temozolomide.19, 69 Much effort has been applied to discovering ways to kill apoptosis‐resistant GBM cells by activating nonapoptotic cell death signaling pathways, such as paraptosis70, 71, 72 or methuosis,73 but none of these attempts have yet successfully translated into effective treatments for GBM patients. Methuosis is a nonapoptotic cell death type associated with vacuolization of macropinosome and endosome compartments.74 Paraptosis is a caspase‐independent cell death type.65, 75 Lee et al.76 recently reviewed the natural products capable of inducing paraptosis in cancer cells.
6. Tumor Metastasis
Metastases are resistant to conventional therapies and remain the major cause of death from cancer.77 Indeed, a great majority of cancer patients (∼90%) die from tumor metastases78, 79 because metastatic cancers are resistant to almost any type of currently available treatment. Belaid et al.61 accordingly state that the survival rates of patients with metastatic or recurrent cancers have remained virtually unchanged in the past 30 years.
The metastatic process is characterized by a complex series of interactions between cancer cells that detach from the primary cancer site, known as circulating cancer cells, and tissue microenvironment.80 As was discussed in a previous section, circulating cancer cells must escape anoikis, and thus resist apoptosis, to be able to form secondary cancer sites (metastases). Of interest, less than 0.01% of circulating cancer cells will succeed in forming metastases81 and initiating cell growth in secondary organs is the most challenging step in this process.82 While some cancer types are capable of forming metastases in virtually every tissue in the body, the most frequent target organs of metastasis are bone, brain, liver, and lung.80 The site preference is described by the “seed” (cancer cells) and “soil” (the tumor microenvironment) theory proposed more than a century ago by Stephen Paget,83 which has been validated by extensive experimental as well as clinical data.80 Part of the seed and soil theory is explained by multiple tumor–stromal interactions (which however represent only part of the tumor microenvironment as explained in the next section) that influence the preference for metastatic spread toward a given organ.80 Fidler and Kripke77 report that targeting these interactions, in addition to the cancer cells themselves, can produce synergistic therapeutic effects against existing metastases. However, Pienta et al.84 state that describing metastasis in terms of a simple one‐way migration of cells from primary to target organs is insufficient to cover the nuances of cancer spread. Pienta et al.84 thus raise the question of whether cancer cells escape the confinement of their original habitat in the primary tumor or are they forced out by ecological changes in their home niche? These authors consequently propose an innovative concept of “diaspora,” which is a term used by social scientists to describe the scattering of people away from an established homeland.84 They argue that invoking the ecological and population science concepts can help understand the biology of tumorigenesis and metastasis, and inspire new ideas for therapy.84
Another problem linked to metastases is the capability of metastatic cancer cell to evade therapies by entering dormancy and resuming proliferation years after primary cancer treatment.85 Ghajar,86 thus, advocates directing therapies toward the niches that harbor dormant disseminated tumor cells to sensitize them to cytotoxic agents.
Tumor‐associated immune cells also play an important role in the promotion of tumor metastasis. Indeed, as discussed by Smith and Kang,87 inflammation and infiltration of the tumor tissue by host immune cells, such as tumor‐associated macrophages, myeloid‐derived suppressor cells, and regulatory T cells, have been shown to support tumor growth, invasion, and metastasis. Smith and Kang87 further describe that each step of tumor development, from initiation through metastatic spread, is promoted by the communication between tumor and immune cells via the secretion of cytokines, growth factors, and proteases that remodel the tumor microenvironment.
7. Tumor Microenvironment
Berns and Pandolfi88 emphasize the major role played by stromal cells in the development of a cancer, citing the example of pancreatic cancer in which stromal cells and their deposited matrices can make up to 90% of the tumor mass. Cancer‐associated fibroblasts actively interact with cancer cells and form a myofibroblastic microenvironment that promotes cancer growth and survival and supports malignancy.89 It appears that specific oncogenes induce cancer‐associated fibroblast phenotype.45 Berns and Pandolfi88 report that stromal components might contribute to drug resistance by creating a physical barrier limiting drug access, secreting growth‐promoting or antiapoptotic factors, providing niches for CSCs, or by mediating immunosuppression. These authors accordingly argue that combining treatments that degrade the tumor stroma with immunomodulation could be effective given the remarkable success of immunomodulation in a number of tumors and the notion that the tumor microenvironment plays an important role in immune suppression.88
Another crucial point in cancer biology is the notion of cancer cell fueling by noncancer cells. For example, autophagic senescent fibroblasts metabolically promote tumor growth and metastasis by paracrine production of high‐energy mitochondrial fuels.90 Migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth.91 Sotgia et al.92 thus state that as metabolic symbiosis promotes drug resistance and may represent an escape mechanism during antiangiogenic therapy, new drugs targeting metabolic symbiosis may also be effective in cancer patients with recurrent and advanced metastatic diseases.
Tabassum and Polyak93 compare tumorigenesis to a village. These authors indeed report that there is growing evidence that cancer cells behave as communities and different cancer subclones manifest cooperative behavior that can influence tumor progression.93 It is in this context that the notion of “cancer cell cannibalism” should also be evoked. Homotypic cell cannibalism, a cell‐death process regulated by the nuclear protein 1, opposes metastasis in pancreatic cancer.94 On the other hand, cancer cell cannibalism also leads to increasing levels of aneuploidy in cancer cells, a cell behavior that could promote tumor progression.95
Another important component of tumor microenvironment is the tumor vasculature network. Indeed, as emphasized by Blazejczyk et al.,96 endothelial cells accompany the malignant cancer cells at almost every stage of the metastatic process. This includes infiltration of tumor cells into the neighboring tissue, transmigration through endothelium (intravasation), survival in the blood stream, and extravasation followed by colonization of the target organ. The concept of antiangiogenic therapies in oncology raised great hopes after the pioneering articles by Folkman in the early 1970s.97, 98 However, 45 years later, the antiangiogenic therapies have not led to improved clinical outcomes, especially of cancers associated with dismal prognoses.99 In addition, once cancers develop resistance to antiangiogenic therapy, they may become more invasive or lead to the metastatic disease.99, 100 Rapisarda and Mellilo99 accordingly discuss several studies, which indicate that inhibitors of vascular endothelial growth factor (VEGF) (and its receptors) can promote an invasive metastatic switch, in part by creating an increasingly hypoxic tumor microenvironment.
For further information on the role of tumor microenvironment in supporting cancer development, the reader is referred to the recent review articles on this topic.101, 102 The above discussion of the tumor microenvironment playing a big role in supporting tumor progression can lead to a great deal of pessimism for the development of successful therapies. However, it is important to mention that recently two marine‐derived compounds that efficiently combat not only cancer cells but also the tumor microenvironment, plitidepsin (aplidin), and trabectedin (Yondelis®) reached late‐stage clinical trials in oncology.103, 104
Due to the success of plitidepsin and trabectedin, anticancer agents targeting tumor microenvironment are actively searched. In marine environment, sessile organisms, especially sponges, make use of bioactive metabolites to prevent the growth of competitors and foulers, and to deter predators from feeding.105, 106, 107, 108 Although such “chemical warfare” frequently represents the main defensive strategy of sessile marine invertebrates, slow moving marine mollusks, including species mentioned in the current review (see Fig. 1 in Section 4.A), are also able to sequester and reuse the chemical weapons from their prey, especially sponges and cnidarians, upon which they feed. In a sense, marine mollusks can thus become effective probes for the selection of bioactive metabolites that evolved in different groups of marine organisms with possible multitargeted effects on predators and competitors. Even though we are still far from having identified the molecular mechanisms behind the natural function of the metabolites from mollusks, it is reasonable to expect that they can interact with more than one molecular target with both critical ecological and pharmacological interest, possibly contributing to the generation of an “antitumor microenvironment.” In other words, the compounds that inhibit the growth of competitors in marine environment can also affect the growth and evolution of cancerous cells in the tumor microenvironment as has been showcased with plitidepsin and trabectedin.
Figure 1.

Illustration of selected mollusks under review. (A) Aplysia dactylomela. (B) Aplysia punctata. (C) Bursatella leachii. (D) Dolabella auricularia. (E) Elysia subornata. (F) Glossodoris quadricolor.(G) Hexabranchus sanguineus. (H) Jorunna funebris. (I) Onchidium sp. 2. (J) Peltodoris atromaculata. (K) Phidiana militaris. (L) Phyllidia coelestis. Table I provides taxonomical, geographical, and ecological information about these mollusks, whose pictures have been taken by one of us.
2. WHAT IS A “PROMISING ANTICANCER AGENT?”
The initial steps in anticancer drug discovery utilizing natural sources (plants, fungi, marine invertebrates) are conducted by chemists or biologists who are neither oncologists nor physicians. After having read hundreds of articles that have led to the writing of the present review, it became obvious to us that the term “promising anticancer agent” has different meanings depending on whether it is used by fundamental (chemists and cell biologists) or more applied researchers (pharmacologists and oncologists). The literature is replete with the use of this term by the fundamental researchers, who provide cancer cell killing data as the only basis for their judgment.
To label a particular compound as promising based on cytotoxicity data alone is to forget that a cancer in a patient is much more complex than a few isolated cancer cells in a plastic flask, growing alone outside of tumor microenvironment, and without the possibility to metastasize. This is, therefore, the reason why in the Introduction section we summarized the major characteristics of cancer cell biology and microenvironment representing a crucial obstacle to successful cancer treatment.109, 113 The biological characteristics that we utilize to label an anticancer compound as promising are described next.
A. In Vitro Activity
1. Antiproliferative Activity
Antiproliferative effects are most commonly expressed as IC50 or GI50 indexes (used interchangeably), representing a compound's concentration that reduces the growth of a given cell population (normal or cancerous) in vitro by 50% as compared to the (untreated) control condition. GI50 values are generally calculated after having cultured the cell population of interest for 48 to 72 hr in the presence (or absence) of the compound of interest. This is commonly done with a colorimetric assay that compares the optical densities (ODs) of a treated cell population to the ODs of a control condition (untreated cells) arbitrarily scaled at 100%. The sole GI50 value obtained by means of such a colorimetric assay (e.g., the popular MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyl tetrazolium bromide) assay) does not represent actual cytotoxic effects, a mistake that is unfortunately so often reported in the literature, but rather only provides relative global growth inhibition information. Indeed, a colorimetric assay does not differentiate cell killing (cytotoxic effects), the inhibition of cell proliferation (cytostatic effects), cell detachment (anti‐adhesive effects), or various combinations of these features. Once the GI50 value has been determined by means of a colorimetric assay for a given compound on a given cell line, it is mandatory to perform complementary morphological and/or biochemical analyses in order to determine whether the compound is cytotoxic, cytostatic, antiadhesive, etc. Galluzzi et al. have comprehensively and regularly reviewed all the assays that are available in this domain, including those that can define the type of cell death.55, 56, 57
The US National Cancer Institute (hereafter referred to as the NCI) established colorimetric assays involving the analyses of compound‐induced growth inhibitory effects in a panel of 60 cancer cell lines belonging to more than ten histopathological types.114 The precise strategy developed by the NCI is detailed by Shoemaker.115 The NCI researchers use three parameters calculated from the colorimetric assays to determine whether a compound is cytotoxic or rather cytostatic. As mentioned above, an NCI‐based GI50 value corresponds to a global growth decrease by 50% induced by a compound on a given cell line after having cultured the cells with the drug for 48 hr in comparison to an untreated cancer cell population (control condition = 100%) grown during the same time.115 An NCI‐based LD50 (or LC50) value corresponds to a global growth decrease by 50% induced by a compound on a given cell line after having cultured the cells with the drug for 48 hr in comparison to the initial number of cells in the untreated control condition.115 A total growth inhibition (TGI) value is used to determine the concentration needed to kill 100% of the treated cells.115 For example, based on the combined analysis of the NCI‐based GI50 and LD50 values, we deduced cytotoxic effects induced by sphaeropsidin A, a rearranged pimarane diterpene of fungal origin, in various cancer cell lines.116
Simple morphological analyses, such as those performed with phase‐contrast microscopy, can be quite useful in interpreting the data obtained with a colorimetric antiproliferative assay and, specifically differentiate cytotoxic from cytostatic effects. For example, such morphological analyses confirmed the cytotoxic nature of sphaeropsidin A that was originally deduced based on the NCI data.116 In contrast, narciclasine, an isocarbostyril isolated from daffodils,117, 118 was cytostatic at its GI50 concentration in melanoma cells as was concluded on the basis of morphological analyses.119
It must also be emphasized that colorimetric assays can lead to false‐positive or false‐negative data with respect to certain types of compounds.120 In addition, for some compounds the growth curves can reach a plateau‐phase in certain cancer cell lines.121 In both cases, morphological analyses can be useful to validate the data generated by a colorimetric assay, not only in cancer, but also in normal cells.
The GI50 indexes that precisely define the ranges “weakly active,” “active,” and “highly active” are not unanimously agreed upon in the literature. The instructions for authors of the Journal of Natural Products define a compound to be inactive if its GI50 value is higher than 10 μM. The NCI also uses 10 μM as the highest dose for compounds tested in the 60 cell line panel. However, some compounds that work through nonapoptotic mechanisms (e.g., inducers of senescence or autophagy) can display GI50 values higher than 10 μM. Thus, we took these issues into account in comments about the “promise” associated with a given anticancer compound.
2. Selectivity
The term “selective” is not a synonym for “bioselective” (see Section 2.A.3). A compound is “selective” when it displays in vitro growth inhibitory selectivity with respect to one or several histological groups of cancer cell lines out of a larger number of histological cancer types. The data obtained in the NCI 60 cell line panel are most useful in determining selectivity.114, 115 For example, using the NCI data we found that sphaeropsidin A displays in vitro selectivity against melanoma and kidney cancer cell lines.116 This type of information is very important in the design of further in vivo experiments, including, for example, the use of nanomedicine approaches to target a specific organ in which a cancer develops.122, 123, 124
3. Bioselectivity
The term “bioselective” is used to refer to a compound that displays differential growth inhibitory effects between normal and cancer cell lines. In the current review, we label a compound as “bioselective” if it displays a GI50 ratio of higher than 3 between normal and cancer cells, which is not highly discriminant but acknowledges the efforts applied by researchers of a given article to perform this kind of analysis. The ratio of 3 is also indicative of the possibility to make more bioselective derivatives of the compound of interest.
4. Characterization of the Mechanism(s) of Action
The NCI researchers have tested so far ∼800,000 compounds and developed an algorithm (the COMPARE software)115 that can assist in identifying a compound's mode of action. The COMPARE software compares the shapes of 60 growth inhibitory curves (one per each of the 60 cancer cell lines) of each of the ∼800,000 compounds available in the NCI database to the growth curves determined for a compound of interest.115 Each growth inhibitory curve is determined using five concentrations ranging from 0.001 to 10 μM for a given compound. The COMPARE software thus provides a statistical probability value that the compound of interest acts as, for example, a tubulin inhibitor, protein synthesis inhibitor, or a topoisomerase II poison. If the statistical probability provided by the COMPARE software is weak (< 0.05), it is potentially indicative of a novel mechanism of action. For example, the COMPARE tool assisted us in identifying a novel compound of interest (a novel trisubstituted harmine derivative) as being a potential protein synthesis inhibitor,125 a fact that was later validated at the biochemical level. The NCI contributed to the characterization of the anticancer activity and/or the characterization of the mechanism(s) of action of major anticancer drugs that are listed in the review by Shoemaker.115 The NCI 60 cell line panel also represents a powerful tool for studying the effects of a given compound at the level of mutations,126 transcript and protein expression profiles,127 microRNA expression profiles,128 and global proteome analysis.129
In the Introduction section, we emphasized why proapoptotic cytotoxic agents are not promising in combating cancers associated with dismal prognoses. Thus, we do not label as “promising” an anticancer compound that kills cancer cells (in vitro) through proapoptotic mechanisms, with no in vivo demonstration of activity. We summed up in the Introduction the extraordinary complexity of the metastatic biological process. It is at first glance difficult to appreciate in vitro the potential antimetastatic effects of compounds that are evaluated in cancer cells that are grown isolated, without cancer‐associated cells, in a 2D environment. We, nevertheless, identify promise in compounds displaying in vitro‐specific antimetastatic properties such as effects on cancer cell adhesion, motility, migration, or invasion. We highlight compounds displaying effects on cell adhesion at the level of cell–cell and/or cell–extracellular matrix interactions, or affecting the adhesive machinery of cancer cells including, for example, the focal adhesion kinase (FAK)130 and beta‐1 integrins.131 We also consider that a compound displays “in vitro antimetastatic effects” if it decreases cancer cell migration132 and/or if it targets cytoskeleton or cytoskeleton components such as actin or tubulin.132 Finally, we regard a compound as able to affect cell invasiveness if it inhibits proteases implicated in the metastatic cascade, such as cysteine cathepsines,133 and/or if it inhibits migration in transwell migration assays. We highlighted in the Introduction the importance of the MDR phenotype in the resistance of cancer cells to chemotherapeutics, especially in relation to natural cytotoxic compounds. Thus, we emphasize compounds that have shown ability to bypass MDR processes. Lastly, we also highlighted in the Introduction section the major roles displayed by hypoxia and CSCs in cancer chemoresistance. We describe promising compounds that can be triggered by hypoxia or that have been shown to affect CSCs.133, 144
B. In Vivo Activity
As emphasized by Cekanova and Rathore,145 cancer is the term used to describe over 100 diseases that share several common hallmarks. There are in vivo models for any type of cancer, which can be studied in a large variety of species ranging from Drosophila flies146, 147 to zebrafish,147 and rodents148, 149, 150, 151, 152 to companion animals such as cats and dogs.145, 153, 154 However, most in vivo preclinical models involve rodents (mostly mice) and generally belong to two groups, cancers developing in immunocompetent rodents and human cancers xenografted in immunocompromised (“nude”) or immunodeficient (SCID) rodents. Cancers developing in immunocompetent mice are obtained by grafting procedures from preexisting solid tumors or cancer cell lines, genetic manipulations, or chemically inducing cancer, among other strategies. Cancers developing in immunocompromised or immunodeficient mice are xenografted into rodents from preexisting solid tumors, cancer cell lines, or cancer cells directly obtained from patients.
There are comprehensive reviews for each type of organ‐specific cancer that analyze the advantages and disadvantages of each model. In the following sections, we highlight compounds that have been evaluated for therapeutic efficacy in vivo in various preclinical models of cancer and in clinical trials.
3. MARINE NATURAL PRODUCTS AS SOURCES OF NEW ANTICANCER DRUGS
Natural products, their potency, selectivity, and mechanisms of action have evolved in nature as critical adaptations, serving as protection from predators, inhibiting competitors, parasites, and pathogens, and influencing reproductive and alimentary behavior. Natural selection is thus the reason behind their importance as drug candidates.155 Even though a detailed discussion of the possible natural functions of the bioactive metabolites is beyond the scope of this review, it is worth noting that chemoecological role of natural products is only available for a limited number of natural compounds. Chemical ecology, the discipline focusing on chemically mediated ecological interactions between organisms, is quite new.156, 157 Instead, the traditional man‐centered perspective on natural products, which is focused on potential medicines, has roots in ancient history and still being a key driver of natural product research.
However, over the past few decades the role of natural products in drug discovery has suffered from several changes. After a boost over the last half of the 20th century, the field experienced a sharp decline as the pharmaceutical industry practically abandoned natural product research in the mid‐1990s and shifted the focus toward building massive compound libraries via combinatorial chemistry.158, 159, 160 Still, against all expectations, in the last 25 years, 70% of all approved drugs have been natural products or their derivatives, while only a few de novo combinatorial compounds have been approved as drugs.160, 161, 162, 163 A special issue on Natural Product‐Based Drug Discovery was published in 2016 in Medicinal Research Reviews. In addition, reviews published on a regular basis by David J. Newman and Gordon M. Cragg are an excellent source of information on product‐based cancer drug discovery.161, 164, 165, 166, 167, 168
In the genomic era, with the advent of a series of novel technologies that speed up the rates of natural product discovery and assist in identifying novel mechanisms of action, there has been a steady reemergence of natural product‐based drug discovery as reviewed by Harvey et al.169 Some of these technologies involve a recapitulation of tumor ecosystems in 3D culture models134 with the possibility to apply quantitative high‐throughput screening,135 patient‐oriented screening,141, 142 the use of cut tissue slices,144 or “organs‐on‐chips” approaches,170 among others.
Traditionally, natural product‐based drug discovery mainly concerned the study of compounds isolated from terrestrial organisms, including bacteria, fungi, and especially higher terrestrial plants (Trachaeophyta), many of which employ some form of chemical defense to escape predation by herbivores.168, 171, 172, 173, 174, 175, 176 In contrast, the history of marine natural products in the context of anticancer drug discovery is relatively young. Oceans and seas constitute 75% of the Earth surface in which nearly one million multicellular (plants and animals) and one billion unicellular (distributed under 100 different phyla) species live. Oceans and seas have already yielded ∼26,000 active compounds.177 Blunt et al.177 report that these ∼26,000 compounds belong to ∼9000 collections obtained worldwide since 1965.
Similar to their terrestrial counterparts, marine organisms have evolved to produce bioactive compounds to adapt to environmental conditions and especially to deter feeding by predators (defensive allomones). Mitsiades et al.178 argued that compounds derived from marine organism may have evolved to be more potent than similar compounds from the above‐water organisms to compensate for the increased diffusion and thus rapidly decreasing protective concentration gradient of these compounds under water. However, although such considerations are certainly appropriate for water‐soluble polar compounds, low water solubility of many bioactive marine natural products, especially terpenes, instead prevents their dilution in the medium, allowing them to act at high doses during the contact with predators.179 In some cases lipophilicity may thus be an important feature both in preventing the dilution of defensive chemical weapons in water and making bioactive compounds of interest in pharmacology capable of permeating lipophilic biomembranes.
The NCI estimates that more than 1% of marine natural products show antitumor properties as compared to the 0.01% among their terrestrial counterparts.180 The marine‐sourced anticancer and cancer pain control agents that have reached late preclinical and clinical development were recently reviewed by Newman and Cragg.166 Gerwick and Moore110 reported in 2012 that the success rate of discovery from the marine world (for any type of clinical indication) with seven clinically useful and approved drugs from 22,000 discovered molecular entities (i.e., one drug per 3140 natural products described) is thus approximately 1.7‐ to 3.3‐fold better than the industry average, which is one in 5000–10,000 tested compounds.181, 185
Skropeta and Wei186 reviewed in 2014 (for the period 2009–2013) 188 novel marine natural products from deep‐water (from 50 to >5000 m) marine fauna including bryozoa, chordata, cnidaria, echinodermata, microorganisms, mollusca, and porifera. They report that 75% of the compounds they reviewed possess bioactivity, with almost half exhibiting low micromolar cytotoxicity toward a range of human cancer cell lines. Gerwick and Moore110 also report that mollusks, sponges, and tunicates are the richest collected sources of clinically useful drugs. However, they emphasize a lot of evidence showing that the actual producers of the bioactive substances are associated microorganisms (mainly heterotrophic bacteria and cyanobacteria). In addition, a trophic transfer of the compounds from sponges to their specialist nudibranch predators is also described.187, 188, 189
The next section covers the anticancer potential of molecules isolated from the phylum Mollusca describing the in vitro, in vivo preclinical and clinical studies published in the literature (PubMed and Scopus databases). As emphasized by Molinski et al.,159 drug discovery from marine natural products has enjoyed a renaissance in the past few years due to the recent marketing of the cancer pain control agent ziconotide (Prialt®) and the anticancer drug trabectedin (Yondelis®).
4. MOLLUSK‐DERIVED ANTICANCER AGENTS
Given the controversial theme of the biosynthetic origin of the metabolites isolated from mollusks, it must be emphasized that in the current review we chose to simply consider the studied mollusks as the natural sources of the compounds under investigation, and not their actual producers. Although some of the bioactive compounds under review are produced by the mollusks themselves, most of them are, in fact, of dietary origin. Noteworthy, the “mollusk‐derived” metabolites that entered or that are in clinical trials are actually produced by microbes. Consequently, the study of the molluscan chemistry has often resulted in the study of compounds produced by organisms at lower trophic levels.
A. Mollusks as Natural Product Sources
The phylum Mollusca shows great morphological, ecological, and chemical variability and it is subdivided in two subphyla, the Auculifera and the Conchifera. The Auculifera, which do not produce a complete shell, comprise the two classes of the Aplacophora, worm‐like in form, and the Polyplacophora, with a dorsal shell divided into eight valves. The shelled Conchifera includes five classes: (i) the Monoplacophora with a cap‐shaped shell; (ii) the Gastropoda including snails, slugs, and limpets; (iii) the Cephalopoda including octopuses, cuttlefish, and squids; (iv) the Bivalvia such as clams, oysters, scallops, and mussels; and (v) the Scaphopoda with a tubular shell.190 Most of the chemical studies on marine natural products have been focused on the gastropods, which are traditionally divided in three major groups, the subclasses Prosobranchia, Pulmonata, and Opisthobranchia,188 and more recently on the bivalves.191
Marine mollusks can be found from tropical seas and temperate waters to Artic‐Antarctic regions, showing different morphologies and occupying a wide range of ecological niches.192 They feed on a wide variety of benthic invertebrates and plants, often accumulating dietary metabolites from their prey to be reused against their own potential predators. Consequently, and as emphasized above, the study of the molluscan chemical diversity involves, at least in part, the exploration of the chemical composition of organisms at lower trophic levels.
Mollusks are also able to accumulate the dietary metabolites in localized anatomical structures.193, 194 This type of defensive strategy, which is particularly common in opisthobranchs, can be reinforced by the presence of visual aposematic patterns, which can be shared by groups of similarly colored species that share the cost of the education of predators within Müllerian mimetic circles.195 Recently, it has been even proposed that a group of nudibranchs belonging to the family Chromodorididae forms a putative Müllerian mimetic circle based on a common chemosensory signal, the anticancer macrolide latrunculin A, which is among the compounds discussed in this review.189 In Table I, we provide taxonomical, geographical, and ecological information for the mollusks under review and in Figure 1, we illustrate a selection of these organisms.
Table I.
Taxonomical, Geographical, and Ecological Information for the Mollusks under Review
| Reported name | Valid name | Class: Family | Photo in Figure 3 | Compounds under review | Distribution | Feeding behavior |
|---|---|---|---|---|---|---|
| Aplysia angasi * Sowerby, 1869 | Aplysia dactylomela (Rang, 1828) | Gastropoda: Aplysiidae | A | 19; 22; 23; 24a‐b; 29a-d; 39; 40 | Circumtropical | Herbivore (on macroalgae) |
| A. dactylomela Rang, 1828 | ||||||
| Aplysia depilans Gmelin, 1791 | Gastropoda: Aplysiidae | – | 42 | Eastern Atlantic and Mediterranean | Herbivore (on macroalgae) | |
| Aplysia fasciata Poiret, 1789 | Gastropoda: Aplysiidae | – | 41 | Eastern Atlantic and Mediterranean | Herbivore (on macroalgae) | |
| Aplysia kurodai Baba, 1937 | Gastropoda: Aplysiidae | – | 21; 64a‐h; 70; 71a‐c | Pacific | Herbivore (on macroalgae) | |
| Aplysia oculifera A. Adams & Reeve, 1850 | Gastropoda: Aplysiidae | – | 27; 28 | Indo‐Pacific | Herbivore (on macroalgae) | |
| Aplysia punctata Cuvier, 1803 | Gastropoda: Aplysiidae | B | 16; 17; 18 | European waters | Herbivore (on macroalgae) | |
| Austrodoris kerguelenensis * Bergh, 1884 | Doris kerguelenensis (Bergh, 1884) | Gastropoda: Dorididae | – | 32a‐f | Antarctic Ocean | Carnivore (on sponges) |
| Bathymodiolus thermophilus Kenk & B. R. Wilson, 1985 | Bivalvia: Mytilidae | – | 80a‐b | Pacific (deep sea) | Suspensivore (also absorbs nutrients synthesized by chemosymbiotic bacteria) | |
| Bursatella leachii Blainville, 1817 | Gastropoda: Aplysiidae | C | 73b‐c; 75a‐b | Indo‐Pacific | Herbivore (on cyanobacteria) | |
| Chelyonotus semperi Bergh 1886 | Gastropoda: Velutinidae | – | 82 | Indo‐Pacific | Carnivore (on tunicates) | |
| Chromodoris annae Bergh, 1877 | Gastropoda: Chromodorididae | – | 67a | Indo/West Pacific | Carnivore (on sponges) | |
| Chromodoris elisabethina Bergh, 1877 | Gastropoda: Chromodorididae | – | Indo/West Pacific | Carnivore (on sponges) | ||
| Chromodoris kuiteri Rudman, 1982 | Gastropoda: Chromodorididae | – | Indo/West Pacific | Carnivore (on sponges) | ||
| Chromodoris magnifica Quoy & Gaimard, 1832 | Gastropoda: Chromodorididae | – | Indo/West Pacific | Carnivore (on sponges) | ||
| Chromodoris inornata * Pease, 1871 | Chromodoris aspersa (Gould, 1852) | Gastropoda: Chromodorididae | – | 35a‐c; 36a‐d; 37 | Indo/West Pacific | Carnivore (on sponges) |
| Chromodoris lochi Rudman, 1982 | Gastropoda: Chromodorididae | – | 63a‐b; 67a | Indo/West Pacific | Carnivore (on sponges) | |
| Chromodoris obsoleta * Rüppell & Leuckart, 1831 | Goniobranchus obsoletus (Rüppell & Leuckart, 1830) | Gastropoda: Chromodorididae | – | 30a‐d; 31a‐g | Indo‐Pacific | Carnivore (on sponges) |
| Coriocella nigra Blainville, 1824 | Gastropoda: Velutinidae | – | 77a‐b | Indo‐Pacific | Carnivore (on tunicates) | |
| Dicathais orbita Gmelin, 1791 | Gastropoda: Muricidae | – | 81 | Coasts of Australia and New Zealand | Carnivore (on mollusks and crustaceans) | |
| Dolabella auricularia Lightfoot, 1786 | Gastropoda: Aplysiidae | D | 1a; 2a; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13a‐b; 14a‐b; 38a; 46; 52a‐b; 56a‐b; 57; 58a‐b; 59a‐d; 74 | Indo‐Pacific | Herbivore (on macroalgae) | |
| Elysia nisbeti Thompson, 1977 | Gastropoda: Plakobranchidae | – | 20 | Caribbean | Herbivore (on macroalgae) | |
| Elysia patina Ev. Marcus, 1980 | Gastropoda: Plakobranchidae | – | 20 | Caribbean | Herbivore (on macroalgae) | |
| Elysia rufescens Pease, 1871 | Gastropoda: Plakobranchidae | – | 15a | Pacific | Herbivore (on macroalgae) | |
| Elysia subornata A. E. Verrill, 1901 | Gastropoda: Plakobranchidae | E | 20 | Atlantic, Caribbean | Herbivore (on macroalgae) | |
| Glossodoris quadricolor * Rüppell & Leuckart, 1828 | Chromodoris quadricolor (Rüppell & Leuckart, 1830) | Gastropoda: Chromodorididae | F | 67b | Red Sea | Carnivore (on sponges) |
| Hexabranchus sanguineus Rüppell & Leuckart, 1830 | Gastropoda: Hexabranchidae | G | 60a‐b; 61a, c‐f; 62a‐b | Indo‐Pacific and Red Sea | Carnivore (on sponges) | |
| Jorunna funebris Kelaart, 1859 | Gastropoda: Discodorididae | H | 84a,85, 86 | Indo‐Pacific and Red Sea | Carnivore (on sponges) | |
| Kelletia kelletii Forbes, 1850 | Gastropoda: Buccinidae | – | 68a‐b | Eastern Pacific | Carnivorous scavenger | |
| Lamellaria sp. Montagu, 1816 | Gastropoda: Velutinidae | – | 76 | Palau | Carnivore (on tunicates) | |
| Leminda millecra Griffiths, 1985 | Gastropoda: Charcotiidae | – | 45 | Coasts of South Africa | Carnivore (on cnidarians) | |
| Mactromeris polynyma Stimpson, 1860 | Bivalvia: Mactrida) | – | 87 | Northeast Pacific | Suspensivore | |
| Onchidium sp.1 Buchannan, 1800 | Gastropoda: Onchidiidae | – | 55a‐b | New Caledonia | Herbivore (on microalgae) | |
| Onchidium sp.2 Buchannan, 1800 | Gastropoda: Onchidiidae | I | 65 | South China Sea | Herbivore (on microalgae) | |
| Peltodoris atromaculata Bergh, 1880 | Gastropoda: Discodorididae | J | 69a | Mediterranean and Atlantic | Carnivore (on sponges) | |
| Phidiana militaris Alder & Hancock, 1864 | Gastropoda: Facelinidae | K | 83a‐b | Indo‐Pacific | Carnivore (on cnidarians) | |
| Philinopsis speciosa Pease, 1860 | Gastropoda: Aglajidae | – | 50; 51a‐b | Indo/West Pacific | Carnivore (on mollusks) | |
| Phyllidia coelestis Bergh, 1905 | Gastropoda: Phyllidiidae | L | 25a‐b | Indo/West‐Pacific | Carnivore (on sponges) | |
| Pleurobranchus albiguttatus Bergh, 1905 | Gastropoda: Pleurobranchidae | – | 33a‐b, d, f, j; 34a‐c | Indo/West‐ Pacific | Carnivore (on tunicates) | |
| Pleurobranchus forskalii Rüppell & Leuckart, 1828 | Gastropoda: Pleurobranchidae | – | 33a‐b, d, f, j; 34a; 53; 54 | Indo/West‐Pacific | Carnivore (on tunicates) | |
| Reticulidia fungia Brunckhorst & Gosliner in Brunckhorst, 1993 | Gastropoda: Phyllidiidae | – | 26a‐b | Indo‐Pacific | Carnivore (on sponges) | |
| Roboastra tigris Farmer, 1978 | Gastropoda: Polyceridae | – | 78a | Gulf of California | Carnivore (on mollusks) | |
| Stylocheilus longicauda Quoy & Gaimard, 1825 | Gastropoda: Aplysiidae | – | 51a‐b; 73a | Circumtropical | Herbivore (on cyanobacteria) | |
| Tambja abdere Farmer, 1978 | Gastropoda: Polyceridae | – | 78a | Eastern Pacific | Carnivore (on bryozoans) | |
| Tambja ceutae Garcia‐Gomez & Ortea, 1988 | Gastropoda: Polyceridae | – | 78c | Eastern Atlantic | Carnivore (on bryozoans) | |
| Tambja eliora Er. Marcus & Ev. Marcus, 1967 | Gastropoda: Polyceridae | – | 78a | Gulf of California | Carnivore (on bryozoans) | |
| Trimusculus costatus Krauss, 1848 | Gastropoda: Ellobiidae | – | 44 | Coasts of South Africa | Filter feeder | |
| Trimusculus peruvianus G. B. Sowerby I, 1835 | Gastropoda: Ellobiidae | – | 43 | Coasts of Chile and Peru | Filter feeder | |
| Turbo stenogyrus P. Fischer, 1873 | Gastropoda: Turbinidae | – | 79 | Indo/West –Pacific | Herbivore | |
| Tylodina perversa Gmelin, 1791 | Gastropoda: Tylodinidae | – | 72 | Northeastern Atlantic and Mediterranean | Carnivore (on sponges) |
The species names reported in the chemical literature are listed by alphabetical order, and marked by asterisks when synonymized. The distribution refers to the native range of the species, rather than new ranges where nonnative species have recently become established.
The species names and distribution was found at [1] World Register of Marine Species. Available from http://www.marinespecies.org at VLIZ; accessed April 8, 2016 [2]. MolluscaBase (2015); accessed at http://www.molluscabase.org on April 8, 2016 [3]. Australian Museum Sea Slug Forum, Sydney; available at http://www.seaslugforum.net.
If reproducibility is a crucial issue in all sciences, it assumes a critical importance in cancer research. When the bioactive compounds are difficult or costly to synthesize, the extraction from natural sources may be the only realistic way to provide sufficient amounts of purified metabolites for preliminary pharmacological screenings. Consequently, the correct species classification and information about the collection sites becomes of utmost importance. Even appropriate taxonomic and geographic information, however, is not always sufficient. Intraspecific variations in secondary metabolites between individuals of a given species, in fact, are widely documented in the literature. In addition, given that the taxonomy evolves, sometimes it is difficult to track the source of a given metabolite in the available chemical literature. The so‐called “chemotaxonomy” adds more confusion to the issue because a secondary metabolite can be of dietary origin, produced by symbionts, or de novo biosynthesized. This generate a sort of “labyrinth” from which we could get out only by providing, in the future, more information about the origin of the metabolites, more biosynthetic studies, more accurate GPS data of the sampling locations, and more complete information about the cases of synonymy and possible misidentification.
The current review highlights the diversity of chemical structures, mechanisms of action, and most importantly the assessment of “promise” of mollusk‐derived metabolites as anticancer agents. We include, as mollusk‐derived metabolites, compounds that were originally identified in mollusks. However, since the initial discovery of certain metabolites with anticancer effects in a given mollusk, it has been demonstrated in certain cases that these compounds are not biosynthesized by the mollusk but instead are of dietary origin. We thus provide in Table II all the compounds under review (alphabetically listed) that were initially discovered in mollusks and we also report the possible actual producer(s) of the compounds of interest when such information is available in the literature.
Table II.
Compounds Under Review That Were Isolated from Mollusks Listed in Alphabetical Order
| Compound | ||||
|---|---|---|---|---|
| Name | No. | Figure | Reported source(s) among mollusks (see Table I) | Possible actual producer(s) |
| 3‐Acetyl‐11‐(3‐methylbutanoyl)‐13‐propanoylilikonapyrone | 65 | 18 | Onchidium sp.2 | – |
| 3‐Epi‐aplykurodinone B | 41 | 12 | Aplysia fasciata | – |
| Aplaminal | 70 | 20 | Aplysia kurodai | – |
| Aplaminones | 71a‐c | 20 | – | |
| Aplyronines | 64a‐h | 19 | – | |
| (‐)‐Aplysin | 21 | 7 | Laurencia tristicha (red algae) | |
| Aplysistatin | 22 | 7 | Aplysia angasi | – |
| Aplysqualenol A | 39 | 12 | Aplysia dactylomela | – |
| Aurilide | 52a‐c | 14 | Dolabella auricularia | Lyngbya (cyanobacteria) |
| Aurilol | 38a | 11 | D. auricularia | Laurencia (red algae) |
| Auripyrones | 58a‐b | 16 | – | |
| Aurisides | 56a‐b | 16 | D. auricularia | Cyanobacteria |
| Bathymodiolamides | 80a‐b | 23 | Bathymodiolus thermophilus | – |
| C‐21‐hydroxylated sterol | 43 | 12 | Trimusculus peruvianus | – |
| Caulerpenyne | 20 | 7 | Elysia subornata, E. patina, E. nisbeti | Caulerpa spp. (green algae) |
| Cycloforskamide | 54 | 15 | Pleurobranchus forskalii | Prochloron didemni (cyanobacteria) |
| Dichlorolissoclimide, chlorolissoclimide | 33 a,b | 10 | Pleurobrancus albiguttatus, P. forskalii | Lissoclinum (tunicate) |
| Dolabelides | 59a‐d | 16 | D. auricularia | – |
| Dolabellin | 74 | 22 | – | |
| Dolastatin 1 | 3 | 3 | – | |
| Dolastatin 3 | 4 | 3 | Lyngbya majuscula (cyanobacteria) | |
| Dolastatin 10 | 1a | 2 | Symploca sp. VP642, S. hydnoides (cyanobacteria) | |
| Dolastatin 11 | 5 | 4 | – | |
| Dolastatin 12 | 6 | 4 | L. majuscula, Schizothrix calcicola (cyanobacteria) | |
| Dolastatin 13 | 7 | 4 | – | |
| Dolastatin 14 | 8 | 4 | – | |
| Dolastatin 15 | 2a | 3 | – | |
| Dolastatin 16 | 9 | 5 | D. auricularia | Lyngbya majuscule, Symploca cf. hydnoides (cyanobacteria) |
| Dolastatin 17 | 10 | 5 | – | |
| Dolastatin 18 | 11 | 5 | – | |
| Dolastatin 19 | 57 | 16 | – | |
| Dolastatin D | 12 | 5 | – | |
| Dolastatin G, Nordolastatin G | 13 a,b | 5 | – | |
| – | ||||
| Dolastatin H, isodolastatin H | 14 a,b | 6 | – | |
| Doliculide | 46 | 13 | Cyanobacteria Sponges | |
| Dorisenones and related spongian diterpenoids | 30 a‐d 31 a‐g | 8‐9 | Chromodoris obsoleta | |
| Elatol | 23 | 7 | A. dactylomela | Laurencia (red algae) |
| Endoperoxide sterol | 42 | 12 | Aplysia depilans | – |
| Enshuol | 38b | 11 | Laurencia (red algae) | |
| Halichondramides | 62a‐c | 18 | Hexabranchus sanguineus | Halichondria sp. (sponges) |
| Halogenated monoterpenes | 16 | 7 | Aplysia punctata | Plocamium (red algae) |
| 17 | 7 | |||
| 18 | 7 | |||
| 19 | 7 | A. dactylomela | – | |
| Haterumaimides | 33c‐q/34a‐c | 10 | P. albiguttatus, P. forskalii | Lissoclinum (tunicates) |
| Hectochlorin | 75a‐b | 22 | Bursatella leachii | L. majuscula (cyanobacterium) |
| Hydroxyl‐dehydroisofulvinol | 69a | 20 | Peltodoris atromaculata | Haliclona fulva (sponges) |
| Inorolides | 35a‐c | 10 | Chromodoris inornata | Hyrtios (sponges) |
| Isofistularin‐3 | 72 | 21 | Tylodina perversa | Aplysina (sponges) |
| Jorumycin | 84a | 24 | Jorunna funebris | Sponges |
| Jorunnamycin C | 85 | 24 | ||
| Kabiramides | 61a‐f | 17 | H. sanguineus | Halichondria (sponges) |
| Kahalalide F | 15a | 6 | Elysia rufescens | Bryopsis pennata (green algae) |
| Keenamide A | 53 | 15 | P. forskalii | – |
| Kelletinins | 68a‐b | 20 | Kelletia kelletii | – |
| Kuanoniamine A | 82 | 23 | Chelynotus semperi | Tunicates |
| Kulokekahilides 1 and 2 | 51 a,b | 14 | Philinopsis speciosa | L. majuscula (cyanobacteria) |
| Kulolide 1 | 50 | 13 | – | |
| Lamellarin D | 76 | 22 | Lamellaria sp. | Tunicates and/or sponges |
| Latrunculin A | 67a | 20 | Chromodoris lochi | Spongia mycifijensis (sponges) |
| Latrunculin B | 67b | 20 | Glossodoris quadricolor | Latrunculia magnifica (sponges) |
| Laulimalides | 63a‐b | 18 | C. lochi | Hyattella sp. (sponges) |
| Malyngamides | 73 a | 21 | Stylocheilus longicauda | L. majuscula (cyanobacteria) |
| 73 b,c | 21 | B. leachii | ||
| Obtusane (epi‐) | 28 | 8 | Aplysia oculifera | Laurencia, Plocanium (red algae) |
| Obtusol | ||||
| Isoobtusol | 24 a,b | 7 | A. dactylomela | Laurencia (red algae) |
| Oculiferane | 27 | 8 | A. oculifera | Laurencia, Plocanium (red algae) |
| Onchidins | 55a‐b | 15 | Onchidium sp.1 | – |
| Palmadorins | 32a‐f | 9 | Austrodoris kerguelenensis | – |
| Parguerol and derivatives | 29a‐e | 8 | A. dactylomela | Jania rubens (red alga) and/or sponges |
| Phidianidines | 83a‐b | 23 | Phidana militaris | – |
| Prenylated hydroquinone | 45 | 12 | Leminda millecra | Leptogorgia (cnidarians) |
| Pupukeanane derivatives | 25 a,b | 7 | Phyllidia coelestis | Sponges |
| Renieramycin M | 86 | 24 | J. funebris | Sponges |
| Reticulidins A and B | 26 a,b | 8 | Reticulia fungia | Sponges |
| Secosterol | 44 | 12 | Trimusculus costatus | – |
| Sesterterpenoids | 36a‐d/37 | 11 | C. inornata | Hyrtios (sponges) |
| Sphinxolide | 66 | 19 | Unidentified nudibranch | Neosiphonia superstes, Reidispongia coreula (sponges) |
| Spisulosine (ES‐285) | 87 | 24 | Mactromeris polymyna | – |
| Staurosporine analogues | 77a‐b | 22 | Coriocella nigra | – |
| Tambjamines D, E, and K | 78a‐c | 22 | Tambja eliora, T. abdere, T. centae, Roboastra tigris | Bryozoans |
| Thyrsiferol | 40 | 12 | A. dactylomela | Laurencia thyrsifera (red algae) |
| Turbostatin A | 79 | 23 | Turbo stenogyrus | – |
| Tyrindoleninone | 81 | 23 | Dicathais orbita | – |
| Ulapualides | 60a‐b | 17 | H. sanguineus | Halichondria (sponges) |
B. Dolastatins
We describe below the cyanobacterial metabolites of the dolastatin type. However, it must be recalled that these types of compounds did not successfully complete clinical trials, usually exiting trials at or before the Phase II level. Thus, though we detail below the use of agents derived from dolastatins as potential anticancer agents, it should be noted that most of the Phase I trials did not proceed any further. Therefore, dolastatins can no longer be labeled as “promising anticancer drugs.”
Dolastatins (1a‐14b; Figs. 2, 3, 4, 5, 6) embody a large family of active compounds originally isolated from Dolabella auricularia (Anaspidea mollusk, Table I; Fig. 1D). From the chemical point of view, dolastatins show high structural heterogeneity including linear and cyclic peptides, depsipeptides, peptides containing the thiazole and oxazole heterocycles, and macrolides. The low concentrations of dolastatins found in sea hares suggested a dietary origin and this was subsequently confirmed by direct isolation of dolastatin 10 from field collections of the marine cyanobacterium Symploca.196 Also, it has been confirmed for a number of other dolastatins that they are not produced by sea hares,197 but ingested and sequestered from the dietary cyanobacteria. In some cases, the true producers of dolastatins have been demonstrated to be cyanobacteria of genus Symploca (Table II) recently revised into the new genus Caldora and species penicillata,198 as well as Lyngbya.
Figure 2.
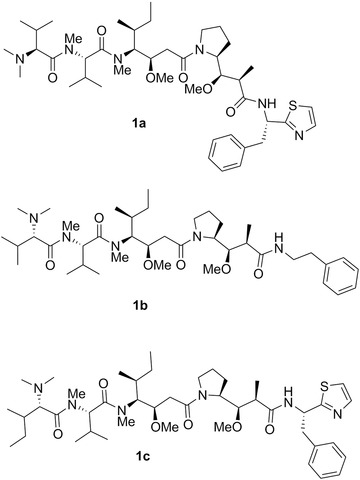
Chemical structures of dolastatin 10 (1a), soblidotin (TZT‐1027; 1b), and symplostatin 1 (1c).
Figure 3.
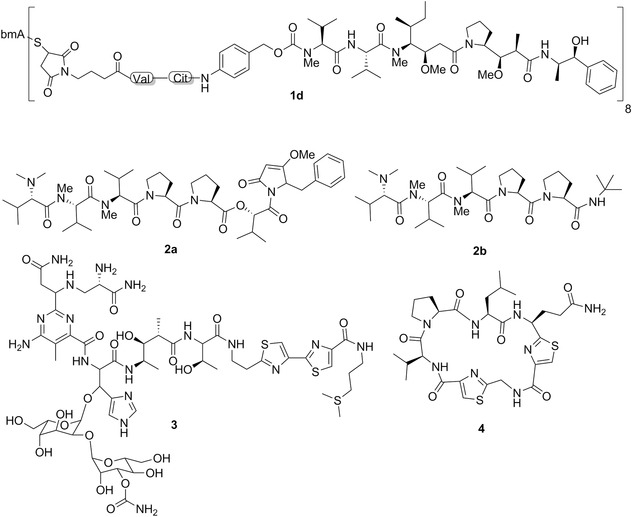
Chemical structures of brentuximab vedotin (SGN‐35; Adcetris®; 1d), dolastatin 15 (2a), tasidotin (ILX‐65; 2b), dolastatin 1 (3), and dolastatin 3 (4). MAB in structure 1d represents the monoclonal antibody portion of this ADC.
Figure 4.
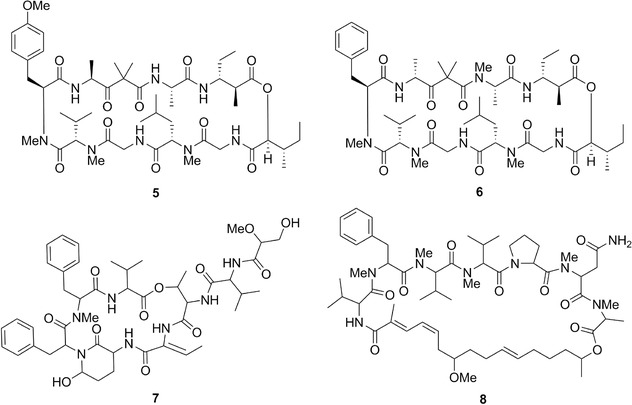
Chemical structures of dolastatin 11 (5), dolastatin 12 (6), dolastatin 13 (7), and dolastatin 14 (8).
Figure 5.
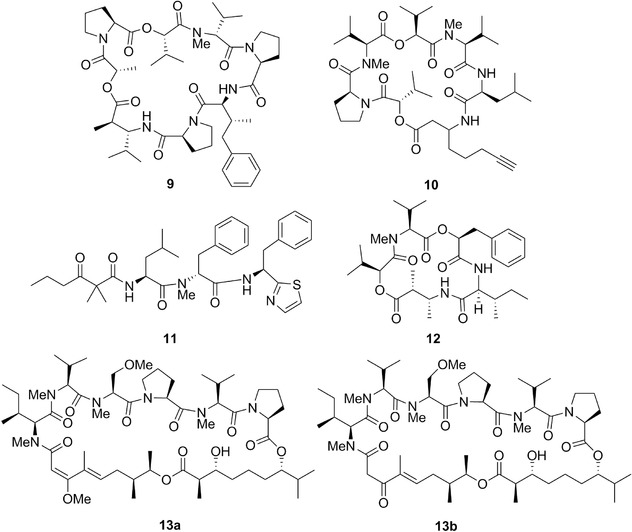
Chemical structures of dolastatin 16 (9), dolastatin 17 (10), dolastatin 18 (11), dolastatin D (12), dolastatin G (13a), and nordolastatin G (13b).
Figure 6.

Chemical structures of dolastatin H (14a), isodolastatin H (14b), kahalalide F (15a), and elipsidepsin (15b).
As reported by Pettit et al.,199 the genus Dolabella has interesting history. The Roman natural scientist Pliny the Elder comprehensively studied and then described (∼60 A.D.) a potent Indian Ocean sea hare of the genus Dolabella. The Romans named the Mollusca of the family Aplysiidae as “sea hare” due to the similarity between the ears of a hare and the auriculate tentacles of these gastropods.199 Extracts from the Dolabella sea hare were already known for their toxic properties during the reign of Nero.199 Nicander (∼150 A.D.) recognized the possibility of using Dolabella extracts to treat certain diseases.199 Pettit et al.199 thus argued that the dolastatin family of compounds they discovered (see below) most probably correspond to the potent D. auricularia constituents recognized by the above‐named Romans. Among these largely diverse compounds, the linear peptides dolastatin 10 and 15 show the most remarkable picomolar GI50 values in most in vitro cancer growth inhibition studies.200
Dolastatin 10 (1a; Fig. 2; Table II) was first isolated from D. auricularia collected from the Indian Ocean201 and then discovered (with its methyl derivative, symplostatin 1) in the cyanobacterium Symploca hydnoides.196 Dolastatin 10 is a linear pentapeptide with four of residues being structurally unique: dolavaline (Dov), dolaisoleuine (Dil), dolaproine (Dap), and dolaphenine (Doe), in addition to valine (Fig. 2). Its absolute configuration was established by synthesis by Pettit et al.202 Singh et al.203 comprehensively reviewed the historical development of dolastatin 10. Briefly, it entered Phase I clinical trials in the early 1990s under the auspices of the NCI. Then, several Phase II clinical studies (advanced and metastatic soft tissue sarcoma, advanced hepatobiliary cancers, pancreatic cancers, among others) did not reveal the efficacy of dolastatin 10 (1a) as a single anticancer agent due to dose‐limiting side effects, such as neuropathy.203
Singh et al.203 explained that although clinical trial results with dolastatin 10 were discouraging, these as well as preclinical studies offered the basis for structure–activity relationship (SAR) analyses and have led to the discovery of soblidotin (TZT‐1027, 1b; Fig. 2), a synthetic analogue of dolastatin 10,203, 204 in which the thiazole moiety of the dolaphenine was replaced by phenylalanine methyl ester. The in vitro growth inhibitory effects of soblidotin decline depending on the amount of P‐gp expressed in cancer cells.205 Soblidotin displayed in vivo anticancer activity in several models including the murine P388 leukemia, Colon26 colon cancer, Lewis Lung carcinoma, B16 melanoma and M5076 sarcoma models as well as human MX‐1 breast cancer, and LX‐1 and SBC‐3 SCLC xenografts.205 Soblidotin entered several Phase II clinical trials (advanced and metastatic soft tissue sarcomas, NSCLCs, among others) but it was also found ineffective as a single agent and its clinical development was no longer pursued.203
The GI50 index of dolastatin 10 in the NCI 60 cell‐line panel is ∼0.2 nM and a COMPARE analysis with a cut‐off correlation at p = 0.7 is negative in the standard agent dataset. This agent is not able to overcome the MDR phenotype mediated by the P‐glycoprotein (P‐gp) in cancer cells206 as is also the case with respect to its analogues soblidotin205 and symplostatin 1 (1c; Fig. 2).207 Dolastatin 10 induces proapoptotic stimuli in cancer cells208, 209 as do soblidotin205, 210 and symplostatin 1.207 Dolastatin 10 was rapidly identified as an inhibitor of tubulin polymerization.211 In addition to the inhibition of tubulin polymerization, it inhibits tubulin‐dependent GTP hydrolysis and the binding of vinblastine, maytansine, and vincristine to tubulin, although its binding site on tubulin is different from that of the vinca alkaloids.210 The tubulin‐binding sites of colchicine, taxol, vinblastine, rhizoxin F, and maytansine are discussed by Prota et al.212 and in the commentary by Field et al.213
Dolastatin 10 displays in vivo anticancer activity in various models, including, for example, MDR diffuse large cell lymphoma WSU‐DLCL2,214 SCLC NCI‐H446215, and ovarian carcinoma216 xenografts. Dolastatin 10 also displayed anticancer activity in various cancer models at the NCI: mouse P388 and L1210 leukemia, B16 melanoma and M5076 sarcoma as well as human LOX melanoma, and MX‐1 breast cancer xenografts.
In addition to soblidotin, the modification of C‐terminus of dolastatin 10 yielded a series of analogs called auristatins.217 However, these were also ineffective in clinical trials.200 An important discovery was made by Miyazaki et al.,218 who found that the removal of one N‐methyl group of dolavaline at the N‐terminus of dolastatin 10 gives an analog showing only slightly attenuated potency. With this secondary amine at their N‐terminus free, the auristatins became suitable for attachment of a linker to facilitate its conjugation to monoclonal antibodies, leading to the generation of highly potent and efficacious antibody drug conjugates (ADCs). This led, for example, to the FDA‐approved ADC brentuximab vedotin (SGN‐35, Adcetris®; 1d; Fig. 3).219 Vedotin is monomethyl auristatin E chemically conjugated to the chimeric anti‐CD30 antibody.220 It was approved for the treatment of relapsed Hodgkin lymphoma and systemic anaplastic large‐cell lymphoma.221 Over 30 ADCs in clinical trials currently employ auristatins as payloads. The reader is referred to recent reviews on this subject.166, 200, 219, 222
After Pettit et al. published the structure of dolastatin 15 (2a; Fig. 3; Table II), also isolated from D. auricularia,223 several groups developed its total synthesis.224, 225 The supply of synthetic dolastatin 15 has allowed its biological evaluation. Dolastatin 15 also interacts with tubulin and may bind in the vinca domain of tubulin, presumably in the same site as dolastatin 10.226 It induces a loss of tension across the kinetochore pairs due to the disruption of normal microtubule assembly dynamics.227 Beckwith et al.228 reported that dolastatin 10 and 15 display in vitro antiproliferative activities that are three to four orders of magnitude more potent than vincristine, a clinically used antiproliferative agent, while dolastatin 15's NCI‐based GI50 is about ten times higher than that of dolastatin 10 (2 vs. 0.2 nM). Dolastatin 15 was also slightly inferior in terms of in vivo anticancer activity in a human ovarian carcinoma xenografted model.216 Dolastatin 15 is a classical inducer of apoptosis in cancer cells and thus behaves as a conventional proapoptotic cytotoxic agent.229, 230 Furthermore, it is not able to overcome the MDR phenotype mediated by the P‐glycoprotein (P‐gp) in cancer cells.216
Like for dolastatin 10, a series of SAR studies have been undertaken with the focus on structural and stereochemical modification at the C terminus (Hiva‐Dpy).217 These studies led to the discovery of tasidotin, which was advanced to clinical trials. Tasidotin (ILX‐651; 2b; Fig. 3) is a peptide analog of dolastatin 15 in which the carboxyl‐terminal ester group is replaced by the tert‐butyl amide.231 Tasidotin, which is orally active, reduces the shortening rate, the switching frequency from growth to shortening (catastrophe frequency), and the fraction of time the microtubules grow.232 Tasidotin is a proapoptotic cytotoxic compound233 and it is also a P‐gp substrate234 as all the other dolastatins. It showed promising in vivo anticancer activity in preclinical models of pediatric sarcomas,233 LOX melanoma xenografts,234 and xenograft models of breast cancer, ovarian cancer, prostate cancer, and colon cancer as reported in the ILX‐651 investigators brochure (Genzyme Corp., 2004) cited by Garg et al.,233 but not in H460 NSCLC xenografts.234 Tasidotin was also efficient in vivo in murine P388 leukemia model.233 But as for soblidotin, the clinical development of tasidotin was not pursued beyond Phase II clinical trials because of lack of efficacy.
As mentioned above, all dolastatins seem to be proapoptotic agents unable to circumvent the MDR phenotype in cancer cells and they all displayed poor anticancer activity in Phase II clinical trials, leading to the discontinuation of their clinical evaluation.203
In 1981, Pettit et al.199 claimed that dolastatin 1 (3; Fig. 3; Table II), which was isolated from D. auricularia, represented by that time the most potent anticancer agent discovered with an 88% life extension (at a dose of 11 μg/kg) in the murine P388 lymphocytic leukemia model, and a curative rate (33% at a dose of ∼2 μg/kg) in the murine B16 melanoma model. The NCI 60 cell‐line panel had not been established yet at that time.115
Dolastatin 3 (4; Fig. 3; Table II), a cyclic peptide containing two thiazole rings, was isolated from a Japanese specimen of D. auricularia (Fig. 1D) and displayed the GI50 of <1 μM in P388 murine leukemia cells.235 Its full structure was confirmed by synthesis some years later.236 This compound was also isolated from a Palauan collection of Lyngbya majuscula.237 Dolastatin 3 induced in vivo a 78% life extension in the murine P388 lymphocytic leukemia model and a 52% life extension in murine colon carcinoma 38.237
Dolastatin 11238 (5; Fig. 4; Table II), dolastatin 12238 (6; Fig. 4; Table II), dolastatin 13239 (7; Fig. 4; Table II), and dolastatin 14240 (8; Fig. 4; Table II) are depsipeptides isolated from D. auricularia. Dolastatin 12 was also isolated from L. majuscula/Schizothrix calcicola cyanobacterial assemblages (Table II).241 Dolastatin 11's mean GI50 was ∼0.07 μM in the NCI 60 cell line panel and the COMPARE analysis with a cut‐off correlation at p = 0.7 was negative in the standard agent dataset. Dolastatin 12's GI50 values ranged from ∼1 (human NCI‐H460 NSCLC) to ∼30 nM (human SF‐295 CNS cancer) when assayed in five cancer cell lines.242 Additionally, its GI50 values in mouse neuro‐2a neuroblastoma243 and P388 leukemia238 cells were ∼0.1 and >1 μM, respectively. Dolastatin 13's and 14's GI50 values were 14 nM239 and 20 nM,240 respectively, in the murine P388 leukemia cell line.
While both dolastatin 10 and 15 affect tubulin as detailed above, dolastatin 11 targets actin and it displays about threefold higher growth inhibitory effects than the sponge‐derived depsipeptide jasplakinolide.244 Dolastatin 11 does not arrest cancer cells in mitosis, as do dolastatin 10 and 15, but induces massive rearrangement of the cellular actin filament network leading to dramatic cytoplasmic retraction and subsequently cell division arrest at the level of cytokinesis.244 It binds actin at a site distinct from phalloidin,244 which is a toxin from the mushroom Amanita phalloides 245 and one of the best known antiactin compounds.246 Specifically, dolastatin 11 connects two long‐pitch strands in F‐actin to stabilize microfilaments.247 Tubulin and actin are not only key players in normal and cancer cell division, but also in cancer cell invasion and metastasis.248
As reported above for dolastatin 11, dolastatin 12 also targets actin microfilaments.241 A dolastatin 12 analog displayed only marginal or no in vivo anticancer activity against murine colon adenocarcinoma 38 and mammary adenocarcinoma 16/C and appeared to be toxic.241
Dolastatin 16 (9; Fig. 5; Table II) is a cyclic depsipeptide containing two new amino acids, dolamethylleuine, and dolaphenvaline collected from D. auricularia in Papua New Guinea.249 The X‐ray crystal structure of dolastatin 16 and syntheses of the new amino acid units dolamethylleuine and dolaphenvaline were published later.250 Dolastatin 16 was also isolated from a Madagascan collection of L. majuscula 251 as well as from Symploca cf. hydnoides collected off Guam.252 Dolastatin 16's GI50 values were in low nanomolar ranges when assayed in a mini panel of four human solid cancer cell lines and in five leukemia cell lines.249 Its mean GI50 in the NCI 60 cell line panel was ∼0.3 μM and COMPARE analyses provided relatively low correlations with dolastatin 10 (p = 0.76) and dolastatin 15 (p = 0.71).249, 253 The total synthesis of dolastatin 16 consisting of 23 steps was recently achieved.254 Interestingly, Pettit et al.254 report that the synthetic dolastatin 16, while otherwise identical (by X‐ray crystal structure and spectral characteristics) with the natural product, did not reproduce the nanomolar cancer cell growth inhibition displayed by the natural isolate. Pettit et al.254 reported that presumably this result can be attributed to the conformation(s) of the synthetic dolastatin 16 or to a chemically undetected component isolated with the natural product.
Additional dolastatins that have been isolated from D. auricularia are dolastatin 17255 (10; Fig. 5), collected in Papua New Guinea specimens; dolastatin 18256 (11, Fig. 5), collected in Indian Ocean specimens; as well as dolastatin D257 (12; Fig. 5), dolastatin G258 (13a; Fig. 5), nordolastatin G258 (13b; Fig. 5), dolastatin H259 (14a; Fig. 6) and isodolastatin H259 (14b; Fig. 6) found in Japanese specimens.
Dolastatin 17 (10) is a cyclodepsipeptide with a novel acetylenic β‐amino acid named dolayne (Doy), similar to that found in onchidin260 (discussed in Section 4.D.2). Dolastatin 17's GI50 values were in submicromolar ranges in the four cancer cell lines in which it was assayed.255 Dolastatin 18 (11) is characterized by the presence of a thiazole ring in its structure. Its GI50 values were in submicromolar ranges in the mouse P388 lymphocytic leukemia and the human NCI‐H460 NSCLC cell lines.256 Dolastatin 19 (57, for structure see Fig. 16), isolated from a different collection of D. auricularia specimens from the Gulf of California,261 is a macrocyclic lactone that is strongly related to aurisides as illustrated in Section 4.D.3 (Table II). Dolastatin 19 displays in vitro growth inhibitory activity with GI50 values of ∼1 μM in breast MCF‐7 and colon KM20L2 cancer cells.261 Dolastatin D is a cyclic depsipeptide with a GI50 value of ∼4 μM in human HeLaS3 cancer cells.257 Dolastatin G and nordolastatin G are cyclic depsipeptides with GI50 values of ∼1 and ∼5 μM, respectively, in human HeLaS3 cancer cells.258 Dolastatin H and isodolastatin H, isolated from Western Indian Ocean specimens of D. auricularia, are linear peptides closely related to dolastatin 10.259 Synthetic dolastatin H displayed a GI50 value of 2 nM in human HeLaS3 cancer cells.259 Isodolastatin H was evaluated in vivo against murine P388 leukemia and shown to exhibit antitumor activity that is slightly weaker (41% of life time extension as compared to control) than that of dolastatin 10 (55% of life time extension as compared to control).259
Figure 16.
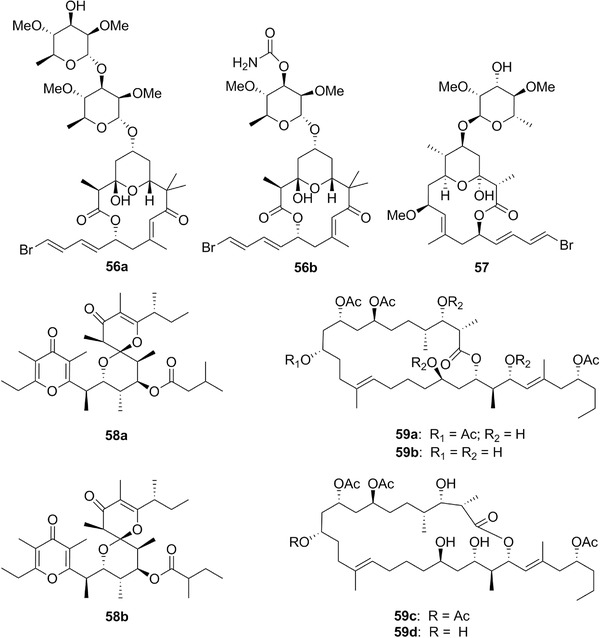
Chemical structures of aurisides A (56a) and B (56b), dolastatin 19 (57), auripyrones A (58a) and B (58b), and dolabelides A (59a), B (59b), C (59c), and D (59d).
C. Kahalalides
Many sacoglossan mollusks feed upon algae from which they acquire biologically active metabolites. For example, several species of Elysia sequester toxic compounds from Bryopsis.262 In particular Elysia rufescens (Table I), E. grandifolia, and E. ornata were found to contain depsipeptides called kahalalides present also in Bryopsis, which are known to be responsible for the deterrent properties of the mollusks.263 Davis et al.264 report that sacoglossan mollusks are characterized by the ability to sequester functional chloroplasts from their algal diet through a process called kleptoplasty, enabling them to photosynthesize. These authors provided experimental evidence showing that a diverse bacterial assemblage is associated with E. rufescens and its mucus, with secreted mucus harboring higher bacterial richness than entire E. rufescens samples. Davis et al.264 have thus shown that the most abundant bacterial groups affiliated with E. rufescens and its mucus are Mycoplasma spp. and Vibrio spp., respectively. They accordingly suggest that kahalalide F, found in E. rufescens (Table I), is possibly of bacterial origin.
Chemically, the size and composition of kahalalides are highly variable, ranging from a C31 tripeptide to a C77 tridecapeptide, and each peptide contains a different fatty acid chain.263 Kahalalide family has reached more than 20 members, of which kahalalide F is the most promising anticancer agent as detailed below. Gao and Hamann263 comprehensively reviewed in 2011 the chemistry and biology of kahalalides.
Kahalalide F (15a; Fig. 6; Table II) is a cyclic peptide connected with an amidic bond to a short chain fatty acid. It was isolated from the herbivorous sacoglossan mollusk E. rufescens (Table I) living in the seas near Hawaii, and then from its green algal diet Brysopsis pennata (Brypsidaceae).262 Before describing its anticancer effects, we would like to draw the reader's attention to the following point. Some articles report on oncosis‐like effects induced by kahalalide F in cancer cells, while others report on the induction of necrosis‐like features. At first glance, it thus seems that kahalalide F could induce either oncosis or necrosis in cancer cells, while in fact this is not the case. Indeed, as clearly stated by Weerasinghe and Buja,265 oncosis is a term that actually relates to a mode of cell injury and cell death, while necrosis relates to a cell population (tissue) degradation process following cell death. In other terms, necrosis should not be used to describe a specific death process occurring in single cells. Weerasinghe and Buja265 comprehensively reviewed the oncosis‐related characteristics at both morphological and biochemical levels and they define that oncotic cell death involves progressive membrane injury involving three stages that they detail in their review. These authors thus define oncosis as a form of cell death accompanied by cellular swelling, organelle swelling, blebbing, and increased membrane permeability caused by the failure of various ionic pumps in the plasma membrane, with increases in concentrations of cytosolic calcium and rearrangement of cytoskeletal proteins.265
The fact remains that (i) oncosis shares no common signaling pathways with apoptosis,265 (ii) lysosomal targeting can lead to cell death types other than oncosis,266, 267, 268 and (iii) altered cancerous lysosomes are involved in promoting cancer progression of metastatic disease.267 Inducing oncosis in cancer cells would thus make it possible to overcome the resistance to proapoptotic stimuli displayed by cancer cells associated with aggressive biological behaviors.65 Suárez et al.269 accordingly observed that kahalalide's F cytotoxicity did not correlate with the expression level of the MDR1 efflux pump. In fact, García‐Rocha et al.270 already reported two decades ago that kahalalide F kills cancer cells via the targeting of lysosomes. Suárez et al.269 then reported that kahalalide F induces oncosis in human prostate and breast cancer cells. Suárez et al.269 thus suspected that kahalalide F does not induce apoptosis in cancer cells. This hypothesis was then experimentally confirmed by Janmaat et al.,271 who observed that several markers of caspase‐dependent apoptosis, such as phosphatidyl‐serine externalization, cytochrome c release, and caspase‐3 and poly‐(ADP‐ribose) polymerase cleavage were negative after exposure of cancer cells to kahalalide F. Also, inhibitors of caspases or cathepsins failed to protect cancer cells against kahalalide's F cytotoxicity. Janmaat et al.271 identified ErbB3, a downstream molecule of the PI3K‐Akt pathway, as an important determinant of the cytotoxic activity of kahalalide F in vitro. Appert‐Collin et al.272 have recently reviewed the roles of ErbB receptors in cancer cell migration and invasion, while Mayer and Arteaga273 comprehensively reviewed the PI3K‐Akt pathway as a target for cancer treatment.
Pardo et al.274 reported that kahalalide F was found to be COMPARE‐negative when it was tested in the NCI 60 cell line panel. Kahalalide's F GI50 values in the NCI 60 cell line panel ranged between 0.2 and 10 μM, with hormone‐independent prostate cancer cells being most sensitive to this compound.274 Ling et al.275 report that kahalalide F was then shown to have a strong correlation between cytotoxicity and high c‐erbB2 mRNA expression levels.275
Suárez et al.269 reported that normal cells are 5–40 times less sensitive to kahalalide F than cancer cells. in vivo, kahalalide F demonstrated activity against human prostate hormone‐independent xenograft models274, 276, 277 and in the hollow fiber assay.263
Kahalalide F underwent several clinical trials in oncology and entered Phase II but without much success.278 For example, Martín‐Algarra et al.279 evaluated the antitumor response of kahalalide F in advanced malignant melanoma patients in a Phase II study; however, while kahalalide F was well tolerated by the patients, this trial was closed after the first stage because of the lack of an objective response. Iso‐kahalalide F, a regioisomer of kahalalide F, also entered Phase II clinical trials for liver cancer, melanoma, and NSCLC patients, but failed to be effective as well.280
Elisidepsin (PM02734, Irvalec®; 15b; Fig. 6) is a synthetic marine‐derived cyclic peptide of the kahalalide F family that also entered clinical development.281, 282, 283 Its GI50 values ranged between 0.4 and ∼9 μM in a panel of 23 cancer cell lines from breast, colon, head and neck, lung, ovary, pancreas, prostate, and melanoma origins.281 Herrero et al.284 showed that elisidepsin induces necrosis‐like cell death in yeast Saccharomyces cerevisiae used as a model system. They also demonstrated that the cell membrane and, in particular, cell membrane components like 2‐hydroxy fatty acid‐containing ceramides, are important for elisidepsin's activity. Elisidepsin‐induced necrosis‐like cell death was also experimentally shown by Váradi et al.285 and also recently by Molina‐Guijarro et al.,286 who demonstrated that elisidepsin interacts directly with glycosylceramides in the plasma membrane of cancer cells. Kiraly et al.287 observed that hypoxia reduces the efficiency of elisidepsin by inhibiting hydroxylation and altering the structure of lipid rafts. In addition, Teixido et al.288 reported that acquired resistance to elisidepsin in cancer cells is associated with a switch to the epithelial‐mesenchymal transition (EMT) state, which may be seen as a hallmark of cancer progression to the invasive and metastatic disease.289 This compound displayed in vivo anticancer activity against human melanoma, liver, pancreas, breast, and prostate cancer xenografts.290
Clinical trials with elisidepsin have so far produced disappointing results. For example, while elisidepsin presented an acceptable safety profile, it was not recommended for further evaluation in advanced or metastatic gastroesophageal cancer due to the absence of activity.291 Similar conclusions were reached by Goldwasser et al.282 for other types of malignancy. Other members of the kahalalide family that display cytotoxic activity against cancer cells are described in the comprehensive review by Gao and Hamann.263
D. Potentially Promising Anticancer Agents Derived from Mollusks
Compounds here considered do not include proteins and other substances with molecular weight higher than 2000 amu. Although we made a significant effort to find mollusk‐derived compounds showing in vitro growth inhibitory activity against cancer cell lines at a concentration of < 10 μM, we are cognizant that some substances could have escaped our attention. Compounds under review are listed alphabetically in Table II along with their possible origin if described in the literature. The information on classification, distribution, and alimentary habits of the mollusks under review is summarized in Table I, while Figure 1 shows photographs of selected species. All the NCI data that we report below (if available) were obtained on the public NCI website at https://dtp.cancer.gov/databases_tools/data_search.htm.
1. Terpenes and Steroids
Unusual acetates of halogenated monoterpenes have been characterized from a Spanish population of Aplysia punctata (Fig. 1B; Table I). Among them, compounds 16–18 (Fig. 7; Table II under halogenated monoterpenes) displayed GI50 values ranging between 4 and 10 μM in four cancer cell lines.292 Another linear halogenated monoterpene (19; Fig. 7; Table II) was isolated from two specimens of Aplysia dactylomela (Fig. 1A; Table I) collected from different sites along the Spanish coast.293 This compound displayed growth inhibitory effects against HM02 (gastric carcinoma), HEP‐G2 (liver carcinoma), and MCF‐7 (breast carcinoma) cancer cells with the GI50 values of ∼3 μM.293
Figure 7.
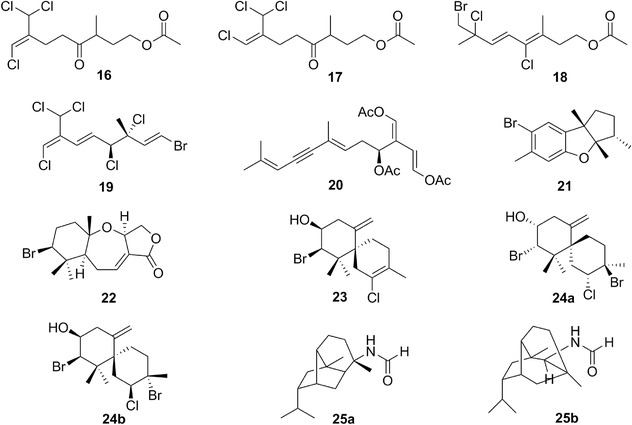
Chemical structures of acetates of halogenated monoterpenes (compounds 16‐19), caulerpenyne (20), (‐)‐aplysin (21), aplysistatin (22), elatol (23), obtusol (24a), isoobtusol (24b), 1‐formamido‐10(1→2)‐abeopupukeanane (25a), and 2‐formamidopupukeanane (25b).
Caulerpenyne (20; Fig. 7; Table II) is a sesquiterpene possessing a 1,4 diacetoxy‐butadiene moiety found in algae of genus Caulerpa and in several sacoglossan mollusks (including Elysia subornata; Fig. 1E; Table I) that feed on these organisms.294, 295 The average GI50 for caulerpenyne is ∼10 μM in various cancer cell lines assayed in academic laboratories.296, 297, 298 The GI50 value reported by the NCI for caulerpenyne in the 60 cell line panel is ∼40 μM and a COMPARE analysis with a cut‐off correlation at p = 0.7 is negative in the standard agent dataset. No selectivity was noted with respect to a given histological type of cancer cell lines in this panel. In the same manner, caulerpenyne does not display bioselectivity between normal (hamster fibroblasts, human keratinocytes, and melanocytes) and cancer cells.298, 299 Barbier et al.298 reported that this compound induces aggregation of tubulin, which may be responsible for the inhibition of tubulin polymerization and bundling of residual microtubules. Bourdron et al.300 then showed that it does not bind to colchicine, taxol, and vinca‐alkaloid binding domains. Caulerpenyne blocks the stimulation of mitogen‐activated protein kinase (MAPK), which participates in the control of cell functions such as proliferation, differentiation, and death.298, 301
One of the first halogenated metabolites isolated from a marine source was (‐)‐aplysin (21; Fig. 7; Table II), which was found in the anaspidean mollusk Aplysia kurodai (Table I).302 Although this algal dietary sesquiterpene was originally described as an antifeedant, later it was reported to induce growth inhibitory effects in various cancer cell lines with GI50 values ranging between 4 and 8 μM.303 The mean GI50 value reported by the NCI in the 60 cell line panel was ∼30 μM and a COMPARE analysis with a cut‐off correlation at p = 0.7 was negative in the standard agent dataset. No selectivity was shown with respect to a given histological type of cancer cell lines in the NCI panel. Aplysin did not display in vivo anticancer activity in the mouse P388 leukemia model used by the NCI. However, it displayed marginal effects in the human A549 NSCLC xenograft with 18% of tumor growth reduction as compared to the control.79 Liu et al.79 showed that aplysin acts as a sensitizer for tumor necrosis factor related apoptosis inducing ligand (TRAIL) induced apoptosis in cancer cells and that these effects occurred via the P38 MAPK/survivin pathway. It sensitized TRAIL‐induced in vivo anticancer activity in the human A549 NSCLC xenograft model.79 As reported by Liu et al.,79 TRAIL has served as a biological agent for cancer treatment for decades. Yet, clinical trials have not shown significant survival benefit in cancer patients. Great hopes are thus placed on innovative compounds that would be able to significantly sensitize apoptosis‐resistant cancer cells to TRAIL. Aplysin also sensitizes the cytotoxic effects of the alkylating drug temozolomide against human glioma cells by increasing miR‐181 expression, which acts as a glioma tumor suppressor.304 In addition, aplysin (21) induces cell cycle arrest and apoptosis in glioma cells through the inhibition of the PI3K/Akt signaling, which plays important roles in glioma cell survival.304 Glioblastoma, the most malignant form of glioma, displays dismal prognoses because of major mechanisms of resistance to pro‐apoptotic stimuli.68
A 2‐propanol extract of the sea hare Aplysia angasi (also named A. dactylomela; Fig. 1A; Table I) displayed growth inhibitory activity against murine lymphocytic leukemia P338 cells.305 The active compound, aplysistatin (22; Fig. 7; Table II), was obtained by bio‐guided fractionation, and identified as a brominated tricyclic 6‐7‐5‐fused sesquiterpene, in which the seven‐membered ring contains a bridging oxygen atom and the five‐membered ring is a lactone. Aplysistatin displayed the GI50 value of ∼8 μM against human KB cancer (an oral cancer cell line but cross‐contaminated by HeLa cervix carcinoma cells115) and mouse P388 leukemia cells.305
Several halogenated chamigrene sesquiterpenes were isolated from A. dactylomela (Fig. 1A; Table I) collected off Canary Islands.293Among these metabolites of dietary algal origin are elatol (23; Fig. 7; Table II) and obtusol (24a; Fig. 7; Table II). Elatol was first isolated from Laurencia elata.306 Lang et al.307 report that several species of Laurencia (red algae) produce elatol as a major secondary metabolite, especially Laurencia microcladia from which elatol was obtained with a yield of ca. 10% (w/w) from the ethanolic extract of the algae. Isoobtusol (24b; Fig. 7; Table II) was isolated for the first time from Laurencia obtusa.308 Elatol displays GI50 values ranging between 1 and 10 μM against about ten cancer cell lines that were assayed by various academic laboratories.293, 309, 310 While normal Vero (African green monkey kidney) cells displayed weaker sensitivity to elatol‐induced growth inhibitory effects (GI50 = 25 μM),309 normal L929 murine fibroblasts displayed sensitivity similar to cancer cells (GI50 = 1 μM).310 Elatol cannot therefore be considered as a bioselective compound. It is a cytotoxic and proapoptotic compound in murine B16F10 melanoma cells through a decrease in Bcl‐x and an increase in Bak, caspase‐9, and p53 expression.310 B16F10 melanoma cells are known to be very sensitive to proapoptotic stimuli.119, 311 It is therefore not surprising that elatol decreased in vivo the growth of B16F10 melanoma growing subcutaneously (s.c.) in syngeneic mice.310 It would interesting to know how efficient elatol is against B16F10 melanoma cells that are intravenously transplanted in syngeneic mice and lead to aggressive lung pseudometastases.311 Indeed, the murine B16F10 melanoma model is not at all representative of the great majority of human melanomas associated with dismal prognoses that display marked resistance to proapoptotic stimuli.67 Obtusol and isoobtusol display weaker in vitro growth inhibitory activity than elatol.293, 307, 309
Two sesquiterpenes named 1‐formamido‐10(1→2)‐abeopupukeanane (25a; Fig. 7; Table II under pupukeanane derivatives) and 2‐formamidopupukeanane (25b; Fig. 7; Table II) were recently isolated and chemically characterized from the nudibranch Phyllidia coelestis (Fig. 1L; Table I).312 As emphasized by Jaisamut and colleagues,312 tubercle nudibranchs of the genus Phyllidia graze on sponges and can sequester specific sponge metabolites. For example, P. varicosa secretes in its mucus 9‐isocyanopupukeanane that is lethal to crustaceans and arises from its sponge prey Ciocalypta sp. (ex. Hymeniacidon sp.). Both compounds 25a and 25b display in vitro growth inhibitory activity in the range between ∼0.1 and ∼7 μM in four distinct human cancer cell lines.312 Also, both of them display some levels of bioselectivity. Specifically, they inhibited by 65 and 25%, respectively, the growth of human gingival fibroblasts when assayed at 5 μM.312
Reticulidin A (26a; Fig. 8; Table II) and B (26b; Fig. 8; Table II) are two carbonimidic chlorinated sesquiterpenes that have been isolated from the Okinawan nudibranch Reticulidia fungia (Table I).313 The in vitro GI50 values displayed by compounds 26a and 26b were ∼1 μM in KB cells, and ∼2 and ∼0.3 μM in mouse L1210 leukemia cells, respectively.313 As mentioned above, tubercle nudibranchs of the genus Phyllidia feed on their sponge prey from the Ciocalypta genus,312 and thus, the possibility remains that the nudibranch R. fungia also feeds on sponges, which could belong to Pseudoaxinyssa pitys, Stylotella aurantium, and Axinyssa sp. genera.313
Figure 8.
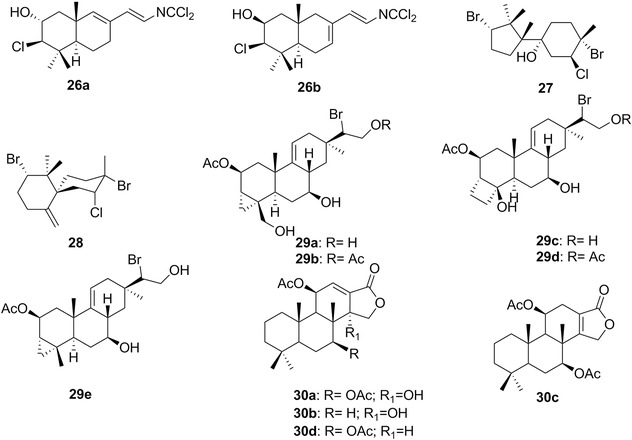
Chemical structures of reticulidins A (26a) and B (26b), oculiferane (27), epi‐obtusane (28), parguerol (29a), parguerol‐16‐acetate (29b), isoparguerol (29c), isoparguerol‐16‐acetate (29d), deoxyparguerol (29e), and dorisenone A (30a), B (30b), C (30c), and D (30d).
Two novel halogenated sesquiterpenes were isolated from the digestive glands of the Egyptian sea hare Aplysia oculifera (Table I).314 These two compounds, oculiferane (27; Fig. 8; Table II) and epi‐obtusane (28; Fig. 8; Table II), displayed GI50 values ranging between ∼2 and ∼8 μM in a minipanel of five human cancer cell lines including the PC‐3 prostate, A549 NSCLC, MCF‐7 breast, HepG2 liver, and HCT116 colon cancer models.314
Parguerol (29a; Fig. 8; Table II), parguerol‐16‐acetate (29b; Fig. 8; Table II), isoparguerol (29c; Fig. 8; Table II), isoparguerol‐16‐acetate (29d; Fig. 8; Table II), and deoxyparguerol (29e; Fig. 8; Table II) are tricyclic monobromoditerpenes isolated from A. dactylomela (Fig. 1A; Table I).315 The algal origin of these metabolites could be strongly suggested as they were isolated from the digestive gland of this anaspidean mollusk.315 Indeed, these metabolites were identified by Awad316 in the red alga Jania rubens (L.) Lamx. collected from the Red Sea coast at Hurghada (Egypt) and it is known that some Aplysia species can feed on sponges.315 It is interesting to note that the complex chemical mixtures obtained from A. dactylomela collected from la Parguera (Puerto Rico) versus the ones from Bimini (Bahamas) do not have any common components.315 The five compounds displayed in vitro growth inhibitory activity in low micromolar ranges but they were tested on one cancer cell line only, the murine P388 leukemia,315 which is highly sensitive to proapoptotic stimuli.317 Awad316 confirmed the data obtained by Schmitz et al.,315 but once more on highly apoptosis‐sensitive cancer cells, Ehrlich ascite carcinoma. Both Schmitz et al.315 and Awad316 showed that isoparguerol derivatives displayed slightly higher in vitro growth inhibition of cancer cells than those of parguerol.
A series of dorisenones (30a‐30d; Fig. 8; Table II) and related spongian diterpenoids (31a‐31g; Fig. 9; Table II) were isolated from the Japanese nudibranch Chromodoris obsoleta (also named Goniobranchus obsoletus; Table I).318 Compounds 30a‐30d and 31a‐31g displayed GI50 values ranging between submicromolar and low micromolar in mouse L1210 leukemia and human KB cells.318 Compounds 31d and 31g were assayed in vivo in the P388 leukemia model but displayed no activity.318
Figure 9.
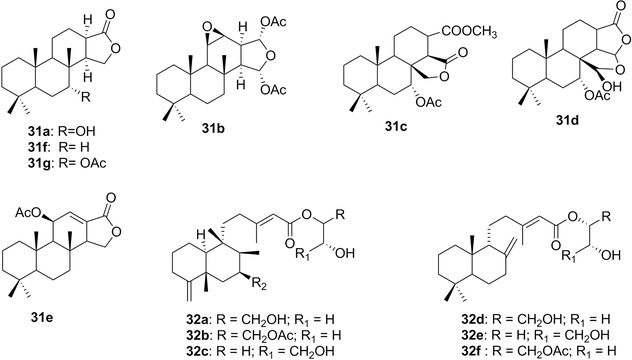
Chemical structures of the other spongian diterpenoids include 7α‐hydroxyspongian‐16‐one (31a), 15α,16α‐diacetoxy‐11,12β‐epoxyspongian (31b), 7α‐acetoxydendrillol‐3 (31c), 7α‐acetoxy‐17β‐hydroxy‐15,17‐oxidospongian‐16‐one (31d), 11β‐hydroxyspongi‐12‐en‐16‐one (31e), spongian‐16‐one (31f), 7α‐acetoxyspongian‐16‐one (31g), and palmadorins (A: 32a; B: 32b; D: 32c; M: 32d; N: 32e; O: 32f).
A chemical study of the Western Antarctic Peninsula nudibranch Austrodoris kerguelenensis (also named Doris kerguelenensis; Table I) led to the characterization of a series of about 20 clerodane, labdane, and halimane diterpene glyceride esters, named palmadorins (palmadorin A to palmadorin Q).319, 320 Palmadorin A (32a), B (32b), D (32c), M (32d), N (32e), and O (32f) (Fig. 9; Table II) inhibit human erythroleukemia (HEL) cell proliferation with low micromolar GI50 values, while palmadorin M was shown to inhibit JAK2, STAT5, and Erk1/2 activation in HEL cells, and cause apoptosis at 5 μM. These HEL cells represent the AML‐M6 model of acute erythroleukemia, a rare (∼3% of cases) form of acute myeloid leukemia (AML), in which erythroblastic precursors cause the myeloproliferation.320
A series of interesting bioactive succinimide‐containing labdane terpenoids have been isolated from ascidians of the genus Lissoclinum since 1991. Dichlorolissoclimide321, 322 (33a; Fig. 10; Table II) and chlorolissoclimide323 (33b; Fig. 10; Table II) were reported from an ascidian collected in New Caledonia, whereas haterumaimides A‐E324 (33c‐33g; Fig. 10; Table II), F‐I325 (33h‐33k; Fig. 10; Table II), J‐K326 (33l‐33m; Fig. 10; Table II), and N‐Q327 (33n‐33q; Fig. 10; Table II) were all isolated from Lissoclinum ascidians collected off Hateruma Islands. In 2004, new haterumamide‐type diterpenes, haterumamides L (34a; Fig. 10; Table II), M (34b; Fig. 10; Table II), and 3β‐hydroxylissoclimide (34c; Fig. 10; Table II) have been found in two Notaspidean mollusks, Pleurobranchus albiguttatus and P. forskalii, along with chlorolissoclimide, dichlorolissoclimide, haterumamides B, D, and H by Fu et al.328 As suggested by the authors,328 these labdanes are presumably metabolites of a Lissoclinum species of the ascidian on which the mollusks feed.
Figure 10.
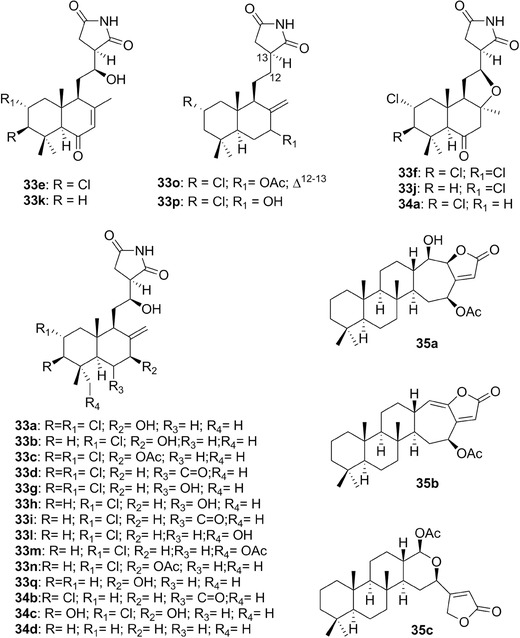
Chemical structures of dichlorolissoclimide (33a), chlorolissoclimide (33b), haterumaimides (A: 33c; B: 33d; C: 33e; D: 33f; E: 33g; F: 33h; G: 33i; H: 33j; I: 33k; J: 33l; K: 33m; N: 33n; O: 33o; P: 33p; Q: 33q; L: 34a; M: 34b), 3β‐hydroxychlorolissoclimide (34c), a lissoclimide‐type alkaloid (34d), and inorolides A (35a), B (35b), and C (35c).
Chlorolissoclimide (33b), dichlorolissoclimide (33a), and haterumaimide D (33f) displayed GI50 values of ∼0.08, ∼0.08, and ∼1 μM in the NCI 60 cell line panel, respectively, and 33a and 33b (but not 33f) showed selectivity toward melanoma cell lines.328, 329 Chlorolissoclimide (33b) and dichlorolissoclimide (33a) also displayed marked growth inhibitory effects in the Corbett–Valeriote soft agar disk diffusion assay, while they showed no solid tumor selectivity.328 In contrast, while less active than 33b and 33a, 3β‐hydroxychlorolissoclimide (33c) displayed solid tumor selectivity (thus potential bioselectivity) in this soft agar assay.328 The Corbett‐Valeriote soft agar disk diffusion assay detects differences in zones of inhibition between a solid tumor cell line (Colon38, ColonH116, LangH125) and either leukemia (L1210 or CEM) or normal (CFU‐GM) cells.330
On the other hand, GI50 values for haterumaimides J (33l) and K (33m), haterumaimides C (33e), G (33i), and I (33k) range between 0.5‐1 nM (33l and 33m) and >20 μM (33e, 33i and 33k).327 Uddin et al.327 report that based on an SAR analysis, it appears that the presence of hydroxyl groups at C‐6, C‐7, C‐12, and C‐18; the chlorine atom at C‐2; and the imido NH in ring C are very important for haterumaimide‐induced in vitro growth inhibitory effects in P388 leukemia cells. A recent synthesis of lissoclimide‐type compounds has been published by Gonzalez et al.331 One of these synthesized compounds, specifically 34d, displayed GI50 values of ∼12 μM in human HeLa cervix epithelia carcinoma and Junket acute lymphoblastic leukemia cells, while the GI50 value of compound 34d was ∼80 μM in normal Vero (African green monkey [Cercopithecus aethiops] kidney) cells, suggesting bioselectivity properties for compound 34d. The synthesis of chlorolissoclimide (33b) was published by Quinn et al in 2016.332 Chlorolissoclimide (33b) and dichlorolissoclimide (33a) are active against murine P388 leukemia cells resistant to adriamycin.328, 333 Robert et al.334 demonstrated that chlorolissoclimide (33b) and dichlorolissoclimide (33a) exert their growth inhibitory effects through the blockade of translation elongation by inhibiting translocation, leading to the accumulation of ribosomes on mRNA. At concentrations relevant to the marked protein synthesis inhibition, DNA synthesis and RNA transcription are slightly or not affected.334 Robert et al.334 also reported that chlorolissoclimide (33b) does not induce a loss of polysomes in contrast to other inhibitors of elongation, such as phyllanthoside and nagilactone C. These authors emphasize that (i) the ribosome recruitment phase of translation initiation is usurped in many human cancers, (ii) several inhibitors of elongation have been previously tested as anticancer agents in preclinical animal models and clinical trials, and (iii) the selectivity of these general inhibitors for cancer cells may stem from the fact that cancer cells have higher translation rates than do normal cells.334 One protein synthesis inhibitor, the Cephalotaxus alkaloid homoharringtonine (an inhibitor of translation elongation), has been approved by the FDA for the treatment of adult patients with chronic myeloid leukemia displaying resistance and/or intolerance to two or more tyrosine kinase inhibitors.335 The clinical data obtained with this compound have been recently reviewed by Heiblig et al.336 and Kantarjian et al.337
Several sesterterpenoids were isolated from Japanese specimens of the nudibranch Chromodoris inornata (also named C. aspersa; Table I).338 These compounds were assayed in vitro for growth inhibition against the human KB and the murine L1210 leukemia cell lines.338 These compounds and their GI50 values are inorolide A (35a; Fig. 10; Table II; GI50 = ∼7 μM), inorolide B (35b; Fig. 10; Table II; GI50 = ∼5 μM), inorolide C (35c; Fig. 10; Table II; GI50 = ∼4 μM), deoxoscalarin (36a; Fig. 11; Table II; GI50 = ∼3 μM), and several of its analogues, namely deoxoscalarin‐3‐one (36b; Fig. 11; Table II under sesterterpenoids; GI50 = ∼2 μM), 21‐hydroxydeoxoscalarin (36c; Fig. 11; Table II; GI50 = ∼9 μM), 21‐acetoxydeoxoscalarin (36d; Fig. 11; Table II; GI50 = ∼1 μM), and 12‐O‐acetyl‐16‐O‐deacetyl‐12,16‐episcalarolbutenolide (37; Fig. 11; Table II under sesterterpenoids; GI50 = ∼5 μM).338 Some of these compounds have also been isolated from marine sponges from the Hyrtios genus.338 We recently reviewed the anticancer activity associated with sesterterpenoids from different origins.339
Figure 11.
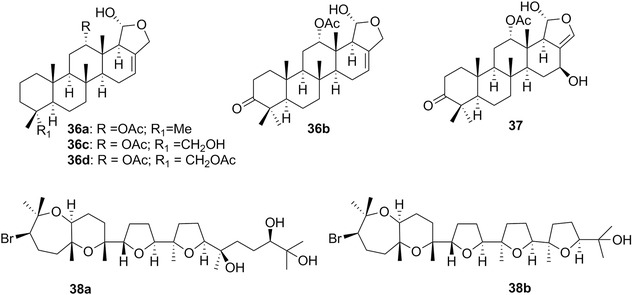
Chemical structures of deoxoscalarin (36a) and several of its analogues, that is, deoxoscalarin‐3‐one (36b), 21‐hydroxydeoxoscalarin (36c), and 21‐acetoxydeoxoscalarin (36d), 12‐O‐acetyl‐16‐O‐deacetyl‐12,16‐episcalarolbutenolide (37), aurilol (38a), and enshuol (38b).
The bromotriterpene aurilol (38a; Fig. 11; Table II ) was isolated from the digestive gland of the sea hare D. auricularia (Fig. 1D; Table I), with a GI50 value of ∼7 μM against human HeLa S3 cancer cells.340 Aurilol is related to enshuol (38b; Fig. 11; Table II ), a bromo triterpenic polyether with a dioxabicyclo(5.4.0) undecane ring system, isolated from the algae Laurencia, suggesting that 38a is most likely dietary.340 The complete synthesis of aurilol (38a) was accomplished in 2005 by Morimoto et al.341
The bromotriterpene polyether aplysqualenol A (39, Fig. 12; Table II ) has been isolated from the Caribbean sea hare A. dactylomela (Fig. 1A; Table I).342 It was submitted to the NCI one‐dose primary assay in the cancer 60 cell line panel and found to display GI50 values of ∼0.4 μM in the human SNB‐19 central nervous system cancer and the T‐47D breast cancer cell lines.342 The GI50 values were >10 μM in the remaining cancer cell lines.342 Vera et al.343 discovered that aplysqualenol is a ligand for the light chain of dynein type 1 (DYNLL1), which is suggestive of potential development of small‐molecule regulators of the dynein complex344 with applications in cancer treatment.345, 346
Figure 12.
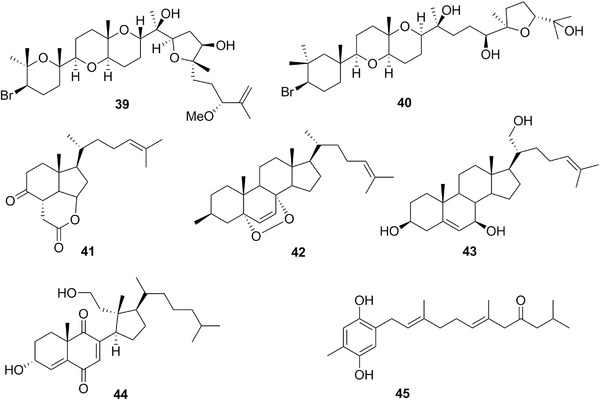
Chemical structures of aplysqualenol A (39), thyrsiferol (40), 3‐epi‐aplykurodinone B (41), an endoperoxide sterol (42), a C‐21 hydroxylated sterol (43), a secosterol (44), and a prenylated hydroquinone (45).
Thyrsiferol (40; Fig. 12; Table II ) is a brominated triterpene polyether first isolated from the tropical marine red algae Laurencia thyrsifera J. Agardh (Rhodomelaceae),347 and more recently also found in the sea hare A. dactylomela (Fig. 1A; Table I) from the South China Sea.348 Mahdi et al.349 stated that thyrsiferol displays potent in vitro growth inhibitory activity in the mouse P388 leukemia cell line, while Fernández et al.350 found that this compound displayed moderate growth inhibitory activity in cell lines originating from solid tumors with GI50 values of ∼0.02 and ∼17 μM in P388 and A549 NSCLC cancer cells, respectively.350 It must be emphasized that P388 leukemia cells display dramatic sensitivity to proapoptotic stimuli,317 while A549 NSCLC cells are moderately resistant.351 These data thus point to the fact that thyrsiferol is indeed more active in vitro against apoptosis‐sensitive cancer cells than their resistant counterparts. Mahdi et al.349 showed that thyrsiferol inhibits hypoxia‐induced HIF‐1 activation in T47D human breast tumor cells and suppressed hypoxic induction of HIF‐1 target genes (VEGF and GLUT‐1) at the mRNA level.349 They also reported that thyrsiferol suppressed mitochondrial respiration at complex I.
The degraded sterol 3‐epi‐aplykurodinone B (41; Fig. 12; Table II ) was isolated from a Spanish collection of Aplysia fasciata (Table I) and it displayed GI50 values of ∼8 μM in four cancer cell lines including mouse P388 leukemia, and human A549 NSCLC, HT‐29 colon cancer, and SKMEl‐28 melanoma.352
An endoperoxide sterol (42; Fig. 12; Table II ) was isolated from the digestive gland of Aplysia depilans (Table I) and its structure confirmed later by synthesis.353 This compound displays a GI50 value of ∼3 μM in human HCT‐116 colorectal cancer cells.354
Trimusculus is a shelled pulmonate mollusk (Table I) living in the intertidal zone of rocky shores.355 An unusual C‐21 hydroxylated sterol (43; Fig. 12; Table II ) was isolated from T. peruvianus (living on Chilean coasts, Table I), and displayed GI50 values of ∼6 μM against human HCT‐116 and HT29 colon cancer cell lines.356 More recently, a novel secosterol (44; Fig. 12; Table II ) was isolated and characterized from a population of T. costatus (Table I) collected in South Africa.357 Compound 44 displays the GI50 value of ∼3 μM in the WHCO1 esophageal cancer cell line.357
Among numerous metabolites isolated from the arminacean nudibranch Leminda millecra (Table I), including among others six sesquiterpenes and eight prenylquinones,358 only one compound, prenylated hydroquinone 45 (Fig. 12; Table II ), displayed moderate in vitro growth inhibitory activity with GI50 values of ∼6 and ∼9 μM in the human WHCO1 and WHCO6 esophageal cancer cell lines, respectively.359 The compounds described above could be from soft coral (Alcyonium fauri, e.g.) or gorgonian (Leptogorgia palma, e.g.) origin because these are the preys of L. millecra.358
2. Peptides and Depsipeptides
Metabolites of this class have been isolated from a wide range of mollusks belonging to different subclasses of gastropods and diverse orders of opisthobranchs as well as other benthic invertebrates.360, 361 These compounds are most likely produced by symbiotic cyanobacteria, as they share very similar structures.362, 363 However, we will describe here all the substances reported from mollusks even if they have been later found to be of cyanobacterial origin, while clearly indicating the origin of each compound in Tables I and II.
Doliculide (46; Fig. 13; Table II ), which has been isolated from Japanese specimens of the anaspidean D. auricularia (Fig. 1D; Table I),364 is a compound of mixed peptide–polyketide biogenesis characterized by the presence of an iodo‐N‐Me‐tyrosine and one unit of glycine. This compound exhibits marked growth inhibitory effects (GI50 = ∼2 nM) against human HeLaS3 cervix carcinoma cells.364 Total enantioselective syntheses of doliculide were reported first by Ishiwata and colleagues,365 and then by Ghosh and Liu366 and Matcha et al.367 Doliculide binds to actin and consequently arrests cancer cells at the G2/M phase of the cell cycle by interfering with normal actin assembly.368 Like jasplakinolide (47; Fig. 13), a cyclodepsipeptide isolated from marine sponges of Jaspis species,366 doliculide causes the hyperassembly of purified actin into F‐actin.368 Doliculide, like jasplakinolide, displaces phalloidin (48; Fig. 13), a cyclic peptide isolated from the mushroom A. phalloides,245 from actin polymer.368 Similar effects were also observed with other depsipeptides known as chondramides.368 Chondramides, such as chondroamide C (49; Fig. 13), are isolated from myxobacteria of the genus Chondromyces.369 Bai et al.368 used a computer‐driven shape descriptor analysis370 to gain insight into a possible pharmacophore shared by doliculide, jasplakinolide, phalloidin, and chondramide C that would explain the apparent binding of this diverse group of substances at the same site on F‐actin. They found that the segment of doliculide that best overlapped with the three other compounds encompassed its phenyl and isopropyl side chains and the portion of the macrocycle between these substituents (Fig. 13). Matcha et al.367 validated these findings by using unbiased computational docking.
Figure 13.
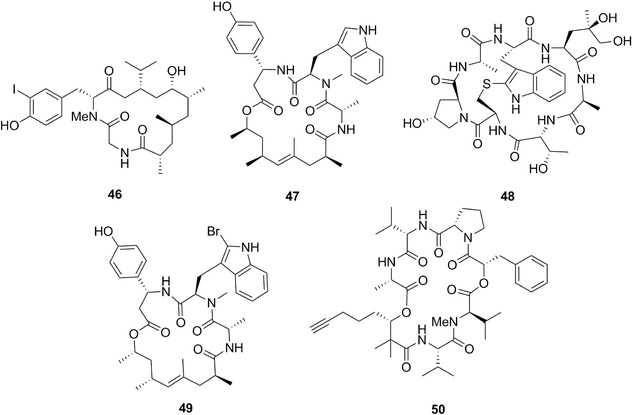
Chemical structures of doliculide (46), jasplakinolide (47), phalloidin (48), chondroamide C (49), and kulolide 1 (50).
Treatment of human MCF‐7 and MDA‐MB‐231 breast cancer cells with doliculide leads to inhibition of proliferation and impairs the migratory potential of these cancer cells.371 Foerster et al.372 quite recently showed that doliculide treatment of p53 wild‐type cancer cells alters up to 13% of senescence‐related genes at nontoxic concentrations. Like tubulin, actin is also implicated in the metastatic process.373, 374, 375
Cyclodepsipeptide kulolide 1 (50; Fig. 13; Table II ) was isolated from the cephalaspidean mollusk Philinopsis speciosa (Table I) collected off Hawaii.376 Kulolide 1 displays growth inhibitory activity in micromolar range against murine L‐1210 (GI50 = ∼1 μM) and P388 (GI50 = ∼3 μM) leukemia cells.376 It must be emphasized once more that L1210 and P388 leukemia cells are highly sensitive to proapoptotic stimuli.317
Kulokekahilide‐1405 (51a; Fig. 14; Table II ) and kulokekahilide‐2378 (51b; Fig. 14; Table II ) are cyclic bidepsipeptides isolated from P. speciosa (Table I), which is a carnivorous mollusk.378 One of the preys of P. speciosa is the sea hare Stylocheilus longicaudus, which feeds on cyanobacteria.379 The possibility thus remains that compounds isolated from P. speciosa could have a cyanobacterial origin.
Figure 14.
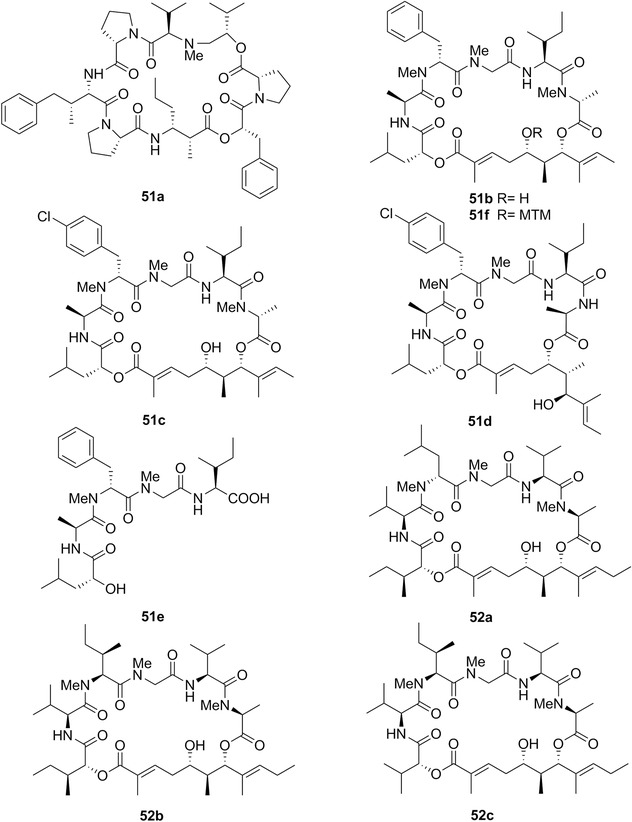
Chemical structures of kulokekahilides 1 (51a) and 2 (51b), kulokekahilide‐2 derivatives (51c‐51f), aurilide (52a), and aurilides B (52b) and C (52c).
Kulokekahilide‐1 displays growth inhibitory effects with the GI50 of ∼2 μM in murine P388 leukemia cells,377 while kulokekahilide‐2 displays higher potency in this cell line with a GI50 value of ∼4 nM.378 In addition, kulokekahilide‐2 displayed GI50 values of ∼8 and ∼15 nM in the human SK‐OV‐3 ovarian and the MDA‐MB‐435 breast cancer cell lines, respectively.378 The mean GI50 value calculated for the three P388, SK‐OV‐3, and MDA‐MB‐435 cancer cell lines is thus ∼9 nM. Because the GI50 value of kulokekahilide‐2 in A‐10 (nontransformed rat [Rattus norvegicus] aortic cells) cell line is ∼60 nM,378 it behaves as a bioselective compound, at least within the few cell lines in which it has been assayed. In the study reported by Umehara et al.,380 the mean GI50 value of kulokekahilide‐2 was ∼0.2 nM in three other cancer cell lines, namely human A549 NSCLC, K562 chronic myelogenous leukemia, and MCF‐7 breast cancer. In the study reported by Takada et al.,381 the GI50 values for kulokekahilide‐2 (51b) were ∼19 and ∼4 nM in murine P388 leukemia and human HeLa cervix carcinoma cells, respectively. Umehara et al.380 used the CellTitler‐Blue Cell Viability Assay with a 72‐hr period of cell‐drug incubation, while Takada et al.381 used the MTT colorimetric assay incubating cells with the compound for 96 hr. The different methods to evaluate the growth inhibition could explain, at least partly, why kulokekahilide‐2 appeared to be at least tenfold more potent in the study of Umehara et al.380
Among the series of kulokekahilide‐2 analogues synthesized by Umehara et al.,380 compounds 51c (Fig. 14) and 51d (Fig. 14) were about 100‐fold more potent than kulokekahilide‐2 with GI50 values of ∼0.001 and ∼0.008 nM, respectively. In contrast, compounds 51e (Fig. 14) and 51f (Fig. 14) were completely devoid of in vitro growth inhibitory activity in A549, K562, and MCF‐7 cancer cell lines with GI50 values >10 μM.380 These authors thus conclude that the addition of halogen at the para position of the phenyl group in the 24‐d‐MePhe residue in kulokekahilide‐2 remarkably increased in vitro growth inhibitory activity. The kulokekahilide‐2 derivatives generated by Takada et al.381 did not display higher in vitro growth inhibitory effects.
Suenaga et al.382 reported the isolation of the cyclic depsipeptide aurilide (52a; Fig. 14; Table II ) from Japanese specimens of D. auricularia (Fig. 1D; Table I). It must be noted that aurilides B (52b; Fig. 14) and C (52c; Fig. 14), which are closely related to aurilide, were isolated from a Papua New Guinea collection of the marine cyanobacterium L. majuscula.383
Aurilide was assayed in the NCI 60 cell line panel and this analysis revealed the mean GI50 value of ∼0.01 μM, with selectivity toward ovarian, renal, and prostate cancer cell lines.384 It also displayed marked in vivo anticancer activity in the NCI hollow fiber assay (detailed by Hollingshead et al.385), while it was inactive in a xenograft model (that was unfortunately not described in detail by Suenaga et al.384) because of high toxicity. Aurilide shows marked microtubule stabilization properties, while it does not seem to interact directly with tubulin; its mechanism of action thus appears to be distinct from that displayed by taxol.384 Aurilide selectively binds to prohibitin 1 (PHB1) in the mitochondria and activates the proteolytic processing of optic atrophy 1 (OPA1) that results in mitochondria‐induced apoptosis.386 Because in the NCI testing aurilide showed no differences in its growth inhibitory effects in apoptosis‐sensitive versus apoptosis‐resistant cancer cell lines,384 it thus appears that this compound can overcome various types of cancer cell resistance to proapoptotic stimuli. Semenzato et al.387 emphasize that proapoptotic drugs targeting the mitochondrial Bcl‐2 rheostat of apoptosis have potential to selectively kill cancer cells. Semenzato et al.387 thus argue that the study by Sato et al.386 adds to the available anticancer strategies by identifying the target of aurilide in the PHB1/OPA1‐dependent apoptotic cristae remodeling. The reader interested in the overview of the various types of resistance patterns to cytotoxic insults displayed by the 60 cancer cell lines used by the NCI is referred to the studies by Shoemaker115 for an overall overview, Ikediobi et al.126 for mutation analysis of 24 known cancer genes, Shankavaram et al.127 for transcript and protein expression profiles, Blower et al.128 for microRNA expression profiles, and Gholami et al.129 for a global proteome analysis. Shankavaram et al.388 also present the CellMiner tool, which is a relational database and query tool for the NCI 60 cancer cell line panel.
Keenamide A (53; Fig. 15; Table II ) is a cyclic peptide isolated from the gastropod notaspidean P. forskalii (Table I) off Manado in Indonesia.389, 390 Its GI50 values range between 4 and 8 μM in four cancer cell lines.389
Figure 15.
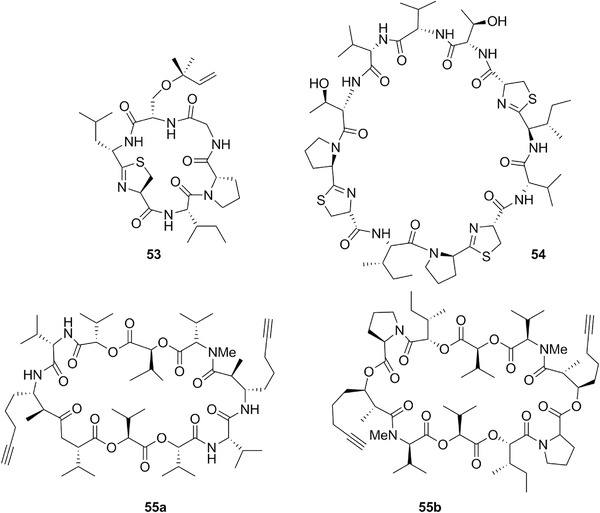
Chemical structures of keenamide A (53), cycloforskamide (54), onchidin (55a), and onchidin B (55b).
Another macrocyclic peptide, cycloforskamide (54; Fig. 15; Table II ), was recently isolated from P. forskalii (Table I), but off Ishigaki Island (Japan).390 Cycloforskamide could be a symbiont‐derived peptide conferring beneficial effects to the host organism because P. forskalii belongs to the family of opportunistic carnivorous mollusks.390 Its GI50 value is ∼6 μM in murine P388 leukemia cells.390 Once more, let us emphasize the high chemosensitivity level of P388 leukemia cells, making therefore this model poorly representative of the clinical cancer,317 which is the reason for why the NCI abandoned this model several decades ago.115
The methanolic extract of the pulmonate mollusk Onchidium sp.1 (Table I) subjected to activity‐guided fractionation using murine P388 leukemia and human KB oral cancer cells afforded cytotoxic onchidin (55a; Fig. 15; Table II ).391 Later, a new metabolite named onchidin B (55b; Fig. 15; Table II ) was isolated from the same animal.392 These two cyclic depsipeptides display GI50 values of ∼7 μM in P388 and KB cancer cells.392
3. Polyketides
Polyketides comprise a wide array of compounds, most commonly sharing a common feature, such as a macrolide ring. As for cyclic depsipeptides, the true origin of macrolidic compounds has been postulated to be microorganisms. For instance bryostatins are complex macrocyclic lactones isolated from bryozoans, whose real origin has been attributed to bacteria after the identification of putative biosynthetic genes in the uncultured symbiotic bacterium “Candidatus Endobugula sertula.”393, 394 As for peptides, we will discuss all polyketide compounds isolated from mollusks (Tables I and II).
A bioguided fractionation of a Japanese collection of the sea hare D. auricularia (Fig. 1D, Table I) led to the isolation of two macrolide glycosides, auriside A (56a; Fig. 16; Table II ) and auriside B (56b; Fig. 16; Table II ).395 From the chemical point of view aurisides have unique structures. Indeed, the aglycon possesses a new type of carbon backbone, 5,7,13‐trihydroxy‐3,9‐dioxoheptadecanoic acid, and contains a bromine‐substituted conjugated diene moiety, a 14‐membered lactone, and a cyclic hemiacetal part.395 Auriside A with the GI50 of ∼0.2 μM displays more potent growth inhibitory effects than auriside B (GI50 = ∼2 μM) in human HeLaS3 cervix cancer cells.395 Various chemical syntheses of aurisides have been reported.396, 397 As mentioned earlier, dolastatin 19 (57) shares significant structural similarities with aurisides A (56a) and B (56b).
Suenaga et al.398 isolated auripyrones A (58a; Fig. 16; Table II) and B (58b; Fig. 16; Table II) from the digestive gland of Japanese specimens of D. auricularia (Fig. 1D; Table I). These two compounds display the GI50 of ∼0.5 μM in human HeLaS3 cells.398 Various chemical syntheses of auripyrones have been reported.399, 400, 401, 402
Dolabelide A (59a; Fig. 16; Table II) and its acetyl derivative dolabelide B (59b; Fig. 16; Table II) are C‐22 macrolides isolated from Japanese specimens of D. auricularia (Fig. 1D; Table I).403 Their GI50 values were ∼8 and ∼2 μM, respectively, in human HeLaS3 cervix cancer cells.403 Dolabelides C (59c; Fig. 16; Table II) and D (59d; Fig. 16; Table II) were also isolated from Japanese specimens of D. auricularia, with GI50 values of ∼2 μM for both compounds in the HeLaS3 cancer cell line.404 Various chemical syntheses of dolabelides have been reported.405, 406
Ulapualide A (60a; Fig. 17; Table II) and ulapualide B (60b; Fig. 17; Table II) were isolated from the red‐colored egg masses of the nudibranch Hexabranchus sanguineus (Fig. 1G; Table 3).407 Though exposed and vulnerable, these eggs have only one known predator, the aeolid nudibranch Favorinus japonicus.407 Ulapualides A and B could be some of the bioactive metabolites protecting these eggs from their predators.407 Ulapualide A can also be found in sponges.408 Ulapualides contain in their structures three contiguous oxazole rings, forming part of a macrolide ring with the attached lipid‐like side chain that terminates in the N‐methyl‐N‐alkenylformamide group.407 The total synthesis of ulapualide A was reported in 1998.408 Ulapualides A and B displayed GI50 values of ∼10 and ∼30 nM, respectively, in murine L1210 leukemia cells.407 Ulapualide A harbors potent actin‐depolymerizing activity.409
Figure 17.
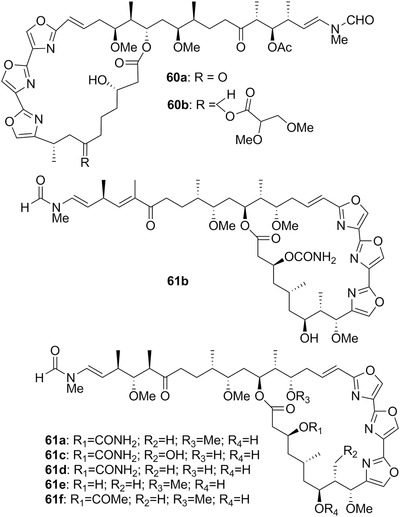
Chemical structures of ulapualides A (60a) and B (60b), and kabiramides C (61a), G (61b), A (61c), B (61d), D (61e), and E (61f).
Another study on the egg masses of an unidentified nudibranch led to the characterization of kabiramide C (61a; Fig. 17; Table II), which contains a macrolide ring possessing contiguous trisoxazole rings.410 Compound 61a was later isolated from the nudibranch H. sanguineus (Fig. 1G; Table I) and from two Halichondria sponges.411 More recently, kabiramide C was found along with other kabiramides, including kabiramide G (61b; Fig. 17; Table II), in the sponge Pachastrissa nux.412 Kabiramide C displays in vitro growth inhibitory effects that are tenfold higher in human MCF‐7 breast cancer cells (GI50 = ∼0.5 μM) than in human fibroblasts (GI50 = ∼8 μM).412 This compound thus appears to display a certain level of bioselectivity. The bioselectivity of kabiramide G is even more pronounced with GI50 values of 0.02 μM in MCF‐7 cancer cells and >2 μM in human fibroblasts.412 Kabiramide C binds to actin and its actin complex, which is formed through a two‐step binding reaction, is extremely stable, and long‐lived.413 Although kabiramide C binds to actin, it nevertheless displays a certain level of bioselectivity between normal and cancer cells as mentioned above.412
Some years later, Matsunaga et al.414 isolated a series of new kabiramides (Table II) including A (61c; Fig. 17), B (61d; Fig. 17), D (61e; Fig. 17), and E (61f; Fig. 17), as well as dihydrohalichondramide (62a; Fig. 18) and 33‐methylhalichondramide (62b; Fig. 18; Table II under halichondramides) from the egg masses of H. sanguineus (Fig. 1G; Table I).414 Halichondramide (62c; Fig. 18) was isolated from the sponge Halichondria sp. collected in Palau.415 Dalisay et al.416 report that H. sanguineus is a specialist predator with a spongeverous diet: in an aquarium, the nudibranch was found to consume only Halichondria containing trisoxazole macrolides and not sponges of other species or genera. Dalisay et al.416 thus argue that these observations provide strong evidence that H. sanguineus acquires its suite of trisoxazole macrolides from a selective sponge diet.
Figure 18.
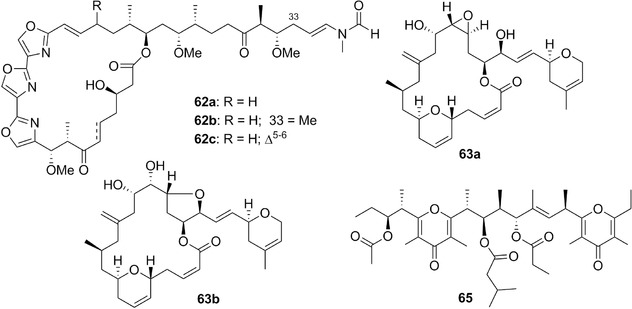
Chemical structures of dihydrohalichondramide (62a), 33‐methylhalichondramide (62b), halichondramide (62c), laulimalide (63a) isolaulimalide (63b), and 3‐acetyl‐11‐(3‐methylbutanoyl)‐13‐propanoylilikonapyrone (65).
Dihydrohalichondramide and 33‐methylhalichondramide displayed growth inhibitory effects in the murine L1210 leukemia cell line with GI50 values of ∼40 and ∼60 nM, respectively.414 Kabiramides A and B were active with GI50 values of ∼10 nM, and kabiramides D and E with GI50 values of ∼30 nM in the same cancer cell line.414 Halichondramide and dihydrohalichondramide disrupt actin microfilaments.417 Halichondramide displays the GI50 value of <1 μM in human PC‐3 prostate cancer cells.418 This compound is cytostatic at low (∼0.5 μM) and cytotoxic at higher concentrations (∼2.5 μM).418 Halichondramide displays both antiproliferative and antimigratory effects in vitro. It induces A549 (NSCLC) cell cycle arrest in the G/2 phase with upregulation of p53 and GADD45α and downregulation of cyclin B1, cyclin A, CDC2, and CDC25C.419 The growth inhibitory effects induced by halichondramide in A549 NSCLC cancer cells were also associated with the suppression of mTOR and its downstream effector 4EBP1 and p70S6K; this process occurs by its effects on the regulation of mTOR upstream proteins, such as downregulation of Akt and p38 MAPK, and also upregulation of AMPK.419 As already mentioned previously, the A549 NSCLC cell line displays various levels of resistance to proapoptotic stimuli.351 In terms of antimigratory effects observed in vitro, halichondramide suppresses the expression of a potential metastatic biomarker, phosphatase of regenerating liver‐3 (PRL‐3), in human PC‐3 prostate cancer cells.418 The suppression of PRL‐3 sequentially downregulates the expression of phosphoinositide 3‐kinase (PI3K) subunits p85 and p110.418 The antimigratory (antimetastatic) effects induced by this compound in PC‐3 cancer cells were also correlated with the downregulation of matrix metalloproteases (MMPs) and the modulation of cadherin switches, N‐cadherin and E‐cadherin.418 It is unfortunate that although halichondramide (NSC622258) exists in the NCI database, no data are available.
Laulimalide (63a; Fig. 18; Table II) and isolaulimalide (63b; Fig. 18; Table II) are macrolides that were isolated from the Indonesian sponge Hyattella sp. and its nudibranch predator, Chromodoris lochi (Table I).420 Laulimalide displays growth inhibitory activity in low nanomolar ranges in about a dozen of cancer cell lines,420, 421 while isolaulimalide is much less potent with GI50 values in low micromolar range.422 Laulimalide is active in P‐gp overexpressing cancer cells421, 423 and against cell lines resistant to paclitaxel or epothilones.423 It has a unique binding site located on two adjacent β‐tubulin units between tubulin protofilaments of a microtubule.212, 424 In addition, although laulimalide is a microtubule stabilizer as are the plant‐derived metabolites taccalonolide and paclitaxel, laulimalide causes the formation of aberrant, but structurally distinct mitotic spindles in contrast to the effects induced by the other two molecules.425 Kanakkanthara et al.426 showed that increased expression of βII‐ and βIII‐tubulin isotypes in ovarian cancer cells confers resistance to laulimalide.426 Downregulation of vimentin (an intermediate filament) expression in human ovarian carcinoma cells also confers resistance to laulimalide.427
Liu et al.421 evaluated the in vivo anticancer activity of laulimalide in two xenograft models, human MDA‐MB‐435 breast cancer and the human HT‐1080 fibrosarcoma models. They found only minimal tumor growth inhibition; rather, severe toxicity accompanied by mortality was observed. In contrast, paclitaxel (positive control) led to the clear and dramatic tumor regression in both studies, without unacceptable toxicities.421 Various teams are actively involved in the syntheses of various fragments of the laulimalide structure with the goal of reaching an efficient total synthesis of this compound.428, 429, 430, 431, 432, 433
An investigation of the chemical composition of A. kurodai (Table I) led to the isolation and characterization of the macrolide family named aplyronines (Table II), specifically aplyronine A (64a; Fig. 19), B (64b; Fig. 19), C (64c; Fig. 19), D (64d; Fig. 19), E (64e; Fig. 19), F (64f; Fig. 19), G (64g; Fig. 19), and H (64h; Fig. 19).434, 435, 436, 437 Yamada et al.438 and Kita and Kigoshi439 reviewed the chemical and pharmacological characteristics of aplyronine A and its analogues.
Figure 19.
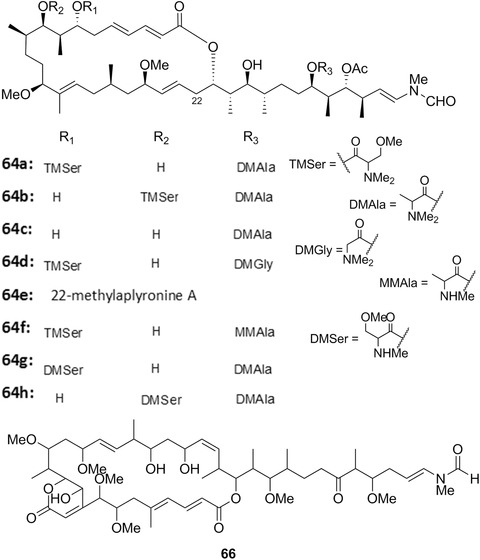
Chemical structures of aplyronines A (64a), B (64b) and C (64c), D (64d), E (64e), F (64f), G (64g), and H (64h), and of sphinxolide (66).
The GI50 values were 0.08, 0.5, 3, 10, and 22 nM for aplyronine D, A, B, H, and C, respectively, in the study by Ojika et al.,437 who used human HeLa‐S3 cancer cells. Yamada et al.434 found GI50 values of ∼0.4 nM for aplyronine A, ∼4 nM for aplyronine B, and ∼20 nM for aplyronine C, respectively. The presence of various methylated amino acids (N,N,O‐trimethylserine or N,N‐dimethylalanine) at the position 22 seems to influence the in vitro growth inhibitory activity of these macrolides.437 Aplyronine A is registered as NSC687160 in the NCI database and it displays the mean GI50 value in the 60 cancer cell line panel of ∼0.2 nM. While highly effective against cancer cells in terms of growth inhibition, it is interesting to note that aplyronine A does not inhibit the 60 cancer cell lines in a homogeneous manner, but rather displays some selectivity toward certain cell lines (but not toward a given histopathological group). Indeed, six cancer cell lines, specifically HOP‐92 (NSCLC), OVCAR‐4 (ovarian cancer), TK‐10 and UO‐31 (renal cancer), and BT‐549 and T47‐D (breast cancer) display GI50 values that are 10‐ to 100‐fold higher than the mean GI50, indicating that these six cancer cell lines are 10‐ to 100‐fold more resistant to aplyronine A. In sharp contrast, the MDR cancer cell line NCI/ADR‐RES displays a GI50 concentration of ∼0.2 nM, indicating that aplyronine A is not a substrate for the P‐gp efflux pump. Thus, aplyronine A does not behave in vitro as a nonspecific poisonous agent. In addition, aplyronine A displayed high in vivo anticancer activity (without obvious limiting toxicity) in various murine syngeneic tumor models, including P388 leukemia, Lewis lung carcinoma, Erhlich carcinoma, colon 26 carcinoma, and B16 melanoma.434 The increases in life span ranged between 201% (B16 melanoma model) and 556% (Lewis lung carcinoma).434 Thus, the life spans of the aplyronine A‐treated tumor‐bearing mice increased by twofold to more than fivefold as compared to those of the control (untreated) tumor‐bearing mice. To our surprise, we did not find any reports describing aplyronine A's in vivo anticancer effects in human xenograft models.
We ran a COMPARE analysis for aplyronine A in the standard agent dataset of the NCI database and found p = 0.62 as a correlation with vincristine sulfate, p = 0.70 with maytansine and 0.87 with vinblastine sulfate. These three compounds are well‐known tubulin inhibitors and both vincristine and vinblastine are routinely used to treat cancer patients whose cancer belongs to various histopathological types. Ado‐trastuzumab emtansine (T‐DM1) is a human epidermal growth factor receptor 2 (HER2) targeted antibody–drug conjugate composed of trastuzumab, a stable linker (4‐(N‐maleimidomethyl)cyclohexane‐1‐carboxylate, MCC), and the cytotoxic agent DM1, which is mertansine, a derivative of maytansine.440 T‐DM1 underwent several successful Phase III clinical trials and it is now proposed as a treatment of choice in second line and beyond for patients with advanced HER2‐expressing breast cancer.440
Aplyronine A was proposed recently as an inhibitor of the actin microfilaments. Hirata et al.441 reported that aplyronine A depolymerizes F‐actin and inhibits the polymerization of actin by forming a 1:1 complex with monomeric actin. These authors441 concluded that, indeed, as expected, aplyronine A binds to a hydrophobic cleft composed of subdomains 1 and 3 of actin by intercalating its aliphatic tail into the actin molecule as do the other reported F‐actin depolymerizing agents. Kita et al.,442 after comparing the amount of abundant actin molecules with the amount of aplyronine A incorporated into the cells, came to the conclusion that the significant antitumor activities of this compound may not be accounted for only by its F‐actin‐severing properties. They elegantly demonstrated recently that, in fact, aplyronine A forms a 1:1:1 heterotrimeric complex with actin and tubulin and inhibits tubulin polymerization. Kita et al.443 thus emphasize that aplyronine A represents a rare type of natural product that binds to two different cytoplasmic proteins to exert its highly potent anticancer activities.
Aplyronine A was reported to exert proapoptotic effects in cancer cells.444 However, these data have been obtained in cancer cells that display very high sensitivity to proapoptotic stimuli (HL‐60 leukemia and HeLa cervix carcinoma). In addition, the fact that this compound could induce proapoptotic effects in some cancer cell type is the logical consequence (and not at all a cause) of cell death occurring when crucial components such as microfilaments (actin) and microtubules (tubulin) are severely damaged by a given compound.
The impressive in vivo anticancer activity observed by Yamada et al.434 in five distinct murine tumor models, as reported above, was obtained with doses ranging from 0.04 to 0.08 mg/kg. Several strategies are already underway to obtain aplyronine A by means of chemical syntheses.445, 446 Paterson et al.447, 448 claimed that aplyronine A has potential as a clinical candidate. However, the in vivo data reported by Yamada et al.434 have never been replicated, even though gram quantities of aplyronine A are currently available. The clinical trial site, https://clinicaltrials.gov, does not mention any ongoing trial with aplyronine A.
A lipophilic extract of the pulmonate mollusk Onchidium sp.2 (Fig. 1I; Table I) collected from the South China Sea contained a series of polypropionates characterized by the presence of bis‐γ‐pyrone rings and structurally related to ilikonapyrones, previously found in a different species of pulmonate.449 The in vitro growth inhibition assay carried out on the metabolites isolated from this mollusk showed growth inhibitory activity for only 3‐acetyl‐11‐(3‐methylbutanoyl)‐13‐propanoylilikonapyrone (65; Fig. 18; Table II) in a minipanel of six human cancer cell lines: A549 NSCLC, MCF‐7 breast cancer, PC‐3 prostate cancer, Hs683 oligodendroglioma, U373 glioblastoma, and SKMEL‐28 melanoma.449 The GI50 values were very similar and ranged from 3 to 9 μM, without any correlation with the level of sensitivity of these cancer cell lines to proapoptotic stimuli, indicating therefore that this metabolite is active against cancer cells displaying resistance to proapoptotic stimuli.449
Sphinxolides bear structural resemblance to scytophycins, which are found in cyanobacteria, such as Scytonema pseudohofmanni.447, 450 Sphinxolide (66; Fig. 19; Table II) is a 26‐membered macrolide that was first isolated from an unidentified Pacific nudibranch451 and later reisolated from the New Caledonian marine sponges Neosiphonia superstes and Reidispongia coreula, along with the congeneric reidispongiolides.452, 453
Like the related families of trisoxazole‐containing marine macrolides (described above), sphinxolide is an actin‐binding molecule that disrupts the actin microfilament organization.447 As previously mentioned and clearly emphasized by Paterson et al.447 and Allingham et al.,454 actin microfilaments exert crucial roles in cancer cell division and metastasis and therefore actin‐binding natural products could be used as leads for novel types of cancer chemotherapy. However, the actin microfilaments also exert crucial roles (as microtubules) on eukaryotic cell shape, cell motility, cell adhesion, and intracellular transportation.447, 454 Antiactin compounds seem at first glance more toxic than antitubulin ones if one takes into account that no antiactin compound is currently marketed as an anticancer drug.
Sphinxolide displays in vitro GI50 value of ∼0.02 nM in human KB cancer cells.451 The actin‐binding properties of sphinxolide in cancer cells lead to subsequent apoptosis,450 as usually occurs with any compounds severely impairing the microfilament and/or the microtubule networks. Sphinxolide causes rapid loss of microfilaments in cultured cells, without affecting microtubule organization, and potently inhibits actin polymerization in vitro as well as the microfilament‐dependent ATPase activity of purified actomyosin, indicating the direct effect on actin.450 It is active in vitro against MDR cancer cells.450, 454 No in vivo studies have been performed for sphinxolide, at least to the best of our knowledge, and it is not registered in the NCI database.
Latrunculins A (67a; Fig. 20; Table II) and B (67b; Fig. 21; Table II) were first reported as spongian metabolites, but were subsequently isolated from different dorid mollusks. In particular, Glossodoris quadricolor (also named Chromodoris quadricolor; Fig. 1F; Table I) accumulates latrunculin B from Latrunculia magnifica,455 whereas latrunculins A was found in C. lochi feeding on Spongia mycifijensis.456 In addition to C. lochi, compound 67a has been recently also found in other four species belonging to the genus Chromodoris: C. elisabethina, C. kuiteri, C. annae, and C. magnifica, showing that it acts as both a feeding deterrent and a toxic chemical weapon.189 These diet‐derived toxins are characterized as 2‐thiazolidinone macrolides and were shown to be strongly ichthyotoxic and cytotoxic.457 Latrunculin A displayed GI50 values of ∼0.5 μM in the study by Longley et al.,458 who utilized three cancer cell lines: murine P388 leukemia, and human HT‐29 colon cancer, and A549 NSCLC.458 Although the P388 leukemia cells115, 317 are much more sensitive to proapoptotic stimuli than the A549 NSCLC ones,351 latrunculin A displayed greater than a fivefold level of in vitro growth inhibitory effects against A549 NSCLC compared to P388 leukemia.458 It thus seems that the marked latrunculin's A growth inhibitory effects are not impaired by the levels of resistance of cancer cells to proapoptotic stimuli. Latrunculin A is registered in the NCI database as NSC613011. Its mean GI50 value in the 60 cancer cell line panel is ∼0.7 μM, and there is more than twofold log magnitude difference between the most sensitive and the most resistant cancer cell lines. Latrunculin A thus does not behave as a nonspecific poison. In addition, this compound is as active against MDR NCI/ADR‐RES as it is against cells that do not display the MDR phenotype.
Figure 20.
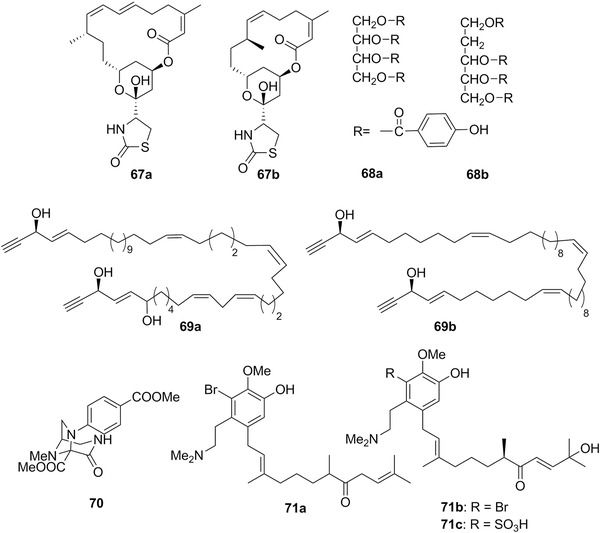
Chemical structures of latrunculins A (67a) and B (67b), kelletins I (68a) and II (68b), hydroxyl‐dehydroisofulvinol (69a), fulvinol (69b), aplaminal (70), aplaminone (71a), neoaplaminone (71b), and neoaplaminone sulfate (71c).
Figure 21.
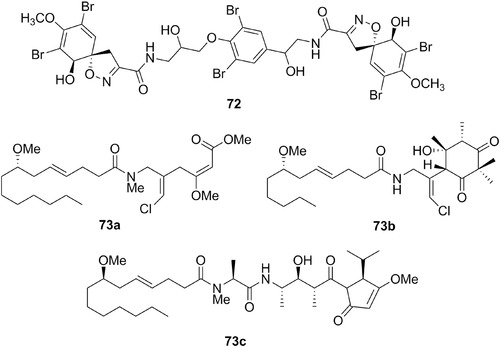
Chemical structures of isofistularin‐3 (72), and malyngamides O (73a), S (73b), and X (73c).
The in vitro growth inhibitory effects of latrunculins, including latrunculin A are related to their ability to reversibly bind actin monomers, forming 1:1 stoichiometric complexes with G‐actin, and therefore disrupting actin polymerization.459, 460 As expected with antitubulin and/or antiactin compounds, apoptosis occurs in latrunculin A treated cancer cells. Konishi et al.461 showed that latrunculin A induced apoptosis in cancer cells occurs via the activation of the caspase‐3/caspase‐7 pathway. We ran a COMPARE analysis on latrunculin A and found no correlation with p ≥ 0.7 with compounds in the standard agent database.
Konishi et al.461 report that latrunculin A has strong anticancer effects in peritoneal dissemination models of MKN45 and NUGC‐4 human gastric cancer in mice. The in vivo anticancer activity of latrunculin A was also evaluated in s.c. A549 NSCLC xenografts in which this compound increased by 46% the life span of the treated tumor‐bearing mice as compared to the control.458
The prosobranch Kelletia kelletii is a hard‐shelled mollusk (Table I) that contained among other compounds kelletinins I (68a; Fig. 20; Table II) and II (68b; Fig. 20; Table II), p‐OH‐benzoic acid tetraester of erithritol, and 2‐deoxy‐d‐ribose, respectively. The two compounds displayed in vitro growth inhibitory effects with GI50 values of ∼0.07 μM in murine L1210 leukemia cells.462
Peltodoris atromaculata (Fig. 1J, Table I) is a Mediterranean nudibranch usually feeding on sponges of genus Petrosia, whose secondary metabolites are mainly long‐chain polyacetylenes called petroformynes.463 This mollusk sequesters the sponge metabolites and accumulates them in their digestive glands.463 No reports about in vitro growth inhibitory effects against cancer cells have yet been published for this type of compounds, at least to the best of our knowledge. However, petroformynes strongly resemble related compounds from sponges, neopetroformynes, which display GI50 values ranging between 0.1 and 0.7 μM in murine P388 leukemia cells.464 Recently, a recollection of the same animal feeding on the sponge Haliclona fulva led to the isolation of new polyacetylenes, including hydroxyl‐dehydroisofulvinol (69a; Fig. 20; Table II), which resembles fulvinol (69b; Fig. 20). Fulvinol (69b), isolated from the sponge Reniera fulva, displays a mean GI50 value of ∼2 μM in four cancer cell lines: murine P388 leukemia, A549 NSCLC, HT‐29 colon cancer, and SKMEL‐28.465 Hydroxyl‐dehydroisofulvinol, isolated from P. atromaculata, displayed a GI50 value of ∼3 μM against SKMEL28 melanoma cells.466
4. Nitrogen Containing Compounds
Kuroda and Kigoshi467 isolated aplaminal (70; Fig. 20; Table II), containing a triazabicyclo [3.2.1] octane framework from A. kurodai. This compound displays a GI50 value of ∼2 μM against human HeLaS3 cervix carcinoma cells.467
Aplaminone (71a; Fig. 20; Table II), neoaplaminone (71b; Fig. 20; Table II), and neoaplaminone sulfate (71c; Fig. 20; Table II) were also isolated from Japanese specimens of A. kurodai.468 From the chemical perspective, these substances are constructed from a bromine‐containing dopamine unit and a sesquiterpenoid part.468 Aplaminone and neoaplaminone sulfate display GI50 values of ∼1 μM against human HeLa cervix cancer cells, while neoaplaminone has the GI50 of ∼1 nM in this cancer cell line.468
Tylodina perversa is a Mediterranean notaspidean mollusk whose dietary preference are sponges of genus Aplysina.469, 470 Brominated isoxazoline alkaloids constitute the metabolite pattern of these porifera with validated feeding deterrent properties against fishes.470 Studies in natural and captivity conditions showed that T. perversa is able to sequester and accumulate these metabolites in different anatomical parts.470 Among all isoxazoline alkaloids found in this mollusk, isofistularin‐3 (72; Fig. 21; Table II) displayed the GI50 value of ∼9 μM against human HeLa cervix carcinoma cells.470
Malyngamides are N‐substituted amides of long chain methoxylated fatty acids, characterized by the presence of a trans double bond and a 7S configuration of the oxygen‐bearing carbon. To date, more than 30 malyngamides have been isolated from cyanobacteria and sea hares. Metabolites of this class have been reported to possess in vitro growth inhibitory properties against cancer cells and include malyngamide O (73a; Fig. 21; Table II), isolated from the anaspidean mollusk Stylocheilus longicauda,471 as well as malyngamide S472 (73b; Fig. 21; Table II) and malyndamide X473 (73c; Fig. 21; Table II), both isolated from Bursatella leachii (Table I). These three compounds displayed GI50 values ranging between ∼4 and ∼8 μM in seven cancer cell lines including murine P388 leukemia and human A549 NSCLC, NCI‐H187 (SCLC), HT‐29 colon cancer, HL60 leukemia, KB and BC breast cancer models.471, 472, 473
An investigation of Indian Ocean specimens of the sea hare D. auricularia (Fig. 1D, Table I) led to the isolation of a bisthiazole metabolite, dolabellin (74; Fig. 22; Table II), which displayed the GI50 value of ∼10 μM in the human HeLaS3 cervix carcinoma cell line.474
Figure 22.
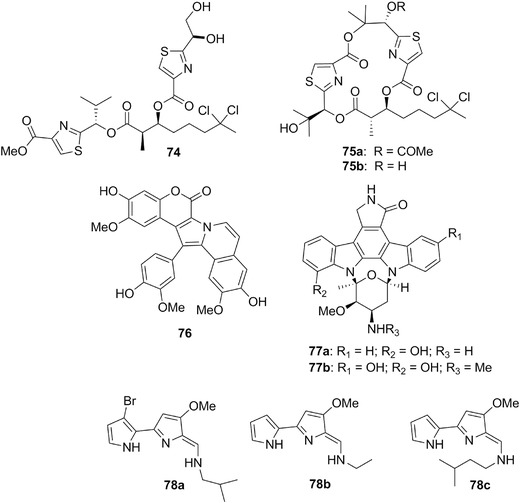
Chemical structures of dolabellin (74), hectochlorin (75a) and its deacetylated form (75b), lamellarin D (76), staurosporine analogues (77a, 77b), and tambjamines D (78a), E (78b), and K (78c).
Although previously isolated from the cyanobacterium L. majuscula, hectochlorin (75a; Fig. 22; Table II) and its deacetylated derivative (75b; Fig. 22; Table II) have also been found in the sea hare B. leachii (Fig. 1C, Table I).475 Hectochlorin structurally resembles dolabellin (74, Fig. 22) as reported by Marquez et al.476 In the study by Suntornchashwej et al.,475 the in vitro growth inhibitory effects of compounds 75a and 75b were determined in the human KB, NCI‐H187 SCLCL, and BC breast cancer cell lines. The deacetylated derivative of hectochlorin 75b was the most active of these two compounds, displaying a mean GI50 value of ∼1 μM.475 Marquez et al.476 showed that hectochlorin is equipotent to jasplakinolide in its ability to promote actin polymerization, however unlike jasplakinolide, it is unable to displace a fluorescent phalloidin analogue from polymerized actin. Hectochlorin has no effect on the polymerization of purified tubulin.476
Marquez et al.476 also report that hectochlorin displays a unique profile in the NCI 60 cancer cell line panel. Indeed, it showed the greatest potency against several cell lines in the colon, melanoma, ovarian, and renal cell line panel with the average GI50 against the 60 cell line panel of ∼5 μM.476 In addition, the flat shape of the dose–response curves of heterochlorin is generally characteristic of compounds that are cytostatic rather than cytotoxic.476 The chemical total synthesis of hectochlorin has been reported by Cetusic et al.477
More than three decades ago, Andersen and et al.478 isolated lamellarins, aromatic alkaloids characterized by the presence of a fused‐pyrrole ring, from the Palauan prosobranch mollusk Lamellaria sp. various. Since the first report, more than 50 lamellarins have been found mainly in Didemnum spp. ascidians as well as in various sponges.479, 480, 481 Of all the lamellarins identified up to date, lamellarin D (76; Fig. 22; Table II) displays the highest growth inhibitory effects on cancer cells,481 with GI50 values ranging between 10 and 20 nM in a large panel of cancer cell lines.478 In addition to the nuclear targeting involving topoisomerase I inhibition with the modulation of several apoptotic mediators,482, 483 lamellarin D accumulates rapidly inside the mitochondria, directly poisoning the mitochondrial topoisomerase I (Top1mt), and thus leading to the dysfunctional mitochondrial respiration in cells.484, 485 As suggested by the sensitivity of topoisomerase I mutated P388‐CPT5 chemoresistant cells,482 lamellarin's D dual mechanism may be effective against drug‐resistant cancers offering an alternative to the conventional topoisomerase I inhibitors, irinotecan, and topotecan, which are inefficient as Top1mt inhibitors due to their inactivation in the mitochondrial pH environment.486 In addition, lamellarin D is not sensitive to P‐gp and its growth inhibitory effects are maintained in MDR cancer cells.487 Due to its intriguing anticancer properties, lamellarin D has been the focus of considerable research and several syntheses and SAR studies of lamellarin derivatives have been reported in the last few years.481 The fact nevertheless remains that despite this research activity, no in vivo efficacy has been demonstrated yet for lamellarins in general and for lamellarin D in particular, at least to the best of our knowledge. A bioguided fractionation of various extracts of the prosobranch mollusk Coriocella nigra resulted in the isolation of two members of the staurosporine488 (selective kinase inhibitor) family: compounds 77a (4’‐N‐demethyl‐11‐hydroxystaurosporine; Fig. 22; Table II under staurosporine analogues) and 77b (3,11‐dihydroxystaurosporine; Fig. 22; Table II).489 Their GI50 values in a minipanel of cancer cell lines ranged between 4 and 130 nM.489
Tambjamines are methoxypyrrolic alkaloids whose chemical features resemble the bacterial prodigiosins.490, 491, 492 All these compounds contain a common 4‐methoxy‐2,2’‐bipyrrole chromophore.491, 492 Tambjamines A–D (tambjamine D; 78a; Fig. 22; Table II) were isolated from the nembrothid nudibranchs Roboastra tigris, Tambja eliora, and Tambja abdere (Table I).490 Roboastra tigris is a large carnivorous nudibranch that preys on two smaller nudibranchs, T. eliora and T. abdere.490 Tambjamines A–D were also traced to a dietary source, the ectoproct (bryozoan) Sessibugula translucens, and are implicated in a chemical defense mechanism.490 Indeed, when attacked by R. tigris, T. abdere produces a yellow mucus from goblet cells in the skin and this defense secretion often causes R. tigris to break off its attack.490 Tambja eliora does not produce a defensive secretion but attempts to escape from R. tigris by using a vigorous writhing motion.490 In laboratory experiments, R. tigris prefers to eat T. eliora rather than T. abdere.490 Tambjamines E (78b; Fig. 22; Table II), F, G, H, I and J were isolated from various bacteria and marine invertebrates.492 Tambjamine K (78c; Fig. 22; Table II) was isolated from the Azorean nudibranch Tambja ceutae.493
The GI50 values of tambjamine K (78c) were determined in four cancer cell lines and ranged between ∼0.004 and 15 μM.493 Tambjamine K displayed the GI50 value of ∼19 μM in mouse 3T3‐L1 fibroblasts.493 At first glance, it thus appears that tambjamine K (78c) is not a bioselective compound. However, it displays a ∼4000‐fold differential sensitivity between human Caco‐2 colon cancer cells and HeLa cervix cancer cells.493 Tambjamine K (78c) has unfortunately not been assayed by the NCI.
Tambjamine C has recently been demonstrated to be an efficient anion carrier, that is, a transmembrane anion transporter similar to prodigiosin, whereas tambjamine E does not share this property and tambjamine K has not yet been assayed as such, at least to the best of our knowledge.494 Transmembrane anion transporters play important roles in cancer cell biology, including cancer cell migration, and they are distinctly expressed in various cancer cell types.495, 496, 497, 498, 499 It is interesting to note that Caco‐2 colon cancer cells, displaying high sensitivity to tambjamine K as mentioned above, have often been used to study the roles of various transmembrane transporters.500, 501 Tambjamine E targets DNA as also does tambjamine D,502 which in addition displays genotoxic effects in normal cells.503 It thus appears that tambjamine K certainly merits additional in depth pharmacological and toxicological preclinical evaluations as a potential anticancer agent.
A chemical investigation of the Asian mollusk Turbo stenogyrus (Table I) led to the isolation of four cerebrosides, turbostatins A–D, whose GI50 values ranged between ∼0.2 and ∼4 μM in murine and human cancer cell lines.504 The chemical structure of turbostatin A (79; Table II) is illustrated in Figure 23.
Figure 23.
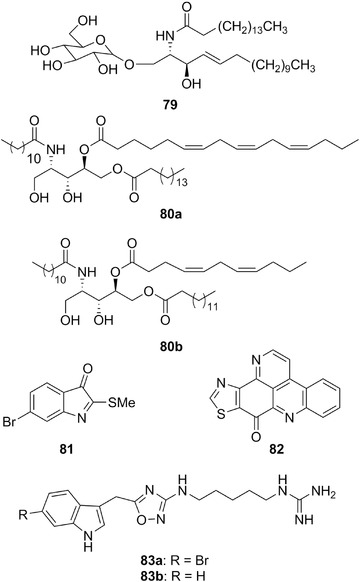
Chemical structures of turbostatin A (79), bathymodiolamides A (80a) and B (80b), tyrindoleninone (81), kuanoniamine A (82), and phidianidines A (83a) and B (83b).
Bathymodiolamides A (80a; Fig. 23; Table II) and B (80b; Fig. 23; Table II) are ceramides isolated from the deep vent mussel Bathymodiolus thermophilus (Table I). Their GI50 values in human HeLa cervix cancer and MCF7 breast cancer cells were <1 μM.505
Tyrindoleninone (81; Fig. 23; Table II) is a brominated indole derivative (6‐bromo‐2‐methylthioindolin‐3‐one) isolated from the egg mass extract of the gastropod Dicathais orbita (Table I).506 The GI50 values of this compound in human colon cancer cells are >100μM.506
A series of pentacyclic alkaloids have been isolated from the prosobranch mollusk Chelynotus semperi (Table I) and its prey, an unidentified colonial tunicate.507 Among these compounds, only kuanoniamine A (82; Fig. 23; Table II) showed weak growth inhibitory activity against KB cancer cells (GI50 = ∼4 μM).507
The indole alkaloids phidianidines A (83a; Fig. 23; Table II) and B (83b; Fig. 23; Table II) were isolated from Phidiana militaris (Fig. 1K; Table I), an aeolid nudibranch from the South China Sea.508 The GI50 values for these compounds range between ∼0.4 and >100 μM in three cancer cell lines. They display no bioselectivity toward mouse 3T3‐L1 fibroblasts and rat H9c2 cardiomyocytes as the GI50 values toward these cells ranged between ∼0.1 and ∼5 μM.508 Interestingly, while human HeLa cervix cancer cells display weak sensitivity to the growth inhibitory effects of tambjamine K,493 they are much more sensitive to the growth inhibitory effects of phidianidines.508 The exactly opposite features are observed with respect to Caco‐2 colon cancer cells.493 These data thus highlight distinct mechanisms of action for tambjamine K and phidianidines A and B as potential anticancer compounds and suggest that tambjamine K as well as phidianidines A and B are not nonspecific poisons. It would be very interesting to have an NCI profiling of these compounds. Phidianidines A and B act as selective and potent ligands with partial agonist activity against the μ opiod receptor (versus δ‐ and κ‐opiod receptors).509 Indeed, the μ opiod receptor is involved in cancer progression.510 They were also demonstrated to act as ligands of CXCR4, a chemokine receptor involved in cancer progression and the metastatic process.511 Total syntheses of phidianidines A and B have been reported.509, 512, 513, 514
Fontana et al.515 isolated jorumycin (84a; Fig. 24; Table II) from the mantle and mucus of the Pacific nudibranch mollusk, Jorunna funebris (Fig. 1H; Table I). Jorumycin displays GI50 values in low nanomolar or even subnanomolar ranges in various cancer cells including cells resistant to proapoptotic stimuli.515, 516 The high potency of jorumycin and the clinical successes of ecteinascidin led to intensive chemical modifications of jorumycin's chemical structure and this campaign thus resulted in PM00104 (Zalypsis®, 84b, Fig. 24).516 Petek and Jones516 recently reviewed the preclinical and clinical data related to PM00104, which has entered several ongoing Phase II clinical trials.
Figure 24.
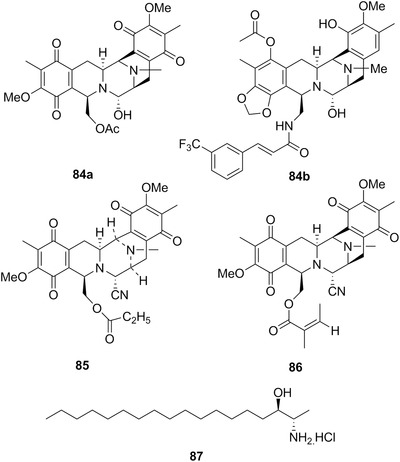
Chemical structures of jorumycin (84a), PM00104 (Zalypsis®, 84b), jorunnamycin C (85), renieramycin M (86), and ES‐285 (spisulosine, 87).
Jorumycin belongs to the class of dimeric isoquinoline alkaloids and it shares the same carbon framework as saframycin and ecteinascidin.515 In fact, ecteinascidin (trabectedin) has three isoquinolines, while the other two alkaloids have only two isoquinolines per molecule, while the two isoquinolines are joined in similar manner in all three compounds.515 The difference in structure between ecteinascidin and the jorumycin analog PM00104 could explain, at least partly, why these two drugs display slightly different DNA‐binding properties and nucleotide excision repair dependencies.516
In 2007, studies on Thai J. funebris (Fig. 1H) specimens led to the isolation from the mantle, viscera, and egg‐ribbons novel tetrahydroisoquinoline alkaloids, jorunnamycins A–C, along with several renieramycins.517 It is interesting to note that certain renieramycins have also been isolated from the Thai sponge Xestospongia sp.518 Indeed, J. funebris is carnivorous and feeds mainly on sponges such a Xestospongia sp., Haliclona sp., Euplacella cf. australis, and Oceanapia sp.517 Jorunnamycin C (85; Fig. 24; Table II) and renieramycin M (86, Fig. 24, Table II) differ only in the C‐22 ester side chain. These two compounds display GI50 values in the low nanomolar ranges against human colon (HCT‐116) and breast (MDA‐MB‐435) cancer cells.518 Charupant et al.518 showed that the downregulation of protein tyrosine phosphatase receptor type K (PTPRK) could be one of the pathways through which jorunnamycin C (85) and renieramycin M (86) exert their anticancer effects, at least in vitro. PTPRK acts as a tumor suppressor gene product519 and could be implicated in the progression of colon cancer.520
Sphingolipid‐related compounds called spisulosines were isolated from the Nord‐Atlantic clam Spisula polynyma (syn. Mactromeris polynima; Table I).521, 522 Sphingolipids constitute a group of natural products characterized by the presence of a long chain 2‐amino‐1,3‐diol scaffold or sphingoid base.522 As emphasized by Abad et al.,522 besides playing a structural role and regulating the physical properties of cell membranes, sphingolipid metabolites also participate in cell signaling and in the control of numerous cellular functions in mammals. Spisulosines constitute a group of 1‐deoxysphingolipids derived from a central core lacking the C1‐OH group present in dihydrosphingosine.522 One of these spisulosines is ES‐285 (87; Fig. 24; Table II under Spisulosine), which resembles the natural sphingolipids in the C18 sphingoid backbone and in the (2S,3R) configuration of the amino and hydroxyl groups, respectively.522 ES‐285 is associated with GI50 values ranging between low nanomolar and low micromolar in the NCI 60 cancer cell line panel.522, 523, 524, 525, 526 ES‐285 triggers an atypical cell death pathway because it does not affect pathways widely implicated in regulating cell survival and apoptosis, such as JNK, ERKs, or Akt, but it nevertheless activates caspase‐3 and caspase‐12.525 Sánchez et al.526 showed that ES‐285 inhibits the growth of human PC‐3 and LNCaP prostate cancer cells through intracellular ceramide accumulation and PKCzeta activation. This compound modulates RHO protein and ceramide signaling and consequently induces disassembly of the actin stress fibers,527 but does not affect the microtubule network.525
ES‐285 is COMPARE‐negative, suggesting therefore a possible novel mechanism of anticancer action as compared to the other anticancer drugs.524 It displayed significant in vivo anticancer activity in a hollow fiber model with s.c. implanted SK‐HEP‐1 hepatoma tumor cells, and xenografted models utilizing human PC‐3 prostate and MRI‐H‐121 renal cancer cells.524 ES‐285 has entered several Phase I clinical trials since the late 2000s. Baird et al.524 report that pharmacologically relevant concentrations of this drug were safely achieved in adult cancer patients, with pharmacogenomics studies indicating changes in the expression of genes of potential mechanistic relevance. Schöffski et al.528 reported hepato‐ and neurotoxicity as schedule independent dose‐limiting adverse events for ES‐285 were noted. Vilar et al.529 also observed liver enzyme elevations as dose limiting for ES‐285 administration to cancer patients. Massard et al.530 also recently conducted a Phase I clinical trial. To the best of our knowledge, there is no available information on its Phase II clinical trials.
5. TARGETED DELIVERY OF MOLLUSK‐DERIVED ANTICANCER AGENTS
Some of the issues that have prevented the advancement of mollusk‐derived anticancer agents to the clinic can be mitigated by employing delivery vehicles that can improve the drugs’ ability to impart a strong therapeutic action while minimizing off‐target effects. Nanomedicine, or the medical application of nanotechnology, has been at the forefront of the anticancer drug delivery field. Nanomedicines are designed to improve the drug's solubility in physiological fluids, improve the drug's biodistribution and pharmacokinetics, passively or actively target tumors, and protect the drug from premature degradation or excretion. Nanomedicines including liposomes, micelles, nanoparticles, nanoemulsions, nanocapsules, dendrimers, and macromolecular conjugates have been utilized to improve the performance of many anticancer agents.531 These drug delivery systems can target tumors passively by taking advantage of the enhanced permeability and retention (EPR) effect which is a result of the high permeability of leaky angiogenic blood vessels and poor lymphatic function at the tumor.532, 533 In addition, nanomedicines can be actively directed to molecular targets overexpressed in tumors by inclusion of high affinity ligands such as antibodies, aptamers, or peptides on their structure.532, 534, 535
Nanomedicines can utilize a number of materials including biocompatible lipids, polyesters, oligosaccharides, polyethers, oligonucleotides, peptides, and even proteins. Among these materials, the polyether poly(ethylene glycol) (PEG) has been widely used in drug delivery applications due to its ability to shield hydrophobic biomaterials and increase the circulation time of macromolecules and nanocarriers.536 Nanomedicines can also include activatable switches that maintain the drug in an inactive, nontoxic state until activated by low pH, reducing conditions, or proteolytic enzymes that are overexpressed in tumors.537 Nanomedicines can enable delivery of high‐drug payloads and have been reported to overcome MDR.531 Several nanomedicine formulations of anticancer agents including Doxil™, Myocet™, DaunoXome™, and Abraxane™ have been approved by the FDA for clinical use. Among the various types of nanomedicines, surprisingly only few types of nanovectors (namely micelles, nanoparticles, and conjugates) have been explored with mollusk‐derived anticancer agents. In all cases, these nanovectos were used as means to increase the solubility of or to minimize dose‐limiting toxicity associated with these highly active anticancer agents by targeting tumors. Next, we summarize the reported design as well as results of in vitro and in vivo evaluation, as available, of nanovectors utilized for the targeted delivery of mollusk‐derived anticancer agents.
A. Lamellarins
Macromolecular covalent conjugates of lamellarin D (76, Fig. 22; Table II) were prepared to enhance the solubility of this hydrophobic alkaloid. A multiarm PEG dendrimer conjugate of lamellarin D (88, Fig. 25) decreased the GI50 value of the drug by 40% in MDA‐MB‐231 breast cancer cells, but showed slightly higher GI50 in HT29 colon cancer cells and A549 lung cancer cells.538 On the other hand, a nuclear localization sequence (NSL) oligopeptide conjugate of lamellarin D (89, Fig. 25) had lower GI50 values in all three cancer cell lines.538 Importantly, both conjugates 88 and 89 showed reduced growth inhibition in noncancerous BJ skin fibroblasts, demonstrating bioselectivity of these drug delivery systems toward cancer cells.538 in vivo evaluation of these conjugates has not been reported.
Figure 25.
Chemical structures of multiarm PEG dendrimer of lamellarin D (88), nuclear localization sequence oligopeptide conjugate of lamellarin D (89), and lamellarin N (90).
Lamellarin N (90, Fig. 25), a hydrophobic compound (logP = 3.37) isolated from mollusks and later found in ascidians and sponges, was incorporated into polymeric micelles as a means to improve its aqueous solubility. Biodegradable micelles are self‐assembled nanostructures made from amphiphilic block copolymers where the hydrophobic polymer segments form an inner core in which hydrophobic drugs can be entrapped, and the hydrophilic segments form a protective shell that interacts with the surrounding aqueous medium. Micelles of the amphiphilic block copolymers poly(ethylene glycol)‐b‐poly(ε‐caprolactone) (PEG‐b‐PCL) and poly(ethylene glycol)‐b‐poly(d,l‐lactide) (PEG‐b‐PLA) were used to encapsulate Lamellarin N.539 Importantly, encapsulation increased the concentration of lamellarin N that could be suspended in water by up to 200‐fold compared to the free drug.539 Neither in vitro nor in vivo evaluation of these micelles was reported.
B. Kahalalides
As previously mentioned, kahalalide F (15a, Fig. 6; Table II) is a depsipeptide that has been evaluated in Phase II clinical trials for the treatment of melanoma, liver cancer, and NSCLC. Unfortunately, these clinical trials failed to demonstrate anticancer response in patients. The activity of two diastereomeric cysteine analogues of kahalalide F (91a and 91b, Fig. 26) conjugated to 20‐ and 40‐nm gold nanoparticles was evaluated in vitro.540 The gold‐drug nanoscaled conjugate aimed to utilize the nanoparticles’ ability to deliver a high payload of the peptide to the cells intracellularly, thereby increasing therapeutic efficacy. Studies in HeLa cervical carcinoma cells showed that although the cell growth inhibition of the 20‐nm gold nanoparticle conjugates was not different than that of the free peptides, the 40‐nm conjugates of 91a and 91b resulted in higher cell growth inhibition.540 Similarly, higher nanoparticle uptake into HeLa cells was observed with 40‐nm nanoconjugates as demonstrated with confocal fluorescence microscopy.540 Neither in vitro nor in vivo therapeutic efficacy was evaluated with these nanoparticle systems.
Figure 26.
Chemical structures of d‐cys (91a) and l‐cys (91b) analogs of Kahalalide F that were conjugated to gold nanoparticles for intracellular delivery.
Elisidepsin (15b, Fig. 6), a cyclic peptide of kahalalide F, also failed clinical trials due to poor therapeutic activity. An elisidepsin oral formulation based on 150‐nm solid lipid nanoparticles of Precirol® ATO 5 was evaluated as a means to increase the oral bioavailability of this drug.541 This delivery system was evaluated in Beagle dogs and compared to a cyclodextrin‐based formulation.541 Despite not being able to increase oral elisidepsin bioavailability above 1%, the nanoparticle formulation enhanced drug absorption and maintained the drug concentration in the blood within measurable levels over a longer timeframe than cyclodextrin.541 The in vivo therapeutic efficacy of this oral delivery system was not reported.
C. Dolastatins
Several dolastatins have been formulated into antibody conjugates as a means to increase their concentration at the tumors while minimizing off‐target effects. The FDA‐approved monomethyl auristatin E (MMAE) conjugate with anti‐CD30 antibody (brentuximab vedotin) has been approved for the treatment of Hodgkin's lymphoma. As previously mentioned, antibody–drug conjugates have been previously reviewed166, 200, 219, 222 and will not be further discussed in this review. Most nanomedicine systems for the delivery of dolastatins have been developed for the highly potent and dose‐limiting dolastatin 10 derivatives MMAE (92, Fig. 27) and monomethyl auristatin F (MMAF, 93, Fig. 27). Here, we summarize current efforts to develop innovative targeted and activatable auristatin nanomedicines that are designed to protect healthy tissue from the drugs’ action while achieving high anticancer effect.
Figure 27.
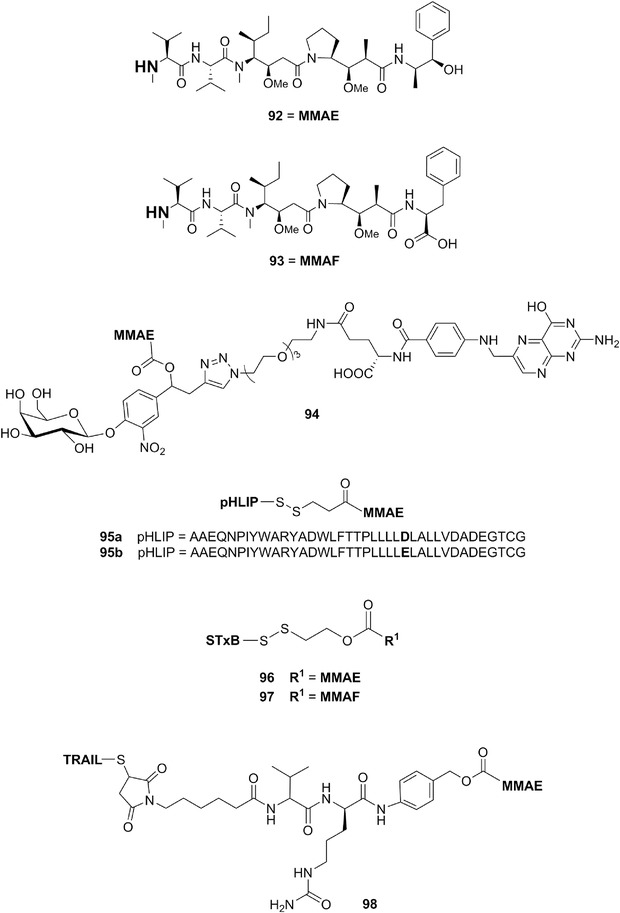
Chemical structures of monomethyl auristatin E (MMAE, 92), monomethyl auristatin F (MMAF, 93), β‐galactosidase activatable folate‐targeted conjugate of MMAE (94), wild type (95a) and modified (95b) pH (low) insertion peptide conjugates of MMAE, Shiga toxin conjugates of MMAE (96) and MMAF (97), and TRAIL‐MMAE conjugate (98). In 94, 95, 96, 97, and 98, MMAE and MMAF are conjugated to the carriers utilizing the monomethyl nitrogen that is bolded in structures 92 and 93.
A folate‐directed conjugate of MMAE (94, Fig. 27) based on a self‐immolative linker that could be activated by lysosomal β‐galactosidase was developed by Legigan et al.542 in vitro tests demonstrated over 20‐fold higher growth inhibition by 94 in folate receptor‐positive KB and HeLa cells compared to folate receptor‐negative A549 cells.542 Conjugate 94 also demonstrated very high anticancer activity, with a GI50 of ∼0.2 nM in KB cells. This system was not evaluated in vivo.
Burns et al.543 demonstrated a pH‐activated system for the delivery of MMAE based on pH (low) insertion peptide (pHLIP) conjugates (95a and 95b, Fig. 27) that undergo conformational changes under the acidic pH typical of tumors. Specifically, at a pH of ∼6, the pHLIP conjugates insert into the membrane and deliver their cargo directly into the cytoplasm of target cancer cells. MMAE was conjugated to the C‐terminus of wild type (95a) and modified (95b) pHLIP via a disulfide bond that could undergo intracellular reduction for delivery of an N‐modified MMAE derivative.543 Studies with HeLa and MDA‐MB‐231 cancer cells demonstrated over 90% inhibition of cell growth after a 2‐hr exposure to both drug conjugates.543 Conjugate translocation and intracellular release of the drug occurred faster than passive diffusion of free drug. Finally, cell growth inhibition with the conjugates was over 11‐fold higher at pH 5.0 versus pH 7.4.543 A pilot in vivo study also showed that conjugate 97a was able to target triple‐negative MDA‐MB‐231 breast cancer xenografts in mice, although the in vivo antitumor effect was not investigated.543
Batisse et al.544 developed delivery systems that targeted MMAE and MMAF to glycosphingolipid globotriaosylceramide (Gb3, also referred to as CD77) positive cancer cells.544 The systems consisted of degradable conjugates of MMAE and MMAF and the cysteine‐terminated B‐subunit of the Shiga toxin (STxB) (96 and 97, respectively, Fig. 27).544 The conjugates were designed to enable release of the active agents under reducing conditions. 544 in vitro studies in Gb3‐positive and Gb3‐negative HT29 colorectal cancer cells showed that the conjugates had approximately 100‐fold higher anticancer activity on Gb3‐positive cells (GI50 in nM range).544 This is in contrast to free MMAE or MMAF, whose cytotoxicities were independent of Gb3 expression. In addition, the activity of the conjugates on Gb3‐positive cells was either similar to (96) or higher (97) than that of the free drugs.544 These conjugates have not yet been tested in vivo.
Pan et al.545 developed a conjugate of MMAE and the TRAIL with the purpose of overcoming TRAIL resistance in cell lines expressing the TRAIL‐targeted DR4 and DR5 death receptors. The conjugate termed N109C‐vcMMAE was prepared by attachment of MMAE and TRAIL through a dipeptide (valine‐citrulline) moiety (98, Fig. 27) that could be cleaved by lysosomal cathepsin to release active MMAE.545 This conjugate showed the highest activity with a GI50 of ∼63 nM in the TRAIL‐resistant MCF‐7 cell line.545 Studies demonstrated that conjugate internalization occurred by TRAIL‐death receptor‐mediated endocytosis, and that cell cycle arrest in the G2‐M phase could be affected within 8 hr of exposure to the conjugate.545 in vivo studies in a mouse NCI‐H460 lung cancer xenograft model demonstrated the ability of the N109C‐vcMMAE conjugate to target the tumors, potentially by taking advantage of the EPR effect.545 The antitumor efficacy of the conjugate was not evaluated in this animal model.
Temming et al.546 developed arginine‐glycine‐aspartic acid (RGD)‐targeted albumin carriers for MMAE. Specifically, MMAE‐human albumin conjugates 99a and 100a (Fig. 28) decorated with cyclic c(RGDfK) peptides were used to target and cause apoptosis of ανβ3‐integrin‐expressing angiogenic endothelial cells, thereby destroying the tumor's blood supply.546 These conjugates also utilized a valine‐citrulline linker to enable intracellular release of MMAE from the albumin carrier upon activation by lysosomal cathepsin.546 The two carriers differed in the use of either a short alkyl chain (99a) or a poly(ethylene glycol) (PEG) spacer (100a) for attachment of the targeting RGD peptide. Both carriers showed high specificity toward, internalization into, and growth inhibition (GI50 in nM range) of ανβ3‐integrin‐expressing human umbilical vein endothelial cells (HUVEC).546 Preliminary evaluation in a C26 murine colon carcinoma mouse xenograft model showed that these carriers could preferentially accumulate on tumor blood vessels as well as inside of tumors within 24 hr of intravenous injection.546 The antitumor effect of these systems was not evaluated in vivo.
Figure 28.
Chemical structures of c(RGDfK)‐targeted conjugates of MMAE and MMAF to albumin using short alkyl chain linker (99a and 99b) or a poly(ethylene glycol) spacer (100a and 100b), and activatable cell‐penetrating peptide conjugate of MMAE (101). In all of these structures, MMAE and MMAF are conjugated to the carriers utilizing the monomethyl nitrogen that is bolded in structures 92 and 93.
This same group also evaluated RGD‐targeted MMAF albumin conjugates.547 As before, two conjugate designs were tested for each of the two drugs: one that used a PEG chain and another a short linker for attachment of the targeting RGD moiety.547 Both MMAF conjugates (99b and 100b, Fig. 28) showed strong ability to reduce HUVEC cell growth, with GI50 values of 22–77 nM compared to 65–420 nM for MMAE conjugates.547 Similar trends were observed in C26 colon cancer cells, with GI50 values of ∼10–150 nM for MMAF conjugates and ∼530–2070 nM for MMAE conjugates.547 Noticeably, RGD‐targeted MMAF conjugates 99b and 100b were more effective than free MMAF, demonstrating that the use of active targeting enabled intracellular delivery of this charged drug into the cells.547 These systems were not evaluated in vivo.
Recently, the use of activatable cell‐penetrating peptides (ACPP) consisting of a polycationic cell penetrating peptide (polyarginine) and a polyanionic (polyglutamate) peptide that are joined by a MMP‐cleavable linker was used for the delivery of MMAE.548 MMAE was covalently conjugated to the polyanionic portion of an ACPP using a linker that could be cleaved by lysosomal cathepsin B.548 The ACPP was further targeted to ανβ3‐integrin‐expressing cells using cyclic RGD (101, Fig. 28). in vitro studies showed that the targeted peptide was readily uptaken by a number of human cancer cell lines.548 In addition, in vivo studies in MDA‐MB‐231 mouse xenograft and in immunocompetent syngenic Py230 murine models demonstrated that the RGD‐targeted MMAE‐ACPP conjugate 101 controlled tumor growth more effectively than either free MMAE or an MMAE‐ACPP conjugate targeted with a sham RAD peptide.548
The use of the MMAE‐ACPP conjugate 101 as a radiosensitizer for combination chemo/radiotherapy was investigated by the same group.549 RGD‐targeted MMAE–ACPP conjugate 101 was shown to sensitize human colorectal and pancreatic cancer cells to radiation, resulting in increased DNA damage upon dual treatment.549 Studies in mouse xenograft models with PANC‐1 and HCT‐116 tumors showed that the targeted conjugate paired with ionizing radiation provided enhanced tumor regression than radiation alone, free MMAE, or MMAE in combination with radiation.549
As described, nanomedicine formulations promise to improve the activity and safety of mollusk‐derived anticancer agents by increasing the payload and specificity of these agents toward cancer cells. While not many types of nanomedicines have been explored as of yet for the targeted delivery of mollusk‐derived anticancer agents, and even fewer evaluated for in vivo therapeutic efficacy, the few systems reported to date all showed improvements in solubility, targeting ability, or therapeutic efficacy compared to the free drugs. We therefore believe that there is a great opportunity for further development in this field that could enable clinical advance of these active therapeutic agents in the future.
6. THE PROBLEM OF SUPPLY OF MOLLUSK‐DERIVED ANTICANCER AGENTS
Usually, chemists working in a marine natural product laboratory isolate only a few milligrams of a promising anticancer compound of novel structure. In the case of marine mollusks, bioactive metabolites are usually available in very small amounts—often less than 1 mg—due either to compound's low natural abundance in these organisms or the difficulty of sampling mollusk biomass. Only small amounts of pure compounds are required for preliminary in vitro screening in cancer cell lines. In contrast, the preclinical evaluation including studies in in vivo rodent models often require over 100 mg of a pure compound. Subsequently, grams to hundreds of grams are usually needed to progress satisfactorily toward clinical trials. Finally, when a compound is commercialized as a pharmaceutical, kilograms of the pure compound are required to supply drug production.220, 550, 551 Due to this, for many years the major perception in the medical and in general scientific communities has been that marine active agents are not directly feasible for further development.552 This was true indeed in the past. In recent years, however, the application and combination of novel techniques in harvest/culture and isolation processes, the increased knowledge of genomics, and major advances in chemical synthesis have enabled the large‐scale production of several marine natural products.110, 220, 550, 551
What strategies can be utilized to scale up the production of mollusk‐derived anticancer compounds? In principle, similar to other invertebrates, this may be performed by large‐scale culture including mariculture (growth of the organism in its natural environment) and aquaculture (under artificial conditions). However, due to critical environmental parameters (temperature, pH, light cycle) and nutrition issues,553, 554, 555 the cultivation or maintenance of mollusk specimens is very difficult and often impossible. Although large‐scale cultivation has been widely applied to bivalves for food production556 and significant advances have been achieved in the larviculture and grow out of marine snails for human consumption,557, 558 the cultivation of mollusks for biotechnological applications is yet to be addressed. To date, the only successful example is the production of the sea slug Aplysia californica, which is the well‐known model organism widely used in neuroscience.559 In addition, if a compound comes from the diet including microorganisms,110 the proper source has to be identified for its production.
Therefore, for almost all mollusk‐derived products, the only feasible route to overcome the supply problem is chemical synthesis. Synthesis provides additional advantages; in fact, it may not only provide corroboration for the structural assignment, but also may lead to the production of valuable intermediates and analogues to be used for SAR studies.560 Synthetic strategies also include the development of derivatives with more manageable properties, or the design of a pharmacophore of reduced complexity, which can be synthesized more easily. For example, the production of kahalalide F for its use in clinical trials was developed using solid‐phase peptide chemistry techniques by PharmaMar and according to the same synthetic procedure, more than 150 analogues were prepared for SAR studies.263, 561, 562 This synthesis reported by Jimenez et al.563 is illustrated in Figure 29 and described next.
Figure 29.
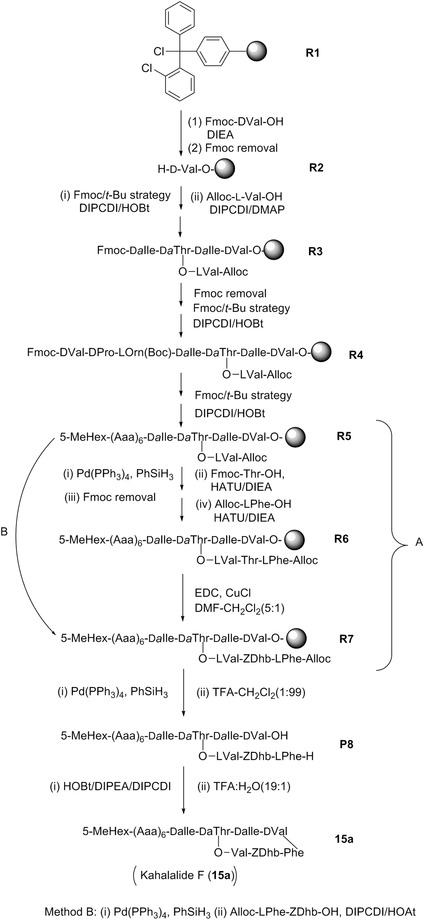
Synthesis of Kahalalide F utilized for its production for clinical trials using solid‐phase peptide chemistry. Alloc, allyloxycarbonyl; DIEA, N,N‐diisopropylethylamine; DIPCDI, N,N’‐diisopropylcarbodiimide; DMAP, 4‐dimethylaminopyridine; EDC, 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide; Fmoc, fluoromethyloxycarbonyl chloride; HATU, 1‐[Bis(dimethylamino)methylene]‐1H‐1,2,3‐triazolo[4,5‐b]pyridinium 3‐oxid hexafluorophosphate, HOAT, 1‐hydroxy‐7‐azabenzotriazole; HOBt, 1‐hydroxybenzotriazole; Pd(PPh3)4, tetrakis(triphenylphosphine)palladium(0); PhSiH3, phenylsilane; t‐Bu, tert‐butyl; TFA, trifluoroacetic acid; (Z)‐Dhb, a,b‐didehydro‐a‐aminobutyric acid.
This solid‐phase synthesis was based on an orthogonal protecting system employing chlorotrityl chloride as resin (Cl‐TrtCl‐resin, R1, Fig. 29). Resin R1, was converted to H‐D‐Val resin R2 using Fmoc‐D‐Val‐OH and DIEA followed by Fmoc removal. A sequential incorporation of D‐allo‐Ile, D‐allo‐Thr, and D‐allo‐Ile derivatives using Fmoc/t‐Bu and DIPCDI/HOBt reagent mixture followed by esterication with allo‐Val‐OH using DIPCDI/DMAP produced tetrapeptide resin R3. This was then converted to decapeptide resin R5 by chain elongation through a sequential addition of six amino acids. The N‐terminus was then capped with 5‐methylhexanoic acid using the Fmoc removal protocol, Fmoc/t‐Bu strategy, and the coupling reagent mixture DIPCDI/HOBt. Resin R7 was synthesized by two methods. In method A, tridecapeptide R6 was prepared from R5 by side chain elongation using first Pd(PPh3)4 and PhSiH3, followed by treatment with FmocThr‐OH and HATU/DIEA and Fmoc removal. Next, the reaction mixture was treated with Alloc‐LPhe‐OH and HATU/DIEA to yield R6. The preparation of R6 was followed by selective synthesis of (Z)‐Dhb on the resin by Fukase's conditions (EDC and CuCl) to give R7. In method B, the dipeptide (Alloc‐Phe‐(Z)‐Dhb‐OH) was directly added to R5 using Pd(PPh3)4, PhSiH3, Alloc‐LPhe‐ZDhb‐OH, and DIPCDI/HOAt to yield R7. Resin R7 was converted to depsipeptide resin R8 by removal of Alloc group in R7 with Pd(PPh3)4 and PhSiH3, followed by peptide release from the resin using TFA/CH2Cl2 (1:99) mixture. The macrocyclization of peptide P8 with HOBt/DIPEA/DIPCDI and the removal of the side chain protecting groups using TFA/H2O (19:1) mixture afforded kahalalide F (15a; Fig. 6; Table II).
The synthetic methods performed to produce dolastatins217 have led to a number of novel intermediate substances such as auristatins and to antibody–drug conjugates currently in Phases I to III clinical trials.166, 222, 564
For an increasing number of synthetically challenging anticancer compounds from mollusks the supply problem has been solved through semisynthesis by starting from a more accessible precursor—typically of microbial origin. An example is Zalypsis® (PM00104, 84b), which was synthesized by workers at PharmaMar (Madrid, Spain) using methodologies related to the trabectedin synthesis from safracin B.565 Moving to more chemically complex molecules, the challenges associated with their total synthesis are significant but not insurmountable. Today, chemists can synthesize organic compounds—natural and designed—of all types of structural architectures.566 In the last few decades a surge in total synthesis endeavors around the world in constructing natural products of medicinal importance against cancer has led to a remarkable collection of achievements that covers a wide ranging landscape of molecular and stereochemical complexity and diversity.566
Of course, scalability of the entire synthetic process leading to the target compound remains a crucial feature.567 Historically, the pharmaceutical industry's acceptance of structurally complex natural products or natural product derivatives supplied commercially through total synthesis has been low for a variety of reasons, but the situation is slowly changing. The accessibility of target active molecules by simple scalable routes is based on a series of factors including step count, level of complexity of the chemistry used, stereochemical control, chemical stability of each process and intermediate, overall yield, and operational complexities, among others. An increasing number of potential anticancer marine natural products are now prepared in a scalable fashion.567 The most significant example of a large‐scale total synthesis that has solved a major supply issue is that of eribulin mesylate (Halaven®) inspired by the marine natural product halichondrin B.568 Eribulin is a macrocyclic ketone with 36 atoms, 19 of which are stereogenic centers, that represents the most structurally complex, nonpeptidic, and fully synthetic drug on the market today. However, it should be taken into consideration that the “supply issue” is only one of the crucial aspects in anticancer drug development. The so‐called “valley of death” that is the gap between the discovery of the active principle and its entering clinical trials is a highly complex problem where numerous driving forces—scientific, financial, and other economic and nontechnical factors—play a role.569 Scientific critical tasks include drug production, of course, but also tumor physiology, drug pharmacokinetics, preclinical models, drug delivery, and clinical translation. The lack of common methods to address these aspects by industrial pharmaceutical companies (Pharma) and academic scientists (Academia) is a fundamental point. Only a greater Pharma‐Academia collaboration could overcome the barrier of the valley of death in the development of new effective anticancer therapies.570
7. CONCLUSIONS: WHAT ARE THE MOST PROMISING MOLLUSK‐DERIVED ANTICANCER COMPOUNDS?
Of note, no “recent” mollusk‐derived compounds have attained late Phase II or Phase III clinical trials in oncology. However, we think that several “mollusk‐derived” compounds merit further consideration.
In terms of in vivo activity, we think that a compound for which clear evidence of activity has been demonstrated in various models (including aggressive metastatic) has a greater probability to move toward clinical trials than a compound that has not been assayed in vivo, displays no activity in vivo, or whose in vivo activity has been demonstrated, for example, in ascitic models of mouse leukemia, which are by far too chemosensitive and not at all representative of the clinical situation.
It is interesting that several dolastatins (or their derivatives) displayed marked in vivo anticancer activity in multiple preclinical models (murine syngeneic as well as human xenograft models), but failed to demonstrate actual anticancer activity in Phase II clinical trials. It is important to keep in mind that tumor doubling times markedly differ between murine tumor models (days to weeks) and human cancers (weeks to years).571 Dolastatins are tubulin inhibitors, which are known to be more effective against rapidly proliferating cells. Human orthotopic xenografts in most cases develop much more rapidly in immunocompromised or immunodeficient mice than their counterparts developing in humans. Furthermore, dolastatins are tubulin inhibitors without other reported targets in the literature (at least to the best of our knowledge). They are cytotoxic proapoptotic compounds and substrates of the P‐gp, which makes them unable to overcome resistance to proapoptotic stimuli or circumvent the MDR phenotype. It is important to emphasize once again that cancers associated with dismal prognoses, including metastatic cancers, display resistance to proapoptotic stimuli.
According to data we report in the current review, we estimate that the following compounds are “promising” and should be pursued further: dolastatin 16 (9), chlorolissoclimide (33b), doliculide (46), ulapaulide A (60a), kabiramide C (61a), halichondramide‐related metabolites (62a‐62c), aplyronine A (64a), sphinxolide (66), latrunculin A (67a), hectochlorin (75a), lamellarin D (76), and spisulosine (87). We urge the readers of the current review to perform experiments designed to address the major “biological cancer characteristics” we detailed in the Introduction to create a stronger basis for the selection of promising anticancer agents derived from marine mollusks.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
R.K. is a director of research with the Fonds National de la Recherche Scientifique (FRS‐FNRS, Belgium). A.K. acknowledges grants from the National Cancer Institute (CA186046‐01A1) and Welch Foundation (AI‐0045). ICB authors thank the CNR bibliographic service; ICB authors also acknowledge dr . G. Villani for providing photos of some mollusks.
Biographies
Maria Letizia Ciavatta studied Pharmacy at the University "Federico II" of Naples, getting graduation "cum laude" in 1988. Since 1999 she is a Researcher at Istituto di Chimica Biomolecolare (ICB) of the Italian Consiglio Nazionale delle Ricerche (CNR). Her research interests are focused on the exploration of the biodiversity in natural products, involving skills in the purification and structural characterisation of new compounds from both marine and terrestrial sources. She is coauthor of more than 70 peer‐reviewed articles. Her profile is available on Research Gate.
Florence Lefranc obtained an M.D. degree in 1995 at the Université Libre de Bruxelles (ULB, Brussels, Belgium) and then she received her neurosurgeon diploma in 2002 (ULB). She received a Ph.D. in tumor cell biology in 2005 in ULB and she is currently neurosurgeon and chief of neuro‐oncology at the Department of Neurosurgery at Hôpital Erasme (ULB, Brussels, Belgium). She was also a M.D. post‐doctoral fellowship with the Fonds National de la Recherche Scientifique (FNRS, Belgium). Her main research area relates to the identification of biologicals or natural compounds aiming to combat glioma, and also to novel semisynthetic or synthetic compounds. She has authored or co‐authored ~150 peer‐reviewed articles and she is currently conducting several clinical trials with glioma and meningioma patients.
Marianna Carbone is Researcher at the Istituto di Chimica Biomolecolare of the Italian Consiglio Nazionale delle Ricerche. She received her PhD in Pharmaceutical Science from the University of Salerno working on the chemistry of marine mollusks. Her research activity is mainly focused on the isolation and structural characterization of secondary metabolites from marine invertebrates. Her studies include the evaluation of the relevant pharmacological properties of marine natural products, and the exploration of their ecological roles.
Ernesto Mollo is Researcher at the Istituto di Chimica Biomolecolare of the Italian Consiglio Nazionale delle Ricerche. He graduated with a Laurea degree in Biological Sciences from the University of Naples "Federico II". The primary focus of his work is on the study of the ecological roles of natural products from marine organisms and their phyletic and geographic distribution in nature. His research campaigns for the exploration of the molluscan biological and chemical diversity have been carried out on a global scale and led to the discovery of many novel bioactive metabolites.
Margherita Gavagnin received her doctoral degree in Organic Chemistry in 1983 from University of Naples. In 1985 she moved to the Institute of Biomolecular Chemistry (ICB), Italian National Council of Research (CNR) where she joined the group of Prof. G. Cimino and started working on the chemical ecology of marine opisthobranch molluscs. Currently, she is Research Director at ICB. Her researches are mainly focused to the definition of the structure of new bioactive molecules from marine organisms and plants. She published more than 150 papers on peer‐reviewed journals.
Tania Betancourt is Assistant Professor in the Department of Chemistry and Biochemistry and in the Materials Science, Engineering, and Commercialization Program at Texas State University in San Marcos, TX (USA). She obtained a Ph.D. in biomedical engineering from the University of Texas at Austin in 2007. Her laboratory's research focuses on the development of stimuli‐responsive biomaterials and nanomedicines for cancer detection and treatment. She has coauthored 17 peer‐reviewed articles and two book chapters. Her profile is available on Research Gate and Google Scholar.
Ramesh Dasari was born in Ambala, Nalgonda district, Telangana, India. He obtained his M.Sc. in organic chemistry from Osmania University, Hyderabad, India and Ph.D. in synthetic organic chemistry from Indian Institute of Chemical Technology (CSIR‐IICT), Hyderabad, India. In 2012, he joined Texas State University as a Postdoctoral Research Associate. During his postdoctoral research (Oct 2012‐present) at Texas State University with Prof. Alexander Kornienko, he has synthesized analogs of various structurally diverse natural products as part of their development as anticancer agents. He has published 33 peer reviewed papers.
Alexander Kornienko was born in a small Arctic town of Vorkuta, Russia. He studied at Mendeleev University in Moscow and then moved to the United States, where he received his Ph.D. in synthetic organic chemistry at Tufts University. After a two‐year postdoctoral fellowship at the University of Montreal, working on the synthesis of novel aminoglycoside antibiotics, he started his independent career at New Mexico Tech in 2001 and then moved to Texas State University where is now Professor of Chemistry Throughout his career he published 86 refereed research papers mostly dealing with natural product‐based cancer drug discovery.
Robert Kiss is Director of Research with the Fonds National de la Recherche Scientifique (FRS‐FNRS, Belgium) at the Laboratoire de Cancérologie et de Toxicologie Expérimentale, Faculté de Pharmacie—Université Libre de Bruxelles (ULB, Brussels, Belgium). He obtained a Ph.D. in tumor cell biology at ULB in 1987. He has characterized, with the various research teams he has headed, the mechanisms of action of numerous novel anticancer drugs, with a particular attention to natural products. He has authored or coauthored more than 520 peer‐reviewed articles. His profile is available on Research Gate.
Contract grant sponsor: National Cancer Institute; Contract grant number: CA186046‐01A1; Contract grant sponsor: Welch Foundation; Contract grant number: AI‐0045.
Contributor Information
Maria Letizia Ciavatta, Email: lciavatta@icb.cnr.it.
Alexander Kornienko, Email: a_k76@txstate.edu.
Robert Kiss, Email: rkiss2012@gmail.com.
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Lubansu A, Ruchoux MM, Brotchi J, Salmon I, Kiss R, Lefranc F. Cathepsin B, D and K expression in adamantinomatous craniopharyngiomas relates to their levels of differentiation as determined by the patterns of retinoic acid receptor expression. Histopathology 2003;43:563–572. [DOI] [PubMed] [Google Scholar]
- 3. Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immune‐oncology. Nat Rev Drug Discover 2015;14:603–622. [DOI] [PubMed] [Google Scholar]
- 4. Carvalho S, Levi‐Schaffer F, Sela M, Yarden Y. Immunotherapy of cancer: From monoclonal to oligoclonal cocktails of anti‐cancer antibodies—IUPHAR review X. Br J Pharmacol 2016;173:1407–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinmann H. Cancer immunotherapy: Selected targets and small‐molecule modulators. ChemMedChem 2016;11:450–466. [DOI] [PubMed] [Google Scholar]
- 6. Lucena SR, Salazar N, Gracia‐Cazaña T, Zamarrón A, González S, Juarranz Á, Gilaberte Y. Combined treatments with photodynamic therapy for non‐melanoma skin cancer. Int J Mol Sci 2015;16:25912–25933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kidane B, Hirpara D, Yasufuku K. Photodynamic therapy in non‐gastrointestinal thoracic malignancies. Int J Mol Sci 2016;17:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doroshow JH, Kummar S. Translational research in oncology—10 Years of progress and future prospects. Nat Rev Clin Oncol 2014;11:649–662. [DOI] [PubMed] [Google Scholar]
- 9. Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer 2008;8:473–480. [DOI] [PubMed] [Google Scholar]
- 10. Blatter S, Rottenberg S. Minimal residual disease in cancer therapy. Small things make all the difference. Drug Resist Updat 2015;21‐22:1–10. [DOI] [PubMed] [Google Scholar]
- 11. Skrbo N, Tenstad E, Mælandsmo GM, Sørlie T, Andersen K. From autonomy to community; new perspectives on tumorigenicity and therapy resistance. Cancer Treat Rev 2015;41:809–813. [DOI] [PubMed] [Google Scholar]
- 12. Khoo BL, Chaudhuri PK, Ramalingam N, Tan DS, Lim CT, Warkiani ME. Single‐cell profiling approaches to probing tumor heterogeneity. Int J Cancer 2016;139:243–255. [DOI] [PubMed] [Google Scholar]
- 13. Webster RM. Combination therapies in oncology. Nat Rev Drug Discov 2016;15:81–82. [DOI] [PubMed] [Google Scholar]
- 14. Atkins MB, Larkin J. Immunotherapy combined or sequenced with targeted therapy in the treatment of solid tumors: Current perspectives. J Natl Cancer Inst 2016;108; doi: 10.1093/jnci/djv414. [DOI] [PubMed] [Google Scholar]
- 15. Nicolini A, Carpi A, Ferrari P, Biava PM, Rossi G. Immunotherapy and hormone‐therapy in metastatic breast cancer: A review and an update. Curr Drug Targets 2016;17:1127–1139. [DOI] [PubMed] [Google Scholar]
- 16. Mullard A. FDA approves first immunotherapy combo. Nat Rev Drug Discov 2015;14:739. [Google Scholar]
- 17. Conroy T, Bachet JB, Ayav A, Huguet F, Lambert A, Caramella C, Maréchal R, Van Laethem JL, Ducreux M. Current standards and new innovative approaches for treatment of pancreatic cancer. Eur J Cancer 2016;57:10–22. [DOI] [PubMed] [Google Scholar]
- 18. Parikh M, Riess J, Lara PN, Jr . New and emerging developments in extensive‐stage small cell lung cancer therapeutics. Curr Opin Oncol 2016;28:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups ; National Cancer Institute of Canada Clinical Trials Group . Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐Year analysis of the EORTC‐NCIC trial. Lancet Oncol 2009;10:459–466. [DOI] [PubMed] [Google Scholar]
- 20. Perret GY, Uzzan B. An anticancer strategic dilemma: To kill or to contain. The choice of the pharmaceutical industry in 2009. Fund Clin Pharmaccol 2011;25:283–295. [DOI] [PubMed] [Google Scholar]
- 21. Xia H, Hui KM. Mechanism of cancer drug resistance and the involvement of noncoding RNAs. Curr Med Chem 2014;21:3029–3941. [DOI] [PubMed] [Google Scholar]
- 22. Bowden NA. Nucleotide excision repair: Why is it not used to predict response to platinum‐based chemotherapy? Cancer Lett 2014;346:163–171. [DOI] [PubMed] [Google Scholar]
- 23. Scott TL, Rangaswamy S, Wicker CA, Izumi T. Repair of oxidative DNA damage and cancer: Recent progress in DNA base excision repair. Antioxid Redox Signal 2014;20:708–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mladenov E, Magin S, Soni A, Iliakis G. DNA double‐strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol 2013;3:113. doi: 10.3389/fonc.2013.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z, Shi T, Zhang L, Zhu P, Deng M, Huang C, Hu T, Jiang L, Li J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett 2016;370:153–164. [DOI] [PubMed] [Google Scholar]
- 26. Dinic J, Podolski‐Renic A, Stankovic T, Bankovic J, Pesic M. New approaches with natural product drugs for overcoming multidrug resistance in cancer. Curr Pharm Des 2015;21:5589–5604. [DOI] [PubMed] [Google Scholar]
- 27. Cui H, Zhang AJ, Chen M, Liu JJ. ABC transporter inhibitors in reversing multidrug resistance to chemotherapy. Curr Drug Targets 2015;16:1356–1371. [DOI] [PubMed] [Google Scholar]
- 28. Cort A, Ozben T. Natural product modulators to overcome multidrug resistance in cancer. Nutr Cancer 2015;67:411–423. [DOI] [PubMed] [Google Scholar]
- 29. Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol 2015. 2016;13:335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim S. New and emerging factors in tumorigenesis: An overview. Cancer Manag Res 2015;7:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pazarentzos E, Bivona TG. Adaptive stress signaling in targeted cancer therapy resistance. Oncogene 2015;34:5599–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Block KI, Gyllenhaal C, Lowe L, Amedei A, Amin AR, Amin A, Aquilano K, Arbiser J, Arreola A, Arzumanyan A, Ashraf SS, Azmi AS, Benencia F, Bhakta D, Bilsland A, Bishayee A, Blain SW, Block PB, Boosani CS, Carey TE, Carnero A, Carotenuto M, Casey SC, Chakrabarti M, Chaturvedi R, GZ, Chen H, Chen S, Chen YC, Choi BK, Ciriolo MR, Coley HM, Collins AR, Connell M, Crawford S, Curran CS, Dabrosin C, Damia G, Dasgupta S, DeBerardinis RJ, Decker WK, Dhawan P, Diehl AM, Dong JT, Dou QP, Drew JE, Elkord E, El‐Rayes B, Feitelson MA, Felsher DW, Ferguson LR, Fimognari C, Firestone GL, Frezza C, Fujii H, Fuster MM, Generali D, Georgakilas AG, Gieseler F, Gilbertson M, Green MF, Grue B, Guha G, Halicka D, Helferich WG, Heneberg P, Hentosh P, Hirschey MD, Hofseth LJ, Holcombe RF, Honoki K, Hsu HY, Huang GS, Jensen LD, Jiang WG, Jones LW, Karpowicz PA, Keith WN, Kerkar SP, Khan GN, Khatami M, Ko YH, Kucuk O, Kulathinal RJ, Kumar NB, Kwon BS, Le A, Lea MA, Lee HY, Lichtor T, Lin LT, Locasale JW, Lokeshwar BL, Longo VD, Lyssiotis CA, MacKenzie KL, Malhotra M, Marino M, Martinez‐Chantar ML, Matheu A, Maxwell C, McDonnell E, Meeker AK, Mehrmohamadi M, Mehta K, Michelotti GA, Mohammad RM, Mohammed SI, Morre DJ, Muralidhar V, Muqbil I, Murphy MP, Nagaraju GP, Nahta R, Niccolai E, Nowsheen S, Panis C, Pantano F, Parslow VR, Pawelec G, Pedersen PL, Poore B, Poudyal D, Prakash S, Prince M, Raffaghello L, Rathmell JC, Rathmell WK, Ray SK, Reichrath J, Rezazadeh S, Ribatti D, Ricciardiello L, Robey RB, Rodier F, Rupasinghe HP, Russo GL, Ryan EP, Samadi AK, Sanchez‐Garcia I, Sanders AJ, Santini D, Sarkar M, Sasada T, Saxena NK, Shackelford RE, Shantha Kumara HM, Sharma D, Shin DM, Sidransky D, Siegelin MD, Signori E, Singh N, Sivanand S, Sliva D, Smythe C, Spagnuolo C, Stafforini DM, Stagg J, Subbarayan PR, Sundin T, Talib WH, Thompson SK, Tran PT, Ungefroren H, Vander Heiden MG, Venkateswaran V, Vinay DS, Vlachostergios PJ, Wang Z, Wellen KE, Whelan RL, Yang ES, Yang H, Yang X, Yaswen P, Yedjou C, Yin X, Zhu J, Zollo M. Designing a broad‐spectrum integrative approach for cancer prevention and treatment. Semin Cancer Biol 2015;35(Suppl):S276–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pierce GB. Neoplasms, differentiations and mutations. Am J Pathol 1974;77:103–118. [PMC free article] [PubMed] [Google Scholar]
- 34. Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS. Cancer across the three of life: Cooperation and cheating in multicellularity. Phil Trans R Soc B 2015;370:2014–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sell S, Nicolini A, Ferrari P, Biava PM. Cancer: A problem of developmental biology. Scientific evidence for reprogramming and differentiation therapy. Curr Drug Targets 2016;17:1103–1110. [DOI] [PubMed] [Google Scholar]
- 36. Nowell PC. The clonal evolution of tumor cell populations. Science 1976;194:23–28. [DOI] [PubMed] [Google Scholar]
- 37. Rycaj K, Tang DG. Cell‐of‐origin of cancer versus cancer stem cells: Assays and interpretations. Cancer Res 2015;75:4003–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morris H. Cancer and its origin. Br Med J 1903;2:1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rous P. The relations of embryonic tissue and tumor in mixed grafts. J Exp Med 1911;13:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watson JD, Crick FH. The structure of DNA. Cold Spring Harb Symp Quant Biol 1953;18:123–131. [DOI] [PubMed] [Google Scholar]
- 41. Watson JD, Crick FH. Molecular structure of nucleic acids. A structure for deoxyribose nucleic acid. Nature 1953;171:737–738. [DOI] [PubMed] [Google Scholar]
- 42. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—What challenges do they pose? Nat Rev Drug Discov 2014;13:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci 2016;107:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu‐Economescu M. Therapies targeting cancer stem cells: Current trends and future challenges. World J Stem Cells 2015;7:1185–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lisanti MP, Martinez‐Outschoorn UE, Sotgia F. Oncogenes induce the cancer‐associated fibroblast phenotype. Cell Cycle 2013;12:2723–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget 2014;5:11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lamb R, Harrison H, Smith DL, Townsend PA, Jackson T, Ozsvari B, Martinez‐Outschoorn UE, Pestell RG, Howell A, Lisanti MP, Sotgia F. Targeting tumor‐initiating cells: Eliminating anabolic cancer stem cells via inhibitors of protein synthesis or by mimicking caloric restriction. Oncotarget 2015;6:4585–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Span PN, Bussink J. Biology of hypoxia. Semin Nucl Med 2015;45:101–109. [DOI] [PubMed] [Google Scholar]
- 49. Dhani N, Fyles A, Hedley D, Milosevic M. The clinical significance of hypoxia in human cancers. Semin Nucl Med 2015;45:110–121. [DOI] [PubMed] [Google Scholar]
- 50. Paolicchi E, Gemignani F, Krstic‐Demonacos M, Dedhar S, Mutti L, Landi S. Targeting hypoxic response for cancer therapy. Oncotarget 2016; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liang D, Miller GH, Tranmer GK. Hypoxia activated prodrugs: Factors influencing design and development. Curr Med Chem 2015;22:4313–4325. [DOI] [PubMed] [Google Scholar]
- 52. Zhao J, Du F, Luo Y, Shen G, Zheng F, Xu B. The emerging role of hypoxia‐inducible factor‐2 involved in chemo/radioresistance in solid tumors. Cancer Treat Rev 2015;41:623–633. [DOI] [PubMed] [Google Scholar]
- 53. Parks SK, Cormerais Y, Marchiq I, Pouyssegur J. Hypoxia optimises tumour growth by controlling nutrient import and acidic metabolite export. Mol Aspects Med 2016;47‐48:3–14. [DOI] [PubMed] [Google Scholar]
- 54. Eales KL, Hollinshead KE, Tennant DA. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016;5:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El‐Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnóczky G, Hardwick JM, Hengartner MO, Ichijo H, Jäättelä M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema JP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nuñez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009;16:1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El‐Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012;19:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Galluzzi L, Bravo‐San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico‐Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M, Candi E, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, Di Daniele N, Dixit VM, Dynlacht BD, El‐Deiry WS, Fimia GM, Flavell RA, Fulda S, Garrido C, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Joseph B, Jost PJ, Kaufmann T, Kepp O, Klionsky DJ, Knight RA, Kumar S, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López‐Otín C, Lugli E, Madeo F, Malorni W, Marine JC, Martin SJ, Martinou JC, Medema JP, Meier P, Melino S, Mizushima N, Moll U, Muñoz‐Pinedo C, Nuñez G, Oberst A, Panaretakis T, Penninger JM, Peter ME, Piacentini M, Pinton P, Prehn JH, Puthalakath H, Rabinovich GA, Ravichandran KS, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Shi Y, Simon HU, Stockwell BR, Szabadkai G, Tait SW, Tang HL, Tavernarakis N, Tsujimoto Y, Vanden Berghe T, Vandenabeele P, Villunger A, Wagner EF, Walczak H, White E, Wood WG, Yuan J, Zakeri Z, Zhivotovsky B, Melino G, Kroemer G. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ 2015;22:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett 2008;272:177–185. [DOI] [PubMed] [Google Scholar]
- 59. Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti‐apoptosis and cell survival: A review. Biochim Biophys Acta 2011;1813:238–259. [DOI] [PubMed] [Google Scholar]
- 60. Speirs CK, Hwang M, Kim S, Li W, Chang S, Varki V, Mitchell L, Schleicher S, Lu B. Harnessing the cell death pathway for targeted cancer treatment. Am J Cancer Res 2011;1:43–61. [PMC free article] [PubMed] [Google Scholar]
- 61. Belaid A, Ndiaye PD, Filippakis H, Roux J, Röttinger É, Graba Y, Brest P, Hofman P, Mograbi B. Autophagy: Moving benchside promises to patient bedsides. Curr Cancer Drug Targets 2015;15:684–702. [DOI] [PubMed] [Google Scholar]
- 62. Huang Z, Zhou L, Chen Z, Nice EC, Huang C. Stress management by autophagy: Implications for chemoresistance. Int J Cancer 2016;139:23–32. [DOI] [PubMed] [Google Scholar]
- 63. Mukhopadhyay S, Sinha N, Das DN, Panda PK, Naik PP, Bhutia SK. Clinical relevance of autophagic therapy in cancer: Investigating the current trends, challenges, and future prospects. Crit Rev Clin Lab Sci 2016;16:1–25. [DOI] [PubMed] [Google Scholar]
- 64. Wang C, Hu Q, Shen HM. Pharmacological inhibitors of autophagy as novel cancer therapeutic agents. Pharmacol Res 2016;105:164–175. [DOI] [PubMed] [Google Scholar]
- 65. Kornienko A, Mathieu V, Rastogi SK, Lefranc F, Kiss R. Therapeutic agents triggering nonapoptotic cancer cell death. J Med Chem 2013;56:4823–4839. [DOI] [PubMed] [Google Scholar]
- 66. Piao S, Amaravadi RK. Targeting the lysosome in cancer. Ann N Y Acad Sci 2016;1371:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene 2003;22:3138–3151. [DOI] [PubMed] [Google Scholar]
- 68. Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: Special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol 2005;23:2411–2422. [DOI] [PubMed] [Google Scholar]
- 69. Tisdale MJ. Antitumour imidazotetrazines—XI: Effect of 8‐carbamoyl‐3‐methylimidazo[5,1‐d]‐1,2,3,5‐tetrazin‐4(3H)‐one [CCRG 81045; M and B 39831 NSC 362856] on poly(ADP‐ribose) metabolism. Br J Cancer 1985;52:789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen Y, Douglass T, Jeffes EW, Xu Q, Williams CC, Arpajirakul N, Delgado C, Kleinman M, Sanchez R, Dan Q, Kim RC, Wepsic HT, Jadus MR. Living T9 glioma cells expressing membrane macrophage colony‐stimulating factor produce immediate tumor destruction by polymorphonuclear leukocytes and macrophages via a “paraptosis”‐induced pathway that promotes systemic immunity against intracranial T9 gliomas. Blood 2002;100:1373–1380. [DOI] [PubMed] [Google Scholar]
- 71. Jadus MR, Chen Y, Boldaji MT, Delgado C, Sanchez R, Douglass T, Al‐Atar U, Schulz W, Lloyd C, Wepsic HT. Human U251MG glioma cells expressing the membrane form of macrophage colony‐stimulating factor (mM‐CSF) are killed by human monocytes in vitro and are rejected within immunodeficient mice via paraptosis that is associated with increased expression of three different heat shock proteins. Cancer Gene Ther 2003;10:411–420. [DOI] [PubMed] [Google Scholar]
- 72. Bury M, Girault A, Mégalizzi V, Spiegl‐Kreinecker S, Mathieu V, Berger W, Evidente A, Kornienko A, Gailly P, Vandier C, Kiss R. Ophiobolin A induces paraptosis‐like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis 2013;4:e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res 2008;6:965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maltese WA, Overmeyer JH. Methuosis: Nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am J Pathol 2014;184:1630–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bröker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: A review. Clin Cancer Res 2005;11:3155–3162. [DOI] [PubMed] [Google Scholar]
- 76. Lee D, Kim IY, Saha S, Choi KS. Paraptosis in the anti‐cancer arsenal of natural products. Pharmacol Ther 2016;162:120–133 [DOI] [PubMed] [Google Scholar]
- 77. Fidler IJ, Kripke ML. The challenge of targeting metastasis. Cancer Metastasis Rev 2015;34:635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mehlen P, Puisieux A. Metastasis: A question of life or death. Nat Rev Cancer 2006;6:449–458. [DOI] [PubMed] [Google Scholar]
- 79. Liu J, Ma L, Wu N, Liu G, Zheng L, Lin X. Aplysin sensitizes cancer cells to TRAIL by suppressing P38 MAPK/survivin pathway. Mar Drugs 2014;12:5072–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Langley RR, Fidler IJ. The seed and soil hypothesis revisited—The role of tumor‐stroma interactions in metastasis to different organs. Int J Cancer 2011;128:2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fidler IJ. Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125‐I‐5‐iodo‐2’‐deoxyuridine. J Natl Cancer Inst 1970;45:773–782. [PubMed] [Google Scholar]
- 82. Chambers AF, MacDonald IC, Schmidt EE, Koop S, Morris VL, Khokha R, Groom AC. Steps in tumor metastasis: new concepts from intravital videomicroscopy. Cancer Metastasis Rev 1995;14:279–301. [DOI] [PubMed] [Google Scholar]
- 83. Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev 1889;8:98–101. [PubMed] [Google Scholar]
- 84. Pienta KJ, Robertson BA, Coffey DS, Taichman RS. The cancer diaspora: metastasis beyond the seed and soil hypothesis. Clin Cancer Res 2013;19:5849–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sosa MS, Bragado P, Aguirre‐Ghiso JA. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat Rev Cancer 2014;14:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer 2015;15:238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Smith HA, Kang Y. The metastasis‐promoting roles of tumor‐associated immune cells. J Mol Med 2013;91:411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Berns A, Pandolfi PP. Tumor microenvironment revisited. EMBO Rep 2014;15:458–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer‐associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res 2012;10:1403–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Capparelli C, Guido C, Whitaker‐Menezes D, Bonuccelli G, Balliet R, Pestell TG, Goldberg AF, Pestell RG, Howell A, Sneddon S, Birbe R, Tsirigos A, Martinez‐Outschoorn U, Sotgia F, Lisanti MP. Autophagy and senescence in cancer‐associated fibroblasts metabolically support tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012;11‐12:2285–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Carito V, Bonuccelli G, Martinez‐Outschoorn UE, Whitaker‐Menezes D, Caroleo MC, Cione E, Howell A, Pestell RG, Lisanti MP, Sotgia F. Metabolic remodeling of the tumor microenvironment: Migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle 2012;11:3403–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sotgia F, Martinez‐Outschoorn UE, Lisanti MP. Cancer metabolism: New validated targets for drug discovery. Oncotarget 2013;4:1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tabassum DP, Polyak K. Tumorigenesis: It takes a village. Nat Rev Cancer 2015;15:473–483. [DOI] [PubMed] [Google Scholar]
- 94. Cano CE, Sandí MJ, Hamidi T, Calvo EL, Turrini O, Bartholin L, Loncle C, Secq V, Garcia S, Lomberk G, Kroemer G, Urrutia R, Iovanna JL. Homotypic cell cannibalism, a cell‐death process regulated by the nuclear protein 1, opposes to metastasis in pancreatic cancer. EMBO Mol Med 2012;4:964–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Krajcovic M, Overholtzer M. Mechanisms of ploidy increase in human cancers: A new role for cell cannibalism. Cancer Res 2012;72:1596–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Blazejczyk A, Papiernik D, Porshneva K, Sadowska J, Wietrzyk J. Endothelium and cancer metastasis: Perspectives for antimetastatic therapy. Pharmacol Rep 2015;67:711–718. [DOI] [PubMed] [Google Scholar]
- 97. Folkman J. Tumor angiogenesis: Therapeutic implications. N Engl J Med 1971;285:1182–1186. [DOI] [PubMed] [Google Scholar]
- 98. Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med 1971;133:275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol 2012;9:378–390. [DOI] [PubMed] [Google Scholar]
- 100. Weathers SP, de Groot J. Resistance to antiangiogenic therapy. Curr Neurol Neurosci Rep 2014;14:443. [DOI] [PubMed] [Google Scholar]
- 101. Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–322. [DOI] [PubMed] [Google Scholar]
- 102. Miles FL, Sikes RA. Insidious changes in stromal matrix fuel cancer progression. Mol Cancer Res 2014;12:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. D'Incalci M, Badri N, Galmarini CM, Allavena P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer 2014;111:646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Galmarini CM, D'Incalci M, Allavena P. Trabectedin and plitidepsin: Drugs from the sea that strike the tumor microenvironment. Mar Drugs 2014;12:719–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Paul VJ, Puglisi MP. Chemical mediation of interactions among marine organisms. Nat Prod Rep 2004;21:189–209. [DOI] [PubMed] [Google Scholar]
- 106. Wulff JL. Ecological interactions of marine sponges. Can J Zool 2006;84:146–166. [Google Scholar]
- 107. Thoms C, Schupp PJ. Chemical defense strategies in sponges: A review In: Custódio MR, Lôbo‐Hajdu G, Hajdu E, Muricy G, Eds. Porifera Research: Biodiversity, Innovation and Sustainability. Série Livros 28. Museu Nacional, Rio de Janeiro, Brazil: 2007. p 627–637. [Google Scholar]
- 108. De Caralt S, Bry D, Bontemps N, Turon X, Uriz MJ, Banaigs B. Sources of secondary metabolite variation in Dysidea avara (Porifera: Demospongiae): The importance of having good neighbors. Mar Drugs 2013;11:489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol 2014;32:40–51. [DOI] [PubMed] [Google Scholar]
- 110. Gerwick WH, Moore BS. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol 2012;19:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ocana A, Tannock IF. When are “positive” clinical trials in oncology truly positive? J Natl Cancer Inst 2011;103:16–20. [DOI] [PubMed] [Google Scholar]
- 112. DiMasi JA, Grabowski HG, Hansen RW. The cost of drug development. N Engl J Med 2015;372:1972. [DOI] [PubMed] [Google Scholar]
- 113. Avorn J. The $2.6 billion pil—Methodologic and policy considerations. N Engl J Med 2015;372:1877–1879. [DOI] [PubMed] [Google Scholar]
- 114. Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 1988;48:589–601. [PubMed] [Google Scholar]
- 115. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813–823. [DOI] [PubMed] [Google Scholar]
- 116. Mathieu V, Chantôme A, Lefranc F, Cimmino A, Miklos W, Paulitschke V, Mohr T, Maddau L, Kornienko A, Berger W, Vandier C, Evidente A, Delpire E, Kiss R. Sphaeropsidin A shows promising activity against drug‐resistant cancer cells by targeting regulatory volume increase. Cell Mol Life Sci 2015;72:3731–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ingrassia L, Lefranc F, Mathieu V, Darro F, Kiss R. Amaryllidaceae isocarbostyril alkaloids and their derivatives as promising antitumor agents. Transl Oncol 2008;1:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kornienko A, Evidente A. Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem Rev 2008;108:1982–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Van Goietsenoven G, Hutton J, Becker JP, Lallemand B, Robert F, Lefranc F, Pirker C, Vandenbussche G, Van Antwerpen P, Evidente A, Berger W, Prévost M, Pelletier J, Kiss R, Kinzy TG, Kornienko A, Mathieu V. Targeting of eEF1A with Amaryllidaceae isocarbostyrils as a strategy to combat melanomas. FASEB J 2010;24:4575–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chan GK, Kleinheinz TL, Peterson D, Moffat JG. A simple high‐content cell cycle assay reveals frequent discrepancies between cell number and ATP and MTS proliferation assays. PLoS One 2013;8:e63583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lefranc F, Nuzzo G, Hamdy NA, Fakhr I, Moreno Y Banuls L, Van Goietsenoven G, Villani G, Mathieu V, van Soest R, Kiss R, Ciavatta ML. in vitro pharmacological and toxicological effects of norterpene peroxides isolated from the Red Sea sponge Diacarnus erythraeanus on normal and cancer cells. J Nat Prod 2013;76:1541–1547. [DOI] [PubMed] [Google Scholar]
- 122. Malakoutikhah M, Teixidó M, Giralt E. Shuttle‐mediated drug delivery to the brain. Angew Chem Int Ed Engl 2011;50:7998–8014. [DOI] [PubMed] [Google Scholar]
- 123. Torchilin VP. Multifunctional, stimuli‐sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov 2014;13:813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Watkins R, Wu L, Zhang C, Davis RM, Xu B. Natural product‐based nanomedicine: Recent advances and issues. Int J Nanomedicine 2015;10:6055–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Frédérick R, Bruyère C, Vancraeynest C, Reniers J, Meinguet C, Pochet L, Backlund A, Masereel B, Kiss R, Wouters J. Novel trisubstituted harmine derivatives with original in vitro anticancer activity. J Med Chem 2012;55:6489–6501. [DOI] [PubMed] [Google Scholar]
- 126. Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Hunter C, Jenkinson A, Jones D, Kosmidou V, Lugg R, Menzies A, Mironenko T, Parker A, Perry J, Raine K, Richardson D, Shepherd R, Small A, Smith R, Solomon H, Stephens P, Teague J, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Reinhold W, Weinstein JN, Stratton MR, Futreal PA, Wooster R. Mutation analysis of 24 known cancer genes in the NCI‐60 cell line set. Mol Cancer Ther 2006;5:2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shankavaram UT, Reinhold WC, Nishizuka S, Major S, Morita D, Chary KK, Reimers MA, Scherf U, Kahn A, Dolginow D, Cossman J, Kaldjian EP, Scudiero DA, Petricoin E, Liotta L, Lee JK, Weinstein JN. Transcript and protein expression profiles of the NCI‐60 cancer cell panel: an integromic microarray study. Mol Cancer Ther 2007;6:820–832. [DOI] [PubMed] [Google Scholar]
- 128. Blower PE, Verducci JS, Lin S, Zhou J, Chung JH, Dai Z, Liu CG, Reinhold W, Lorenzi PL, Kaldjian EP, Croce CM, Weinstein JN, Sadee W. MicroRNA expression profiles for the NCI‐60 cancer cell panel. Mol Cancer Ther 2007;6:1483–1491. [DOI] [PubMed] [Google Scholar]
- 129. Gholami MA, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M, Kuster B. Global proteome analysis of the NCI‐60 cell line panel. Cell Rep 2013;4:609–620. [DOI] [PubMed] [Google Scholar]
- 130. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: Mechanistic findings and clinical applications. Nat Rev Cancer 2014;14:598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Blandin AF, Renner G, Lehmann M, Lelong‐Rebel I, Martin S, Dontenwill M. β1 integrins as therapeutic targets to disrupt hallmarks of cancer. Front Pharmacol 2015;6:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Decaestecker C, Debeir O, Van Ham P, Kiss R. Can anti‐migratory drugs be screened in vitro? A review of 2D and 3D assays for the quantitative analysis of cell migration. Med Res Rev 2007;27:149–176. [DOI] [PubMed] [Google Scholar]
- 133. Olson OC, Joyce JA. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat Rev Cancer 2015;15:712–729. [DOI] [PubMed] [Google Scholar]
- 134. Kim J, Tanner K. Recapitulating the tumor ecosystem along the metastatic cascade using 3D culture models. Front Oncol 2015;5:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kenny HA, Lal‐Nag M, White EA, Shen M, Chiang CY, Mitra AK, Zhang Y, Curtis M, Schryver EM, Bettis S, Jadhav A, Boxer MB, Li Z, Ferrer M, Lengyel E. Quantitative high throughput screening using a primary human three‐dimensional organotypic culture predicts in vivo efficacy. Nat Commun 2015;6:6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. White EA, Kenny HA, Lengyel E. Three‐dimensional modeling of ovarian cancer. Adv Drug Deliv Rev 2014;79–80:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Mastro AM, Vogler EA. A three‐dimensional osteogenic tissue model for the study of metastatic tumor cell interactions with bone. Cancer Res 2009;69:4097–4100. [DOI] [PubMed] [Google Scholar]
- 138. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, Sgroi D, Kapur R, Shioda T, Ting DT, Ramaswamy S, Getz G, Iafrate AJ, Benes C, Toner M, Maheswaran S, Haber DA. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014;345:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kolostova K, Zhang Y, Hoffman RM, Bobek V. in vitro culture and characterization of human lung cancer circulating tumor cells isolated by size exclusion from an orthotopic nude‐mouse model expressing fluorescent protein. J Fluoresc 2014;24:1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. López‐Lázaro M. A simple and reliable approach for assessing anticancer activity in vitro. Curr Med Chem 2015;22:1324–1334. [DOI] [PubMed] [Google Scholar]
- 142. López‐Lázaro M. Two preclinical tests to evaluate anticancer activity and to help validate drug candidates for clinical trials. Oncoscience 2015;2:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kang J, Hsu CH, Wu Q, Liu S, Coster AD, Posner BA, Altschuler SJ, Wu LF. Improving drug discovery with high‐content phenotypic screens by systematic selection of reporter cell lines. Nat Biotechnol 2016;34:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Davies EJ, Dong M, Gutekunst M, Närhi K, van Zoggel HJ, Blom S, Nagaraj A, Metsalu T, Oswald E, Erkens‐Schulze S, Delgado San Martin JA, Turkki R, Wedge SR, Af Hällström TM, Schueler J, van Weerden WM, Verschuren EW, Barry ST, van der Kuip H, Hickman JA. Capturing complex tumour biology in vitro: Histological and molecular characterisation of precision cut slices. Sci Rep 2015;5:17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cekanova M, Rathore K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des Devel Ther 2014;8:1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Gonzalez C. Drosophila melanogaster: A model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer 2013;13:172–183. [DOI] [PubMed] [Google Scholar]
- 147. Gao G, Chen L, Huang C. Anti‐cancer drug discovery: Update and comparisons in yeast, Drosophila, and zebrafish. Curr Mol Pharmacol 2014;7:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ottewell PD, Coleman RE, Holen I. From genetic abnormality to metastases: Murine models of breast cancer and their use in the development of anticancer therapies. Breast Cancer Res Treat 2006;96:101–113. [DOI] [PubMed] [Google Scholar]
- 149. Dutt A, Wong KK. Mouse models of lung cancer. Clin Cancer Res 2006;12:4396s–4402s. [DOI] [PubMed] [Google Scholar]
- 150. De Minicis S, Kisseleva T, Francis H, Baroni GS, Benedetti A, Brenner D, Alvaro D, Alpini G, Marzioni M. Liver carcinogenesis: Rodent models of hepatocarcinoma and cholangiocarcinoma. Dig Liver Dis 2013;45:450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, Nephew KP, Hales DB, Stack MS. Epithelial ovarian cancer experimental models. Oncogene 2014;33:3619–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Oh BY, Hong HK, Lee WY, Cho YB. Animal models of colorectal cancer with liver metastasis. Cancer Lett 2016; In press. [DOI] [PubMed] [Google Scholar]
- 153. Axiak‐Bechtel SM, Maitz CA, Selting KA, Bryan JN. Preclinical imaging and treatment of cancer: The use of animal models beyond rodents. Q J Nucl Med Mol Imaging 2015;59:303–316. [PubMed] [Google Scholar]
- 154. Lawrence J, Cameron D, Argyle D. Species differences in tumour responses to cancer chemotherapy. Philos Trans R Soc Lond B Biol Sci 2015;370:1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Paterson I, Anderson EA. The renaissance of natural products as drug candidates. Science 2005;310:451–453. [DOI] [PubMed] [Google Scholar]
- 156. Eisner T, Meinwald J. Chemical Ecology. The Chemistry of Biotic Interaction. National Academy Press, Washington, DC: 1995. [Google Scholar]
- 157. Raguso RA, Agrawal AA, Douglas AE, Jander G, Kessler A, Poveda K, Thaler JS. The raison d'être of chemical ecology. Ecology 2015;96:617–630. [DOI] [PubMed] [Google Scholar]
- 158. Carter GT. Natural products and Pharma: Strategic changes spur new opportunities. Nat Prod Rep 2011;28:1783‒1789. [DOI] [PubMed] [Google Scholar]
- 159. Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Drug development from marine natural products. Nat Rev Drug Discov 2010;8:69‒85. [DOI] [PubMed] [Google Scholar]
- 160. Newman DJ. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem 2008;51:2589‒2599. [DOI] [PubMed] [Google Scholar]
- 161. Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 2012;75:311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Montaser R, Luesch H. Marine natural products: A new wave of drugs? Future Med Chem 2011;3:1475‒1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Butler MS, Robertson AAB, Cooper MA, Natural product and natural product derived drugs in clinical trials. Nat Prod Rep 2014;31:1612‒1661. [DOI] [PubMed] [Google Scholar]
- 164. Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti‐cancer agents. Chem Rev 2009;109:3012–3043. [DOI] [PubMed] [Google Scholar]
- 165. Newman DJ, Cragg GM. Marine natural products and related compounds in clinical and advanced preclinical trials. J Nat Prod 2004;67:1216‒1238. [DOI] [PubMed] [Google Scholar]
- 166. Newman DJ, Cragg GM. Marine‐sourced anti‐cancer and cancer pain control agents in clinical and late preclinical development. Mar Drugs 2014;12:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 2016;79:629–661. [DOI] [PubMed] [Google Scholar]
- 168. Cragg GM, Newman DJ. Natural products: A continuing source of novel drug leads. Biochim Biophys Acta 2013;1830:3670–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Harvey AL, Edrada‐Ebel R, Quinn RJ. The re‐emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov 2015;14:111–129. [DOI] [PubMed] [Google Scholar]
- 170. Esch EW, Bahinski A, Huh D. Organs‐on‐chips at the frontiers of drug discovery. Nat Rev Drug Discov 2015;14:248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Beutler JA. Natural products as a foundation for drug discovery. Curr Protoc Pharmacol 2009;46:9.11.1–9.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Butler MS, Newman DJ. Mother nature's gifts to diseases of man: The impact of natural products on anti‐infective, anticholestemics and anticancer drug discovery. In: Petersen F, Amstutz R, Eds.Progress in Drug Research, Vol. 65 2008. p 1–44, Natural Compounds as Drugs Vol 1; Birkhäuser Verlag, Basel, Springer (Switzerland). [DOI] [PubMed] [Google Scholar]
- 173. Butler MS. The role of natural product chemistry in drug discovery. J Nat Prod 2004;67:2141‒2153. [DOI] [PubMed] [Google Scholar]
- 174. Carlson EE. Natural products as chemical probes. ACS Chem Biol 2010;5:639‒653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Clardy J, Walsh C. Lessons from natural molecules. Nature 2004;432:829‒837. [DOI] [PubMed] [Google Scholar]
- 176. Bucar F, Wube A, Schmid M. Natural product isolation—How to get from biological material to pure compounds. Nat Prod Rep 2013;30:525‒545. [DOI] [PubMed] [Google Scholar]
- 177. Blunt JW, Copp BR, Keyzers RA, Munro MH,Prinsep MR. Marine natural products. Nat Prod Rep 2016;33:382–431. [DOI] [PubMed] [Google Scholar]
- 178. Mitsiades CS, Ocio EM, Pandiella A, Maiso P, Gajate C, Garayoa M, Vilanova D, Montero JC, Mitsiades N, McMullan CJ, Munshi NC, Hideshima T, Chauhan D, Aviles P, Otero G, Faircloth G, Mateos MV, Richardson PG, Mollinedo F, San‐Miguel JF, Anderson KC. Aplidin, a marine organism‐derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res 2008;68:5216–5225. [DOI] [PubMed] [Google Scholar]
- 179. Mollo E, Fontana A, Roussis V, Polese G, Amodeo P, Ghiselin MT. Sensing marine biomolecules: smell, taste, and the evolutionary transition from aquatic to terrestrial life. Front Chem 2014;2:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Vinothkumar S, Parameswaran PS. Recent advances in marine drug research. Biotechnol Adv 2013;31:1826‒1845. [DOI] [PubMed] [Google Scholar]
- 181. Mayer AMS, Glaser KB, Cuevas C, Jacobs RS, Kem W, Little DR, McIntosh JM, Newman DJ, Potts BC, Shuster DE. The odyssey of marine pharmaceutical: A current pipeline perspective. Trends Pharmacol Sci 2010;31:255‒265. [DOI] [PubMed] [Google Scholar]
- 182. Li R. Natural product‐based drug discovery. Editorial. Med Res Rev 2016;36:3. [DOI] [PubMed] [Google Scholar]
- 183. Kalimuthu S, Se‐Kwon K. Cell survival and apoptosis signaling as therapeutic target for cancer: Marine bioactive compounds. Int J Mol Sci 2013;14:2334‒2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Mayer AMS, Rodriguez AD, Taglialatela‐Scafati O, Fusetani N. Marine pharmacology in 2009–2011: Marine compounds with antibacterial, antidiabetic, antifungal, anti‐inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar Drugs 2013;11:2510‒2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Hu Y, Chen J, Hu G, Yu J, Zhu X, Lin Y, Chen S, Yuan J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar Drugs 2015;13:202‒221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Skropeta D, Wei L. Recent advances in deep‐sea natural products. Nat Prod Rep 2014;31:999–1025. [DOI] [PubMed] [Google Scholar]
- 187. Proksch P. Defensive roles for secondary metabolites from marine sponges and sponge‐feeding nudibranchs. Toxicon 1994;32:639–655. [DOI] [PubMed] [Google Scholar]
- 188. Cimino G, Ghiselin MT. Chemical defense and the evolution of opisthobranch gastropods. Proc Calif Acad Sci 2009;60:175‒422. [Google Scholar]
- 189. Cheney KL, White A, Mudianta IW, Winters AE, Quezada M, Capon RJ, Mollo E, Garson MJ. Choose your weaponry: Selective storage of a single toxic compound, latrunculin A, by closely related nudibranch molluscs. PLoS One 2016;11:e0145134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Ponder W F. Classification of Mollusca In: Beesley PL, Ross GJB, Wells A, Eds. Mollusca: The Southern Synthesis. Fauna of Australia. . Melbourne: CSIRO Publishing; 1998, p 1–6. [Google Scholar]
- 191. Benkendorff K. Molluscan biological and chemical diversity: Secondary metabolites and medicinal resources produced by marine molluscs. Biol Rev 2010;85:757‒775. [DOI] [PubMed] [Google Scholar]
- 192. Paul VJ, Ritson‐Williams R, Sharp K, Marine chemical ecology in benthic environments. Nat Prod Rep 2011;28:345‒387. [DOI] [PubMed] [Google Scholar]
- 193. Wägele H, Ballesteros M, Avila C. Defensive glandular structures in opisthobranch molluscs—from hystologyy to ecology. Oceanogr Mar Biol Ann Rev 2006;44:197‒276. [Google Scholar]
- 194. Carbone M, Gavagnin M, Haber M, Guo YW, Fontana A, Manzo E, Genta‐Jouve G, Tsoukatou M, Rudman WB, Cimino G, Ghiselin MT, Mollo E. Packaging and delivery of chemical weapons: A defensive trojan horse stratagem in chromodorid nudibranchs. PlosOne 2013;8:e62075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Haber M, Cerfeda S, Carbone C, Calado G, Gaspar H, Neves R, Maharajan V, Cimino G, Gavagnin M, Ghiselin MT, Mollo E. Coloration and defense in the nudibranch gastropod Hypselodoris fontandraui . Biol Bull 2010;218:181–188. [DOI] [PubMed] [Google Scholar]
- 196. Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ, Mooberry SL, Corbett TH, Valeriote FA. Symplostatin 1: A dolastatin 10 analogue from the marine cyanobacterium Symploca hydnoides . J Nat Prod 1998;61:1075–1077. [DOI] [PubMed] [Google Scholar]
- 197. Luesch H, Harrigan G, Goetz G, Horgen F. The cyanobacterial origin of potent anticancer agents originally isolated from sea hares. Curr Med Chem 2002;9:1791–1806. [DOI] [PubMed] [Google Scholar]
- 198. Engene N, Tronholm A, Salvador‐Reyes LA, Luesch H, Paul VJ. Caldora penicillata gen.nov., comb.nov. (Cyanobacteria), a pantropical marine species with biomedical relevance. J Phycol 2015;51:670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Pettit GR, Kamano Y, Fujii Y, Herald CL, Inoue M, Brown P, Gust D, Kitahara K, Schmidt JM, Doubek DL, Michel C. Marine animal biosynthetic constituents for cancer chemotherapy. J Nat Prod 1981;44:482–485. [DOI] [PubMed] [Google Scholar]
- 200. Maderna A, Leverett CA. Recent advances in the development of new auristatins: structural modifications and application in antibody drug conjugates. Mol Pharm 2015;12:1798–1812. [DOI] [PubMed] [Google Scholar]
- 201. Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J Am Chem Soc 1987;109:6883–6885. [Google Scholar]
- 202. Pettit GR, Singh SB, Hogan F, Lloyd‐Williams P, Herald DL, Burkett DD, Clewlow PJ. The absolute configuration and synthesis of natural (‐)‐dolastatin 10. J Am Chem Soc 1989; 111:5463‒5466. [Google Scholar]
- 203. Singh R, Sharma M, Joshi P, Rawat DS. Clinical status of anti‐cancer agents derived from marine sources. Anticancer Agents Med Chem 2008;8:603–617. [PubMed] [Google Scholar]
- 204. Kobayashi M, Natsume T, Tamaoki S, Watanabe J, Asano H, Mikami T, Miyasaka K, Miyazaki K, Gondo M, Sakakibara K, Tsukagoshi S. Antitumor activity of TZT‐1027, a novel dolastatin 10 derivative. Jpn J Cancer Res 1997;88:316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Watanabe J, Minami M, Kobayashi M. Antitumor activity of TZT‐1027 (Soblidotin). Anticancer Res 2006;26:1973–1981. [PubMed] [Google Scholar]
- 206. Toppmeyer DL, Slapak CA, Croop J, Kufe DW. Role of P‐glycoprotein in dolastatin 10 resistance. Biochem Pharmacol 1994;48:609–612. [DOI] [PubMed] [Google Scholar]
- 207. Mooberry SL, Leal RM, Tinley TL, Luesch H, Moore RE, Corbett TH. The molecular pharmacology of symplostatin 1: A new antimitotic dolastatin 10 analog. Int J Cancer 2003;104:512–521. [DOI] [PubMed] [Google Scholar]
- 208. Pathak S, Multani AS, Ozen M, Richardson MA, Newman RA. Dolastatin‐10 induces polyploidy, telomeric associations and apoptosis in a murine melanoma cell line. Oncol Rep 1998;5:373–376. [DOI] [PubMed] [Google Scholar]
- 209. Madden T, Tran HT, Beck D, Huie R, Newman RA, Pusztai L, Wright JJ, Abbruzzese JL. Novel marine‐derived anticancer agents: a phase I clinical, pharmacological, and pharmacodynamic study of dolastatin 10 (NSC 376128) in patients with advanced solid tumors. Clin Cancer Res 2000;6:1293–1301. [PubMed] [Google Scholar]
- 210. Watanabe J, Natsume T, Fujio N, Miyasaka K, Kobayashi M. Induction of apoptosis in human cancer cells by TZT‐1027, an antimicrotubule agent. Apoptosis 2000;5:345–353. [DOI] [PubMed] [Google Scholar]
- 211. Bai R, Pettit GR, Hamel E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem Pharmacol 1990;39:1941–1949. [DOI] [PubMed] [Google Scholar]
- 212. Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann KH, Miller JH, Díaz JF, Steinmetz MO. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem Int Ed 2014;5:1621‒1625. [DOI] [PubMed] [Google Scholar]
- 213. Field JJ, Waight AB, Senter PD. A previously undescribed tubulin binder. Proc Natl Acad Sci USA 2014;111:13684–13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Mohammad RM, Pettit GR, Almatchy VP, Wall N, Varterasian M, Al‐Katib A. Synergistic interaction of selected marine animal anticancer drugs against human diffuse large cell lymphoma. Anticancer Drugs 1998;9:149–156. [DOI] [PubMed] [Google Scholar]
- 215. Kalemkerian GP, Ou X, Adil MR, Rosati R, Khoulani MM, Madan SK, Pettit GR. Activity of dolastatin 10 against small‐cell lung cancer in vitro and in vivo: Induction of apoptosis and bcl‐2 modification. Cancer Chemother Pharmacol 1999;43:507–515. [DOI] [PubMed] [Google Scholar]
- 216. Aherne GW, Hardcastle A, Valenti M, Bryant A, Rogers P, Pettit GR, Srirangam JK, Kelland LR. Antitumour evaluation of dolastatins 10 and 15 and their measurement in plasma by radioimmunoassay. Cancer Chemother Pharmacol 1996;38:225–232. [DOI] [PubMed] [Google Scholar]
- 217. Flahive E, Srirangam J. The dolastatins. Novel antitumor agents from Dolabella auricularia In: Cragg GM, Kingston DGI, Newmann DJ, Eds. Anticancer Agents from Natural Products. 2nd ed Boca Raton FL: CRC Press, Taylor & Francis Group; 2012. p 263–289. [Google Scholar]
- 218. Miyazaki K, Kobayashi M, Natsume T, Gondo M, Mikami T, Sakakibara K, Tsukagoshi S. Synthesis and antitumor activity of novel dolastatin 10 analogs. Chem Pharm Bull 1995;43:1706–1718. [DOI] [PubMed] [Google Scholar]
- 219. Martins A, Vieira H, Gaspar H, Santos S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar Drugs 2014;12:1066–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Xiao Z, Morris‐Natschke S, Lee KH. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med Res Rev 2016;36:32–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Naumovski L, Junutula JR. Glembatumumab vedotin, a conjugate of an anti‐glycoprotein non‐metastatic melanoma protein B mAb and monomethyl auristatin E for the treatment of melanoma and breast cancer. Curr Opin Mol Ther 2010;12:248–257. [PubMed] [Google Scholar]
- 222. Newmann DJ, Cragg GM. Drugs and drug candidates from marine sources: An assessment of the current “state of play.” Planta Med 2016;82:775–789. [DOI] [PubMed] [Google Scholar]
- 223. Pettit GR, Kamano Y, Dufresne C, Cerny RL, Herald CL, Schmidt JM. Isolation and structure of the cytostatic linear depsipeptide dolastatin 15. J Org Chem 1989;54:6005–6006. [Google Scholar]
- 224. Pettit GR, Herald DL, Singh SB, Thornton TJ, Mullaney JT. Antineoplastic agents. 220. Synthesis of natural (‐)‐dolastatin 15. J Am Chem Soc 1991;113:6692–6693. [Google Scholar]
- 225. Akaji K, Hayashi Y, Kiso Y, Kuriyama N. Convergent synthesis of dolastatin 15 by solid phase coupling of an N‐methylamino acid. J Org Chem 1999;64:405–411. [Google Scholar]
- 226. Bai R, Friedman SJ, Pettit GR, Hamel E. Dolastatin 15, a potent antimitotic depsipeptide derived from Dolabella auricularia. Interaction with tubulin and effects of cellular microtubules. Biochem Pharmacol 1992;43:2637–2645. [DOI] [PubMed] [Google Scholar]
- 227. Lopus M. Mechanism of mitotic arrest induced by dolastatin 15 involves loss of tension across kinetochore pairs. Mol Cell Biochem 2013;382:93–102. [DOI] [PubMed] [Google Scholar]
- 228. Beckwith M, Urba WJ, Longo DL. Growth inhibition of human lymphoma cell lines by the marine products, dolastatins 10 and 15. J Natl Cancer Inst 1993;85:483–488. [DOI] [PubMed] [Google Scholar]
- 229. Ali MA, Rosati R, Pettit GR, Kalemkerian GP. Dolastatin 15 induces apoptosis and BCL‐2 phosphorylation in small cell lung cancer cell lines. Anticancer Res 1998;18:1021–1026. [PubMed] [Google Scholar]
- 230. Sato M, Sagawa M, Nakazato T, Ikeda Y, Kizaki M. A natural peptide, dolastatin 15, induces G2/M cell cycle arrest and apoptosis of human multiple myeloma cells. Int J Oncol 2007;30:1453–1459. [PubMed] [Google Scholar]
- 231. Bai R, Edler MC, Bonate PL, Copeland TD, Pettit GR, Ludueña RF, Hamel E. Intracellular activation and deactivation of tasidotin, an analog of dolastatin 15: Correlation with cytotoxicity. Mol Pharmacol 2009;75:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Ray A, Okouneva T, Manna T, Miller HP, Schmid S, Arthaud L, Luduena R, Jordan MA, Wilson L. Mechanism of action of the microtubule‐targeted antimitotic depsipeptide tasidotin (formerly ILX651) and its major metabolite tasidotin C‐carboxylate. Cancer Res 2007;67:3767–3776. [DOI] [PubMed] [Google Scholar]
- 233. Garg V, Zhang W, Gidwani P, Kim M, Kolb EA. Preclinical analysis of tasidotin HCl in Ewing's sarcoma, rhabdomyosarcoma, synovial sarcoma, and osteosarcoma. Clin Cancer Res 2007;13:5446–5454. [DOI] [PubMed] [Google Scholar]
- 234. Bonate PL, Beyerlein D, Crawford J, Roth S, Krumbholz R, Schmid S. Pharmacokinetics in mice implanted with xenografted tumors after intravenous administration of tasidotin (ILX651) or its carboxylate metabolite. AAPS J 2007;9:E378–E387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Pettit GR, Kamano Y, Brown P, Gust D, Inoue M, Herald CL. Structure of the cyclic peptide dolastatin 3 from Dolabella auricularia . J Am Chem Soc 1982;104:905–907. [Google Scholar]
- 236. Pettit GR, Kamano Y, Holzapfel CW, van Zyl WJ, Tuinman AA, Herald CL, Baczynskyj L, Schmidt JM. The structure and synthesis of dolastatin 3. J Am Chem Soc 1987;109:7581–7582. [Google Scholar]
- 237. Mitchell SS, Faulkner DJ, Rubins K, Bushman FD. Dolastatin 3 and two novel cyclic peptides from a Palauan collection of Lyngbya majuscula . J Nat Prod 2000;63:279–282. [DOI] [PubMed] [Google Scholar]
- 238. Pettit GR, Kamano Y, Kizu H, Dufresne C, Herald CL, Bontems RJ, Schmidt JM, Boettner FE, Nieman RA. Isolation and structure of the cell growth inhibitory depsipeptides dolastatins 11 and 12. Heterocycles 1989;28:553‒558. [Google Scholar]
- 239. Pettit GR, Kamano Y, Herald CL, Dufresne C, Cerny RL, Herald DL, Schmidt JM, Kizu H. Isolation and structure of the cytostatic depsipeptide dolastatin 13 from the sea hare Dolabella auricularia . J Am Chem Soc 1989;111:5015‒5017. [Google Scholar]
- 240. Pettit GR, Kamano Y, Herald CL, Dufresne C, Bates RB, Schmidt JM, Cerny RL, Kizu H. Antineoplastic agents. 190. Isolation and structure of the cyclodepsipeptide dolastatin 14. J Org Chem 1990;55:2989‒2990. [Google Scholar]
- 241. Harrigan GG, Yoshida WY, Moore RE, Nagle DG, Park PU, Biggs J, Paul VJ, Mooberry SL, Corbett TH, Valeriote FA. Isolation, structure determination, and biological activity of dolastatin 12 and lyngbyastatin 1 from Lyngbya majuscula/Schizothrix calcicola cyanobacterial assemblages. J Nat Prod 1998;61:1221–1225. [DOI] [PubMed] [Google Scholar]
- 242. Ali MA, Bates RB, Crane ZD, Dicus CW, Gramme MR, Hamel E, Marcischak J, Martinez DS, McClure KJ, Nakkiew P, Pettit GR, Stessman CC, Sufi BA, Yarick GV. Dolastatin 11 conformations, analogues and pharmacophore. Bioorg Med Chem 2005;13:4138–4152. [DOI] [PubMed] [Google Scholar]
- 243. Thornburg CC, Thimmaiah M, Shaala LA, Hau AM, Malmo JM, Ishmael JE, Youssef DT, McPhail KL. Cyclic depsipeptides, grassypeptolides D and E and Ibu‐epidemethoxylyngbyastatin 3, from a Red Sea Leptolyngbya cyanobacterium . J Nat Prod 2011;74:1677–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Bai R, Verdier‐Pinard P, Gangwar S, Stessman CC, Mcclure KJ, Sausville EA, Pettit GR, Bates RB, Hamel E. Dolastatin 11, a marine depsipeptide, arrests cells at cytokinesis and induces hyperpolymerization of purified actin. Mol Pharmacol 2001;59:462‒469. [DOI] [PubMed] [Google Scholar]
- 245. Wehland J, Osborn M, Weber K. Phalloidin‐induced actin polymerization in the cytoplasm of cultured cells interferes with cell locomotion and growth. Proc Natl Acad Sci USA 1977;74:5613–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol 1987;105:1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Oda T, Crane ZD, Dicus CW, Sufi BA, Bates RB. Dolastatin 11 connects two long‐pitch strands in F‐actin to stabilize microfilaments. J Mol Biol 2003;328:319–324. [DOI] [PubMed] [Google Scholar]
- 248. Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev 2009;28:5–14. [DOI] [PubMed] [Google Scholar]
- 249. Pettit GR, Xu JP, Hogan F, Williams MD, Doubek DL, Schmidt JM, Cerny RL, Boyd MR. Isolation and structure of the human cancer cell growth inhibitory cyclodepsipeptide dolastatin 16. J Nat Prod 1997;60:752‒754. [DOI] [PubMed] [Google Scholar]
- 250. Pettit GR, Smith TH, Xu JP, Herald DL, Flahive EJ, Anderson CR, Belcher PE, Knight JC. Antineoplastic agents. 590. X‐ray crystal structure of dolastatin 16 and syntheses of the dolamethylleuine and dolaphenvaline units. J Nat Prod 2011;74:1003‒1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Nogle LM, Gerwick WH. Isolation of four new cyclic depsipeptides, antanapeptins A‐D, and dolastatin 16 from a Madagascan collection of Lyngbya majuscula . J Nat Prod 2002;6:21–24. [DOI] [PubMed] [Google Scholar]
- 252. Salvador LA, Biggs JS, Paul VJ, Luesch H. Veraguamides A‐G, cyclic hexadepsipeptides from a dolastatin 16‐producing cyanobacterium Symploca cf. hydnoides from Guam. J Nat Prod 2011;74:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro‐Wolff A, Gray‐Goodrich M, Campbell H, Mayo J, Boyd M. Feasibility of a high‐flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–766. [DOI] [PubMed] [Google Scholar]
- 254. Pettit GR, Smith TH, Arce PM, Flahive EJ, Anderson CR, Chapuis JC, Xu JP, Groy TL, Belcher PE, Macdonald CB. Antineoplastic agents. 599. Total synthesis of dolastatin 16. J Nat Prod 2015;78:476–485. [DOI] [PubMed] [Google Scholar]
- 255. Pettit GR, Xu JP, Hogan F, Cerny RL. Isolation and structure of dolastatin 17. Heterocycles 1998;47:491‒496. [Google Scholar]
- 256. Pettit GR, Xu JP, Williams MD, Hogan F, Schmidt JM, Cerny RL. Antineoplastic agents 370. Isolation and structure of dolastatin 18. Bioorg Chem Med Lett 1997;7:827‒832. [Google Scholar]
- 257. Sone H, Nemoto T, Ishiwata H, Ojika M, Yamada K. Isolation, structure, and synthesis of dolastatin D, a cytotoxic cyclic depsipeptide from the sea hare Dolabella auricularia . Tetrahedron Lett 1993;34:8449‒8452. [Google Scholar]
- 258. Mutou T, Kondo T, Ojika M, Yamada K. Isolation and stereostructures of dolastatin G and nordolastatin G, cytotoxic 35‐membered cyclodepsipeptides from the Japanese sea hare Dolabella auricularia . J Org Chem 1996;61:6340‒6345. [DOI] [PubMed] [Google Scholar]
- 259. Sone H, Shibata T, Fujita T, Ojika M, Yamada K. Dolastatin H and isodolastatin H, potent cytotoxic peptides from the sea hare Dolabella auricularia. Isolation, stereostructures, and synthesis. J Am Chem Soc 1996;118:1874‒1880. [Google Scholar]
- 260. Kobayashi S, Kobayashi J, Yazaki R, Ueno M. Toward the total synthesis of onchidin, a cytotoxic cyclic depsipeptide from a mollusc. Chem Asian J 2007;2:135–144. [DOI] [PubMed] [Google Scholar]
- 261. Pettit GR, Xu JP, Doubek DL, Chapuis JC, Schmidt JM. Antineoplastic agents. 510. Isolation and structure of dolastatin 19 from the gulf of California sea hare Dolabella auricularia . J Nat Prod 2004;67:1252‒1255. [DOI] [PubMed] [Google Scholar]
- 262. Hamann MT, Scheuer, PJ . Kahalalide F: a bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J Am Chem Soc 1993;115:5825‒5826. [Google Scholar]
- 263. Gao J, Hamann MT. Chemistry and biology of kahalalides. Chem Rev 2011;111:3208‒3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Davis J, Fricke WF, Hamann MT, Esquenazi E, Dorrestein PC, Hill RT. Characterization of the bacterial community of the chemically defended Hawaiian sacoglossan Elysia rufescens . Appl Environ Microbiol 2013;79:7073–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Weerasinghe P, Buja LM. Oncosis: An important non‐apoptotic mode of cell death. Exp Mol Pathol 2012;93:302–308. [DOI] [PubMed] [Google Scholar]
- 266. Mijatovic T, Mathieu V, Gaussin JF, De Nève N, Ribaucour F, Van Quaquebeke E, Dumont P, Darro F, Kiss R. Cardenolide‐induced lysosomal membrane permeabilization demonstrates therapeutic benefits in experimental human non‐small cell lung cancers. Neoplasia 2006;8:402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Hämälistö S, Jäättelä M. Lysosomes in cancer—living on the edge (of the cell). Curr Opin Cell Biol 2016;39:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Solitro AR, MacKeigan JP. Leaving the lysosome behind: Novel developments in autophagy inhibition. Future Med Chem 2016;8:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Suárez Y, González L, Cuadrado A, Berciano M, Lafarga M, Muñoz A. Kahalalide F, a new marine‐derived compound, induces oncosis in human prostate and breast cancer cells. Mol Cancer Ther 2003;2:863–872. [PubMed] [Google Scholar]
- 270. García‐Rocha M, Bonay P, Avila J. The antitumoral compound kahalalide F acts on cell lysosomes. Cancer Lett 1996;99:43–50. [DOI] [PubMed] [Google Scholar]
- 271. Janmaat ML, Rodriguez JA, Jimeno J, Kruyt FA, Giaccone G. Kahalalide F induces necrosis‐like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol Pharmacol 2005;68:502–510. [DOI] [PubMed] [Google Scholar]
- 272. Appert‐Collin A, Hubert P, Crémel G, Bennasroune A. Role of ErbB Receptors in cancer cell migration and invasion. Front Pharmacol 2015;6:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med 2016;67:11–28. [DOI] [PubMed] [Google Scholar]
- 274. Pardo B, Paz‐Ares L, Tabernero J, Ciruelos E, García M, Salazar R, López A, Blanco M, Nieto A, Jimeno J, Izquierdo MA, Trigo JM. Phase I clinical and pharmacokinetic study of kahalalide F administered weekly as a 1‐hour infusion to patients with advanced solid tumors. Clin Cancer Res. 2008;14:1116–1123. [DOI] [PubMed] [Google Scholar]
- 275. Ling YH, Aracil M, Jimeno J, Perez‐Soler R, Zou Y. Molecular pharmacodynamics of PM02734 (elisidepsin) as single agent and in combination with erlotinib; synergistic activity in human non‐small cell lung cancer cell lines and xenograft models. Eur J Cancer 2009;45:1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Rademaker‐Lakhai JM, Horenblas S, Meinhardt W, Stokvis E, de Reijke TM, Jimeno JM, Lopez‐Lazaro L, Lopez Martin JA, Beijnen JH, Schellens JH. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin Cancer Res 2005;11:1854–1862. [DOI] [PubMed] [Google Scholar]
- 277. Sewell JM, Mayer I, Langdon SP, Smyth JF, Jodrell DI, Guichard SM. The mechanism of action of Kahalalide F: Variable cell permeability in human hepatoma cell lines. Eur J Cancer 2005;41:1637–1644. [DOI] [PubMed] [Google Scholar]
- 278. Miguel‐Lillo B, Valenzuela B, Peris‐Ribera JE, Soto‐Matos A, Pérez‐Ruixo JJ. Population pharmacokinetics of kahalalide F in advanced cancer patients. Cancer Chemother Pharmacol 2015;76:365–374. [DOI] [PubMed] [Google Scholar]
- 279. Martín‐Algarra S, Espinosa E, Rubió J, López JJ, Manzano JL, Carrión LA, Plazaola A, Tanovic A, Paz‐Ares L. Phase II study of weekly Kahalalide F in patients with advanced malignant melanoma. Eur J Cancer 2009;45:732–735. [DOI] [PubMed] [Google Scholar]
- 280. Shilabin AG, Hamann MT. in vitro and in vivo evaluation of select kahalalide F analogs with antitumor and antifungal activities. Bioorg Med Chem 2011;19:6628–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Serova M, de Gramont A, Bieche I, Riveiro ME, Galmarini CM, Aracil M, Jimeno J, Faivre S, Raymond E. Predictive factors of sensitivity to elisidepsin, a novel Kahalalide F‐derived marine compound. Mar Drugs 2013;11:944–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Goldwasser F, Faivre S, Alexandre J, Coronado C, Fernández‐García EM, Kahatt CM, Paramio PG, Dios JL, Miguel‐Lillo B, Raymond E. Phase I study of elisidepsin (Irvalec®) in combination with carboplatin or gemcitabine in patients with advanced malignancies. Invest New Drugs 2014;32:500–509. [DOI] [PubMed] [Google Scholar]
- 283. Ratain MJ, Geary D, Undevia SD, Coronado C, Alfaro V, Iglesias JL, Schilsky RL, Miguel‐Lillo B. First‐in‐human, phase I study of elisidepsin (PM02734) administered as a 30‐min or as a 3‐hour intravenous infusion every three weeks in patients with advanced solid tumors. Invest New Drugs 2015;33:901–910. [DOI] [PubMed] [Google Scholar]
- 284. Herrero AB, Astudillo AM, Balboa MA, Cuevas C, Balsinde J, Moreno S. Levels of SCS7/FA2H‐mediated fatty acid 2‐hydroxylation determine the sensitivity of cells to antitumor PM02734. Cancer Res 2008;68:9779–9787. [DOI] [PubMed] [Google Scholar]
- 285. Váradi T, Roszik J, Lisboa D, Vereb G, Molina‐Guijarro JM, Galmarini CM, Szöllosi J, Nagy P. ErbB protein modifications are secondary to severe cell membrane alterations induced by elisidepsin treatment. Eur J Pharmacol 2011;667:91–99. [DOI] [PubMed] [Google Scholar]
- 286. Molina‐Guijarro JM, García C, Macías Á, García‐Fernández LF, Moreno C, Reyes F, Martínez‐Leal JF, Fernández R, Martínez V, Valenzuela C, Lillo MP, Galmarini CM. Elisidepsin interacts directly with glycosylceramides in the plasma membrane of tumor cells to induce necrotic cell death. PLoS One 2015;10:e0140782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Király A, Váradi T, Hajdu T, Rühl R, Galmarini CM, Szöllősi J, Nagy P. Hypoxia reduces the efficiency of elisidepsin by inhibiting hydroxylation and altering the structure of lipid rafts. Mar Drugs 2013;11;4858–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Teixidó C, Marés R, Aracil M, Ramón Y, Cajal S, Hernández‐Losa J. Epithelial‐mesenchymal transition markers and HER3 expression are predictors of elisidepsin treatment response in breast and pancreatic cancer cell lines. PLoS One 2013;8:e53645. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 289. Aneta G, Tomáš V, Daniel R, Jan B. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget 2016;7:25022–25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Salazar R, Jones RJ, Oaknin A, Crawford D, Cuadra C, Hopkins C, Gil M, Coronado C, Soto‐Matos A, Cullell‐Young M, Iglesias Dios JL, Evans TR. A phase I and pharmacokinetic study of elisidepsin (PM02734) in patients with advanced solid tumors. Cancer Chemother Pharmacol 2012;70:673–681. [DOI] [PubMed] [Google Scholar]
- 291. Petty R, Anthoney A, Metges JP, Alsina M, Gonçalves A, Brown J, Montagut C, Gunzer K, Laus G, Iglesias Dios JL, Miguel‐Lillo B, Bohan P, Salazar R. Phase Ib/II study of elisidepsin in metastatic or advanced gastroesophageal cancer (IMAGE trial). Cancer Chemother Pharmacol 2016;77:819–827. [DOI] [PubMed] [Google Scholar]
- 292. Ortega MJ, Zubìa E, Salvá J. New polyhalogenated monoterpenes from the sea hare Aplysia punctata . J Nat Prod 1997;60:482‒484. [DOI] [PubMed] [Google Scholar]
- 293. Wessels M, König GM, Wright AD. New natural product isolation and comparison of the secondary metabolite content of three distinct samples of the sea hare Aplysia dactylomela from Tenerife. J Nat Prod 2000;63:920‒928. [DOI] [PubMed] [Google Scholar]
- 294. Gavagnin M, Mollo E, Montanaro D, Ortea J, Cimino G. Chemical studies of caribbean sacoglossans: Dietary relationships with green algae and ecological implications. J Chem Ecol 2000;26:1563‒1578. [Google Scholar]
- 295. Ciavatta ML, Lopez‐Gresa MP, Gavagnin M, Manzo E, Mollo E, D'Souza L, Cimino G. New caulerpenyne‐derived metabolites of an Elysia sacoglossan from the south Indian coast. Molecules 2006;11:806‒816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Fischel JL, Lemee R, Formento P, Caldani C, Moll JL, Pesando D, Meinesz A, Grelier P, Pietra F, Guerriero A, Milano G. Cell growth inhibitory effects of caulerpenyne, a sesquiterpenoid from the marine algae Caulerpa taxifolia . Anticancer Res 1995;15:2155‒2160. [PubMed] [Google Scholar]
- 297. Cavas L, Baskin Y, Yurdakoc K, Olgun N. Antiproliferative and newly contributed apoptotic activities from an invasive marine alga: Caulerpa racemosa var. cylindracea . J Exp Mar Biol Ecol 2006;339:111‒119. [Google Scholar]
- 298. Barbier P, Guise S, Huitorel P, Amade P, Pesando D, Briand C, Peyrot V. Caulerpenyne from Caulerpa taxifolia has an antiproliferative activity on tumor cell line SK‐N‐SH and modifies the microtubule network. Life Sci 2001;70:415–429. [DOI] [PubMed] [Google Scholar]
- 299. Parent‐Massin D, Fournier V, Amade P, Lemée R, Durand‐Clément M, Delescluse C, Pesando D. Evaluation of the toxicological risk to humans of caulerpenyne using human hematopoietic progenitors, melanocytes, and keratinocytes in culture. J Toxicol Environ Health 1996;47:47–59. [DOI] [PubMed] [Google Scholar]
- 300. Bourdron J, Barbier P, Allegro D, Villard C, Lafitte D, Commeiras L, Parrain JL, Peyrot V. Caulerpenyne binding to tubulin: Structural modifications by a non‐conventional pharmacological agent. Med Chem 2009;5:182–190. [DOI] [PubMed] [Google Scholar]
- 301. Pesando D, Pesci‐Bardon C, Huitorel P, Girard JP. Caulerpenyne blocks MBP kinase activation controlling mitosis in sea urchin eggs. Eur J Cell Biol 1999;78:903–910. [DOI] [PubMed] [Google Scholar]
- 302. Yamamura S, Hirata Y. Structures of aplysin and aplysinol, naturally occurring bromo‐compounds. Tetrahedron 1963;19:1485‒1496. [Google Scholar]
- 303. Ryu G, Park SH, Choi BW, Lee NH, Hwang HJ, Ryu SY, Lee BH. Cytotoxic activities of brominated sesquiterpenes from the red alga Laurencia okamurae . Nat Prod Sci 2002;8:103‒107. [Google Scholar]
- 304. Gong AJ, Gong LL, Yao WC, Ge N, Lu LX, Liang H. Aplysin induces apoptosis in glioma cells through HSP90/AKT pathway. Exp Biol Med 2015;240:639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Pettit GR, Herald CH, Allen MS, Von Dreele RB, Vanell LD, Kao JPY, Blake W. The isolation and structure of aplysistatin. J Am Chem Soc 1977;99:262‒263. [DOI] [PubMed] [Google Scholar]
- 306. Sims JJ, Lin GHY, Wing RM. Marine natural products: Elatol, a halogenated sesquiterpene alcohol from the red alga Laurencia elata . Tetrahedron Lett 1974;39:3487–3490. [Google Scholar]
- 307. Lang KL, Silva IT, Zimmermann LA, Lhullier C, Mañalich Arana MV, Palermo JA, Falkenberg M, Simões CM, Schenkel EP, Durán FJ. Cytotoxic activity of semi‐synthetic derivatives of elatol and isoobtusol. Mar Drugs 2012;10:2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Gonzalez AG, Darias J, Diaz A, Fourneron JD, Martin JD, Perez C. Evidence for the biogenesis of halogenated chamigrenes from the red alga Laurencia obtusa . Tetrahedron Lett 1976;17:3051–3054. [Google Scholar]
- 309. Dias T, Brito I, Moujir L, Paiz N, Darias J, Cueto M. Cytotoxic sesquiterpenes from Aplysia dactylomela . J Nat Prod 2005;68:1677‒1679. [DOI] [PubMed] [Google Scholar]
- 310. Campos A, Souza CB, Lhullier C, Falkenberg M, Schenkel EP, Ribeiro‐do‐Valle RM, Siqueira JM. Anti‐tumour effects of elatol, a marine derivative compound obtained from red algae Laurencia microcladia . J Pharm Pharmacol 2012;64:1146–1154. [DOI] [PubMed] [Google Scholar]
- 311. Mathieu V, Le Mercier M, De Neve N, Sauvage S, Gras T, Roland I, Lefranc F, Kiss R. Galectin‐1 knockdown increases sensitivity to temozolomide in a B16F10 mouse metastatic melanoma model. J Invest Dermatol 2007;127:2399–2410. [DOI] [PubMed] [Google Scholar]
- 312. Jaisamut S, Prabpai S, Tancharoen C, Yuenyongsawad S, Hannongbua S, Kongsaeree P, Plubrukarn A. Bridged tricyclic sesquiterpenes from the tubercle nudibranch Phyllidia coelestis Bergh. J Nat Prod 2013;76:2158‒2161. [DOI] [PubMed] [Google Scholar]
- 313. Tanaka J, Higa T. Two new cytotoxic carbonimidic dichlorides from the nudibranch Reticulidia fungia . J Nat Prod 1999;62:1339‒1340. [DOI] [PubMed] [Google Scholar]
- 314. Hegazy MEF, Moustfa AY, Mohamed AEHH, Alhammady MA, Elbehairi SEI, Ohta S, Paré PW. New cytotoxic halogenated sesquiterpenes from the Egyptian sea hare Aplysia oculifera . Tetrahedron Lett 2014;55:1711‒1714. [Google Scholar]
- 315. Schmitz FJ, Michaud DP, Schmidt PG. Marine natural products: Parguerol, deoxyparguerol, and isoparguerol. new brominated diterpenes with modified pimarane skeletons from the sea hare Aplysia dactylomela . J Am Chem Soc 1982;104:6415‒6423. [Google Scholar]
- 316. Awad NE. Bioactive brominated diterpenes from the marine red alga Jania rubens (L.) Lamx. Phytother Res 2004;18:275‒279. [DOI] [PubMed] [Google Scholar]
- 317. Darro F, Decaestecker C, Gaussin JF, Mortier S, Van Ginckel R, Kiss R. Are syngeneic mouse tumor models still valuable experimental models in the field of anti‐cancer drug discovery? Int J Oncol 2005;27:607‒616. [PubMed] [Google Scholar]
- 318. Miyamoto T, Sakamoto K, Arao K, Komori T, Higuchi R, Sasaki T. Dorisenones, cytotoxic spongian diterpenoids, from the nudibranch Chromodoris obsoleta . Tetrahedron 1996;52:8187‒8198. [Google Scholar]
- 319. Diyabalanage T, Iken KB, McClintock JB, Amsler CD, Baker BJ. Palmadorins A‐C, diterpene glycerides from the Antarctic nudibranch Austrodoris kerguelenensis . J Nat Prod 2010;73:416‒421. [DOI] [PubMed] [Google Scholar]
- 320. Maschek JA, Mevers E, Diyabalanage T, Chen L, Ren Y, McClintock JB, Amsler CD, Wu J, Baker BJ. Palmadorin chemodiversity from the Antarctic nudibranch Austrodoris kerguelenensis and inhibition of Jak2/STAT5‐dependent HEL leukemia cells. Tetrahedron 2012;68:9095‒9104. [Google Scholar]
- 321. Malochet‐Grivois C, Cotelle P, Biard JF, Henichart JP, Débitus C, Roussakis C, Verbist JF. Dichlorolissoclimide, a new cytotoxic labdane derivative from Lissoclinum voeltzkowi Michaelson (Urochordata).Tetrahedron Lett 1991;32:6701–6702. [Google Scholar]
- 322. Toupet L, Biard JF, Verbist JF. Dichlorolissoclimide from Lissoclinum voeltzkowi Michaelson (Urochordata): Crystal structure and absolute stereochemistry. J Nat Prod 1996;59:1203–1204. [Google Scholar]
- 323. Biard JF, Malochet‐Grivois C, Roussakis C, Cotelle P, Henichart JP, Débitus C, Verbist JF. Lissoclimides, cytotoxic diterpenes from Lissoclinum voeltzkowi Michaelsen. Nat Prod Lett 1994;4:43–50. [Google Scholar]
- 324. Uddin MJ, Kokubo S, Suenaga K, Ueda K, Uemura D. Haterumaimides A‐E, five new dichlorolissoclimide‐type diterpenoids from an ascidian, Lissoclinum sp. Heterocycles 2001;54:1039–1047. [Google Scholar]
- 325. Uddin MJ, Kokubo S, Suenaga K, Ueda K, Uemura D. Haterumaimides F‐I, four new cytotoxic diterpene alkaloids from an ascidian Lissoclinum species. J Nat Prod 2001;64:1169–1173. [DOI] [PubMed] [Google Scholar]
- 326. Uddin MJ, Kokubo S, Suenaga K, Ueda K, Uemura D. Haterumaimides J and K, potent cytotoxic diterpene alkaloids from the ascidian Lissoclinum species. Chem Lett 2002;10:1028–1029. [DOI] [PubMed] [Google Scholar]
- 327. Uddin J, Ueda K, Siwu ER, Kita M, Uemura D. Cytotoxic labdane alkaloids from an ascidian Lissoclinum sp.: Isolation, structure elucidation, and structure‐activity relationship. Bioorg Med Chem 2006;14:6954–6961. [DOI] [PubMed] [Google Scholar]
- 328. Fu X, Palomar AJ, Hong EP, Schmitz FJ, Valeriote FA. Cytotoxic lissoclimide‐type diterpenes from the molluscs Pleurobranchus albiguttatus and Pleurobranchus forskalii . J Nat Prod 2004;67:1415‒1418. [DOI] [PubMed] [Google Scholar]
- 329. Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev Res 1995;34:91‒109. [Google Scholar]
- 330. Valeriote F, Grieshaber CK, Media J, Pietraszkewicz H, Hoffman J, Pan M, McLaughin S. Discovery and development of anticancer agents from plants. J Exp Ther Oncol 2002;2:228–236. [DOI] [PubMed] [Google Scholar]
- 331. Gonzalez MA, Romero D, Zapata B, Betancur‐Galvis L. First synthesis of lissoclimide‐type alkaloids. Lett Org Chem 2009;6:289–292. [Google Scholar]
- 332. Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. Site‐selective aliphatic C‐H chlorination using N‐chloroamides enables a synthesis of chlorolissoclimide. J Am Chem Soc 2016;138:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Malochet‐Grivois C, Roussakis C, Robillard N, Biard JF, Riou D, Débitus C, Verbist JF. Effects in vitro of two marine substances, chlorolissoclimide and dichlorolissoclimide, on a non‐small‐cell bronchopulmonary carcinoma line (NSCLC‐N6). Anticancer Drug Des 1992;7:493–502. [PubMed] [Google Scholar]
- 334. Robert F, Gao HQ, Donia M, Merrick WC, Hamman MT, Pelletier J. Chlorolissoclimides: New inhibitors of eukaryotic protein synthesis. RNA 2006;12:717‒724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Alvandi F, Kwitkowski VE, Ko CW, Rothmann MD, Ricci S, Saber H, Ghosh D, Brown J, Pfeiler E, Chikhale E, Grillo J, Bullock J, Kane R, Kaminskas E, Farrell AT, Pazdur R. U.S. Food and Drug Administration approval summary: Omacetaxine mepesuccinate as treatment for chronic myeloid leukemia. Oncologist 2014;19:94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Heiblig M, Sobh M, Nicolini FE. Subcutaneous omacetaxine mepesuccinate in patients with chronic myeloid leukemia in tyrosine kinase inhibitor‐resistant patients: Review and perspectives. Leuk Res 2014;38:1145–1153. [DOI] [PubMed] [Google Scholar]
- 337. Kantarjian H, O'Brien S, Jabbour E, Barnes G, Pathak A, Cortes J. Effectiveness of homoharringtonine (omacetaxine mepesuccinate) for treatment of acute myeloid leukemia: A meta‐analysis of Chinese studies. Clin Lymphoma Myeloma Leuk 2015;15:13–21. [DOI] [PubMed] [Google Scholar]
- 338. Miyamoto T, Sakamoto K, Amano H, Arakawa Y, Nagarekawa Y, Komori T, Higuchi R, Sasaki T. New cytotoxic sesterterpenoids from the nudibranch Chromodoris inornata . Tetrahedron 1999;55:9133‒9142. [Google Scholar]
- 339. Evidente A, Kornienko A, Lefranc F, Cimmino A, Dasari R, Evidente M, Mathieu V, Kiss R. Sesterterpenoids with anticancer activity. Curr Med Chem 2015;22:3502–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Suenaga K, Shibata T, Takada N, Kigoshi H, Yamada K. Aurilol, a cytotoxic bromotriterpene isolated from the sea hare Dolabella auricularia . J Nat Prod 1998;61:515‒518. [Google Scholar]
- 341. Morimoto Y, Nishikawa Y, Takaishi M. Total synthesis and complete assignment of the stereostructure of a cytotoxic bromotriterpene polyether (+)‐aurilol. J Am Chem Soc 2005;127:5806‒5807. [DOI] [PubMed] [Google Scholar]
- 342. Vera B, Rodríguez AD, Avilés E, Ishikawa Y. Aplysqualenols A and B: Squalene‐derived polyethers with antitumoral and antiviral activity from the caribbean sea slug Aplysia dactylomela . Eur J Org Chem 2009;5327‒5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Vera B, Rodríguez A., AD La Clair JJ. Aplysqualenol A binds to the light chain of dynein type 1 (DYNLL1). Angew Chem Int Ed 2011;50:8134‒8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Zhu G, Yang F, Balachandran R, Hççk P, Vallee RB, Curran DP, Day BW. Synthesis and biological evaluation of purealin and analogues as cytoplasmic dynein heavy chain inhibitors. J Med Chem 2006;49:2063‒2076. [DOI] [PubMed] [Google Scholar]
- 345. Alberti C. Cytoskeleton structure and dynamic behaviour: Quick excursus from basic molecular mechanisms to some implications in cancer chemotherapy. Eur Rev Med Pharmacol Sci 2009;13:13–21. [PubMed] [Google Scholar]
- 346. Wong DM, Li L, Jurado S, King A, Bamford R, Wall M, Walia MK, Kelly GL, Walkley CR, Tarlinton DM, Strasser A, Heierhorst J. The transcription factor ASCIZ and its target DYNLL1 are essential for the development and expansion of MYC‐driven B cell lymphoma. Cell Rep 2016;14:1488–1499. [DOI] [PubMed] [Google Scholar]
- 347. Blunt JW, Hartshorn MP, McLennan TJ, Munro MHG, Robinson WT, Yorke SC. Thyrsiferol: A squalene‐derived metabolite of Laurencia thyrsifera . Tetrahedron Lett 1978;19:69‒72. [Google Scholar]
- 348. Manzo E, Gavagnin M, Bifulco G, Cimino P, Di Micco S, Ciavatta ML, Guo YW, Cimino G. Aplysiols A and B, squalene‐derived polyethers from the mantle of the sea hare Aplysia dactylomela . Tetrahedron 2007;63:9970‒9978. [Google Scholar]
- 349. Mahdi F, Falkenberg M, Ioannou E, Roussis V, Zhou YD, Nagle DG. Thyrsiferol inhibits mitochondrial respiration and HIF‐1 activation. Phytochem Lett 2011;4:75‒78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Fernández JJ, Souto ML, Norte M. Evaluation of the cytotoxic activity of polyethers isolated from Laurencia . Bioorg Med Chem 1998;6:2237–2243. [DOI] [PubMed] [Google Scholar]
- 351. Mathieu A, Remmelink M, D'Haene N, Penant S, Gaussin JF, Van Ginckel R, Darro F, Kiss R, Salmon I. Development of a chemoresistant orthotopic human nonsmall cell lung carcinoma model in nude mice: Analyses of tumor heterogenity in relation to the immunohistochemical levels of expression of cyclooxygenase‐2, ornithine decarboxylase, lung‐related resistance protein, prostaglandin E synthetase, and glutathione‐S‐transferase‐alpha (GST)‐alpha, GST‐mu, and GST‐pi. Cancer 2004;101:1908–1918. [DOI] [PubMed] [Google Scholar]
- 352. Ortega MJ, Zubìa E, Salvà J. 3‐Epi‐aplykurodinone B, a new degraded sterol from Aplysia fasciata . J Nat Prod 1997;60:488‒489. [DOI] [PubMed] [Google Scholar]
- 353. Jiménez C, Quiňoá E, Castedo L, Riguera R. Epidioxy sterols from the tunicates Dendrodoa grossularia and Ascidiella aspersa and the gastropoda Aplysia depilans and Aplysia punctata . J Nat Prod 1986;49:905‒909. [Google Scholar]
- 354. Mun B, Wang W, Kim H, Hahn D, Yang I, Won DH, Kim EH, Lee J, Han C, Kim H, Ekins M, Nam SJ, Choi H, Kang H. Cytotoxic 5α,8α‐epidioxy sterols from the marine sponge Monanchora sp. Arch Pharm Res 2015;38:18–25. [DOI] [PubMed] [Google Scholar]
- 355. Walsby JR, Morton JE, Croxall JP. The feeding mechanism and ecology of the New Zealand pulmonate limpet Gadinalea nivea . J Zool Lond 1973;171:257‒283. [Google Scholar]
- 356. Díaz‐Marrero AR, Dorta E, Cueto M, Rovirosa J, San Martín A, Loyola A, Darias J. New polyhydroxylated steroids from the marine pulmonate Trimusculus peruvianus . Arkivoc 2003;10:107‒117. [Google Scholar]
- 357. van Wyk AW, Gray CA, Whibley CE, Osoniyi O, Hendricks DT, Caira MR, Davies‐Coleman MT. Bioactive metabolites from the south african marine mollusk Trimusculus costatus . J Nat Prod 2008;71:420‒425. [DOI] [PubMed] [Google Scholar]
- 358. McPhail KL, Davies‐Coleman MT, Starmer J. Sequestered chemistry of the arminacean nudibranch Leminda millecra in Algoa bay, South Africa. J Nat Prod 2001;64:1183‒1190. [DOI] [PubMed] [Google Scholar]
- 359. Whibley CE, McPhail KL, Keyzers RA, Maritz MF, Leaner VD, Birrer MJ, Davies‐Coleman MT, Hendricks DT. Reactive oxygen species mediated apoptosis of esophageal cancer cells induced by marine triprenyl toluquinones and toluhydroquinones. Mol Cancer Ther 2007;6:2535‒2543. [DOI] [PubMed] [Google Scholar]
- 360. Rinehart KL, Gloer JB, Cook JC, Mizsak SA, Scahill TA. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a Caribbean tunicate. J Am Chem Soc 1981;103:1857‒1859. [Google Scholar]
- 361. Zabriskie TM, Klocke JA, Ireland CM, Marcus AH, Molinski TF, Faulkner DJ, Xu C, Clardy JC. Jaspamide, a modified peptide from a Jaspis sponge, with insecticidal and antifungal activity. J Am Chem Soc 1986;108:3123‒3124. [Google Scholar]
- 362. Harrigan GG, Goetz G. Symbiotic and dietary marine microalgae as a source of bioactive molecules‐experience from natural products research. J Appl Phycol 2002;14:103‒108. [Google Scholar]
- 363. Tan LT. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007;68:954‒979. [DOI] [PubMed] [Google Scholar]
- 364. Ishiwata H, Nemoto T, Ojika M, Yamada K. Isolation and stereostructure of doliculide, a cytotoxic cyclodepsipeptide from the Japanese sea hare Dolabella auricularia . J Org Chem 1994;59:4710‒4711. [Google Scholar]
- 365. Ishiwata H, Sone H, Kigoshi H, Yamada K. Enantioselective total synthesis of doliculide, a potent cytotoxic cyclodepsipeptide of marine origin and structure‐cytotoxicity relationships of synthetic doliculide congeners. Tetrahedron 1994;50:12853‒12882. [Google Scholar]
- 366. Ghosh AK, Liu C. Total synthesis of antitumor depsipeptide (‐)‐doliculide. Org Lett 2001;3:635–638. [DOI] [PubMed] [Google Scholar]
- 367. Matcha K, Madduri AV, Roy S, Ziegler S, Waldmann H, Hirsch AK, Minnaard AJ. Total synthesis of (‐)‐doliculide, structure–activity relationship studies and its binding to F‐actin. ChemBioChem 2012;13:2537‒2548. [DOI] [PubMed] [Google Scholar]
- 368. Bai R, Covell DG, Liu C, Ghosh AK, Hamel E. (‐)‐Doliculide, a new macrocyclic depsipeptide enhancer of actin assembly. J Biol Chem 2002;277:32165‒32171. [DOI] [PubMed] [Google Scholar]
- 369. Kunze B, Jansen R, Sasse F, Höfle G, Reichenbach H. Chondramides A approximately D, new antifungal and cytostatic depsipeptides from Chondromyces crocatus (myxobacteria). Production, physico‐chemical and biological properties. J Antibiot 1995;48:1262–1266. [DOI] [PubMed] [Google Scholar]
- 370. Mansfield ML, Covell DG, Jernigan RL. A new class of molecular shape descriptors. 1. Theory and properties. J Chem Inf Comput Sci 2002;42:259–273. [DOI] [PubMed] [Google Scholar]
- 371. Foerster F, Braig S, Chen T, Altmann KH, Vollmar AM. Pharmacological characterization of actin‐binding (‐)‐doliculide. Bioorg Med Chem 2014;22:5117‒5122. [DOI] [PubMed] [Google Scholar]
- 372. Foerster F, Chen T, Altmann KH, Vollmar AM. Actin‐binding doliculide causes premature senescence in p53 wild type cells. Bioorg Med Chem 2016;24:123–129. [DOI] [PubMed] [Google Scholar]
- 373. Spuul P, Ciufici P, Veillat V, Leclercq A, Daubon T, Kramer IJ, Génot E. Importance of RhoGTPases in formation, characteristics, and functions of invadosomes. Small GTPases 2014;5:e28195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Collins RJ, Jiang WG, Hargest R, Mason MD, Sanders AJ. EPLIN: A fundamental actin regulator in cancer metastasis? Cancer Metastasis Rev 2015;34:753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Frugtniet B, Jiang WG, Martin TA. Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer (Dove Med Press) 2015;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Reese MT, Gulavita NK, Nakao Y, Hamann MT, Yoshida WY, Coval SJ, Scheuer PJ. Kulolide: A cytotoxic depsipeptide from a cephalaspidean mollusk, Philinopsis speciosa . J Am Chem Soc 1996;118:11081‒11084546. [Google Scholar]
- 377. Kimura J, Takada Y, Inayoshi T, Nakao Y, Goetz G, Yoshida WY, Scheuer PJ. Kulokekahilide‐1, a cytotoxic depsipeptide from the Cephalaspidean mollusk Philinopsis speciosa . J Org Chem 2002;67:1760‒1767. [DOI] [PubMed] [Google Scholar]
- 378. Nakao Y, Yoshida WY, Takada Y, Kimura J, Yang L, Mooberry SL, Scheuer PJ. Kulokekahilide‐2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa . J Nat Prod 2004;67:1332‒1340. [DOI] [PubMed] [Google Scholar]
- 379. Nakao Y, Yoshida WY, Szabo CM, Baker BJ, Scheuer PJ. More peptides and other diverse constituents of the marine mollusk Philinopsis speciosa . J Org Chem 1998;63:3272‒3280. [Google Scholar]
- 380. Umehara M, Negishi T, Tashiro T, Nakao Y, Kimura J. Structure‐related cytotoxic activity from kulokekahilide‐2, a cyclodepsipeptide of Hawaiian marine mollusk. Bioorg Med Chem Lett 2012;22:7422‒7425. [DOI] [PubMed] [Google Scholar]
- 381. Takada Y, Umehara M, Katsumata R, Nakao Y, Kimura J. The total synthesis and structure‐activity relationships of a highly cytotoxic depsipeptide kulokekahilide‐2 and its analogs. Tetrahedron 2012;68:659‒669. [Google Scholar]
- 382. Suenaga K, Mutou T, Shibata T, Itoh T, Kigoshi H, Yamada K. Isolation and stereostructure of aurilide, a novel cyclodepsipeptide from the Japanese sea hare Dolabella auricularia . Tetrahedron Lett 1996;37:6771‒6774. [Google Scholar]
- 383. Han B, Gross H, Goeger DE, Mooberry SL, Gerwick WH. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula . J Nat Prod 2006;69:572–575. [DOI] [PubMed] [Google Scholar]
- 384. Suenaga K, Mutou T, Shibata T, Itoh T, Fujita T, Takada N, Hayamizu K, Takagi M, Irifune T, Kigoshi H, Yamada K. Aurilide, a cytotoxic depsipeptide from the sea hare Dolabella auricularia: Isolation, structure determination, synthesis, and biological activity. Tetrahedron 2004;60:8509‒8527. [Google Scholar]
- 385. Hollingshead MG, Alley MC, Camalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. In vivo cultivation of tumor celles in hollow fibers. Life Sci 1995;57:131–141. [DOI] [PubMed] [Google Scholar]
- 386. Sato SI, Murata A, Orihara T, Shirakawa T, Suenaga K, Kigoshi H, Uesugi M. Marine natural product aurilide activates the OPA1‐mediated apoptosis by binding to prohibitin. Chem Biol 2011;18:131‒139. [DOI] [PubMed] [Google Scholar]
- 387. Semenzato M, Cogliati S, Scorrano L. Prohibitin(g) cancer: Aurilide and killing by Opa1‐dependent cristae remodeling. Chem Biol 2011;18:8–9. [DOI] [PubMed] [Google Scholar]
- 388. Shankavaram UT, Varma S, Kane D, Sunshine M, Chary KK, Reinhold WC, Pommier Y, Weinstein JN. CellMiner: A relational database and query tool for the NCI‐60 cancer cell lines. BMC Genomics 2009;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Wesson KJ, Hamann MT. Keenamide A, a bioactive cyclic peptide from the marine mollusk Pleurobranchus forskalii . J Nat Prod 1996;59:629‒631. [DOI] [PubMed] [Google Scholar]
- 390. Co Tan K, Wakimoto T, Takada K, Ohtsuki T, Uchiyama N, Goda Y, Abe I. Cycloforskamide, a cytotoxic macrocyclic peptide from the sea slug Pleurobranchus forskalii . J Nat Prod 2013;76:1388‒1391. [DOI] [PubMed] [Google Scholar]
- 391. Rodriguez J, Fernández R, Quiñoá E, Riguera R, Debitus C, Bouchet P. Onchidin: A cytotoxic depsipeptide with c2 symmetry from a marine mollusc. Tetrahedron Lett 1994;35:9239‒9242. [Google Scholar]
- 392. Fernández R, Rodríguez J, Quiñoá E, Riguera R, Muñoz L, Fernández‐Suárez M, Debitus C. Onchidin B: A new cyclodepsipeptide from the mollusc Onchidium sp. J Am Chem Soc 1996;118:11635‒11643. [Google Scholar]
- 393. Sudek S, Lopanek NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina . J Nat Prod 2007;70:67‒74. [DOI] [PubMed] [Google Scholar]
- 394. Napolitano JG, Daranas AH, Norte M, Fernández JJ. Marine macrolides, a promising source of antitumor compounds. Anti‐cancer Agents Med Chem 2009;9:122–137. [DOI] [PubMed] [Google Scholar]
- 395. Sone H, Kigoshi H, Yamada K. Aurisides A and B, cytotoxic macrolide glycosides from the Japanese sea hare Dolabella auricularia . J Org Chem 1996;61:8956‒8960. [DOI] [PubMed] [Google Scholar]
- 396. Paterson I, Florence GJ, Heimann AC, Mackay AC. Stereocontrolled total synthesis of (‐)‐aurisides A and B. Angew Chem Int Ed Engl 2005;44:1130–1133. [DOI] [PubMed] [Google Scholar]
- 397. Tello‐Aburto R, Olivo HF. A formal synthesis of the auriside aglycon. Org Lett 2008;10:2191–2194. [DOI] [PubMed] [Google Scholar]
- 398. Suenaga K, Kigoshi H, Yamada K. Auripyrones A and B, cytotoxic polypropionates from the sea hare Dolabella auricularia. Isolation and Structures. Tetrahedron Lett 1996;37:5151‒5154. [Google Scholar]
- 399. Lister T, Perkins MV. Total synthesis of auripyrone A. Angew Chem Int Ed Engl 2006;45:2560–2564. [DOI] [PubMed] [Google Scholar]
- 400. Jung ME, Chaumontet M, Salehi‐Rad R. Total synthesis of auripyrone B using a non‐aldol aldol‐cuprate opening process. Org Lett 2010;12:2872–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Jung ME, Salehi‐Rad R. Total synthesis of auripyrone A using a tandem non‐aldol aldol/Paterson aldol process as a key step. Angew Chem Int Ed Engl 2009;48:8766–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Hayakawa I, Takemura T, Fukasawa E, Ebihara Y, Sato N, Nakamura T, Suenaga K, Kigoshi H. Total synthesis of auripyrones A and B and determination of the absolute configuration of auripyrone B. Angew Chem Int Ed Engl 2010;49:2401–2405. [DOI] [PubMed] [Google Scholar]
- 403. Ojika M, Nagoya T, Yamada K. Dolabelides A and B, cytotoxic 22‐membered macrolides isolated from the sea hare Dolabella auricularia . Tetrahedron Lett 1995;36:7491‒7494. [Google Scholar]
- 404. Suenaga K, Nagoya T, Shibata T, Kigoshi H, Yamada K. Dolabelides C and D, cytotoxic macrolides isolated from the sea hare Dolabella auricularia . J Nat Prod 1997;60:155‒157. [Google Scholar]
- 405. Park PK, O'Malley SJ, Schmidt DR, Leighton JL. Total synthesis of dolabelide D. J Am Chem Soc 2006;128:2796–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Hanson PR, Chegondi R, Nguyen J, Thomas CD, Waetzig JD, Whitehead A. Total synthesis of dolabelide C: A phosphate‐mediated approach. J Org Chem 2011;76:4358–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Roesener JA, Scheuer PJ. Ulapualide A and B, extraordinary antitumor macrolides from nudibranch eggmasses. J Am Chem Soc 1986;108:846‒847. [Google Scholar]
- 408. Chattopadhyay SK, Pattenden G. Total synthesis of ulapualide A, a novel tris‐oxazole containing macrolide from the marine nudibranch Hexabranchus sanguineus . Tetrahedron Lett 1998;39:6095‒6098. [Google Scholar]
- 409. Vincent E, Saxton J, Baker‐Glenn C, Moal I, Hirst JD, Pattenden G, Shaw PE. Effects of ulapualide A and synthetic macrolide analogues on actin dynamics and gene regulation. Cell Mol Life Sci 2007;64:487‒497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Matsunaga S, Fusetani N, Hashimoto K. Kabiramide C, a novel antifungal macrolide from nudibranch eggmasses. J Am Chem Soc 1986;108:847‒849. [Google Scholar]
- 411. Kernan MR, Molinski TF, Faulkner DJ. Macrocyclic antifungal metabolites from the Spanish dancer nudibranch Hexabranchus sanguineus and sponges of the genus Halichondria . J Org Chem 1988;53:5014‒5020. [Google Scholar]
- 412. Sirirak T, Kittiwisut S, Janma C, Yuenyongsawad S, Suwanborirux K, Plubrukarn A. Kabiramides J and K, trisoxazole macrolides from the sponge Pachastrissa nux . J Nat Prod 2011;74:1288‒1292. [DOI] [PubMed] [Google Scholar]
- 413. Tanaka J, Yan Y, Choi J, Bai J, Klenchin VA, Rayment I, Marriott G. Biomolecular mimicry in the actin cytoskeleton: Mechanisms underlying the cytotoxicity of kabiramide C and related macrolides. Proc Natl Acad Sci 2003;100:13851‒13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Matsunaga S, Fusetani N, Hashimoto K, Koseki K, Noma M, Noguchi H, Sankawa U. Further kabiramides and halichondramides, cytotoxic macrolides embracing trisoxazole, from the Hexabranchus egg masses. J Org Chem 1989;54:1360–1363. [Google Scholar]
- 415. Kernan MR, Faulner DJ. Halichondramide, an antifungal macrolide from the spongia Halichondria sp. Tetrahedron Lett 1987;28:2809–2812. [Google Scholar]
- 416. Dalisay DS, Rogers EW, Edison AS, Molinski TF. Structure elucidation at the nanomole‐scale. Trioxazole macrolides and thiazole‐containing cyclic peptides from the nudibranch Hexabranchus sanguineus . J Nat Prod 2009;72:732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Braet F, Spector I, Shochet N, Crews P, Higa T, Menu E, de Zanger R, Wisse E. The new anti‐actin agent dihydrohalichondramide reveals fenestrae‐forming centers in hepatic endothelial cells. BMC Cell Biol 2002;3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Shin Y, Kim GD, Jeon JE, Shin J, Lee SK. Antimetastatic effect of halichondramide, a trisoxazole macrolide from the marine sponge Chondrosia corticata, on human prostate cancer cells via modulation of epithelial‐to‐mesenchymal transition. Mar Drugs 2013;11:2472–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Bae SY, Kim GD, Jeon JE, Shin J, Lee SK. Anti‐proliferative effect of (19Z)‐halichondramide, a novel marine macrolide isolated from the sponge Chondrosia corticata, is associated with G2/M cell cycle arrest and suppression of mTOR signaling in human lung cancer cells. Toxicol in vitro 2013;27:694–699. [DOI] [PubMed] [Google Scholar]
- 420. Corley DG, Herb R, Moore RE, Scheuer PJ, Paul VJ. Laulimalides: New potent cytotoxic macrolides from a marine sponge and a nudibranch predator. J Org Chem 1988;53:3644‒3646. [Google Scholar]
- 421. Liu J, Towle MJ, Cheng H, Saxton P, Reardon C, Wu J, Murphy EA, Kuznetsov G, Johannes CW, Tremblay MR, Zhao H, Pesant M, Fang FG, Vermeulen MW, Gallagher BM, Jr , Littlefield BA. in vitro and in vivo anticancer activities of synthetic (‐)‐laulimalide, a marine natural product microtubule stabilizing agent. Anticancer Res 2007;27:1509–1518. [PubMed] [Google Scholar]
- 422. Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Laulimalide and isolaulimalide, new paclitaxel‐like microtubule‐stabilizing agents. Cancer Res 1999;59:653‒660. [PubMed] [Google Scholar]
- 423. Pryor DE, O'Brate A, Bilcer G, Díaz JF, Wang Y, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 2002;41:9109‒9115. [DOI] [PubMed] [Google Scholar]
- 424. Churchill CD, Klobukowski M, Tuszynski JA. The unique binding mode of laulimalide to two tubulin protofilaments. Chem Biol Drug Des 2015;86:190–199. [DOI] [PubMed] [Google Scholar]
- 425. Rohena CC, Peng J, Johnson TA, Crews P, Mooberry SL. Chemically diverse microtubule stabilizing agents initiate distinct mitotic defects and dysregulated expression of key mitotic kinases. Biochem Pharmacol 2013;85:1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Kanakkanthara A, Northcote PT, Miller JH. βII‐tubulin and βIII‐tubulin mediate sensitivity to peloruside A and laulimalide, but not paclitaxel or vinblastine, in human ovarian carcinoma cells. Mol Cancer Ther 2012;11:393–404. [DOI] [PubMed] [Google Scholar]
- 427. Kanakkanthara A, Rawson P, Northcote PT, Miller JH. Acquired resistance to peloruside A and laulimalide is associated with downregulation of vimentin in human ovarian carcinoma cells. Pharm Res 2012;29:3022–3032. [DOI] [PubMed] [Google Scholar]
- 428. Ahmed A, Hoegenauer EK, Enev VS, Hanbauer M, Kaehlig H, Ohler E, Mulzer J. Total synthesis of the microtubule stabilizing antitumor agent laulimalide and some nonnatural analogues: The power of Sharpless’ asymmetric epoxidation. J Org Chem 2003;68:3026–3042. [DOI] [PubMed] [Google Scholar]
- 429. Uenishi J, Ohmi M. Total synthesis of (‐)‐laulimalide: Pd‐catalyzed stereospecific ring construction of the substituted 3,6‐dihydro[2H] pyran units. Angew Chem Int Ed Engl 2005;44:2756–2760. [DOI] [PubMed] [Google Scholar]
- 430. Gollner A, Altmann KH, Gertsch J, Mulzer J. The laulimalide family: Total synthesis and biological evaluation of neolaulimalide, isolaulimalide, laulimalide and a nonnatural analogue. Chemistry 2009;15:5979–5997. [DOI] [PubMed] [Google Scholar]
- 431. Trost BM, Amans D, Seganish WM, Chung CK. Evaluating transition‐metal‐catalyzed transformations for the synthesis of laulimalide. J Am Chem Soc 2009;131:17087–17089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Trost BM, Seganish WM, Chung CK, Amans D. Total synthesis of laulimalide: Synthesis of the northern and southern fragments. Chemistry 2012;18:2948–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Trost BM, Amans D, Seganish WM, Chung CK. Total synthesis of laulimalide: Assembly of the fragments and completion of the synthesis of the natural product and a potent analogue. Chemistry 2012;18:2961–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Yamada K, Ojika M, Ishigaki T, Yoshida Y. Aplyronine A, a potent antitumor substance, and the congeners aplyronines B and C isolated from the sea hare Aplysia kurodai . J Am Chem Soc 1993;115:11020‒11021. [Google Scholar]
- 435. Kigoshi H, Suenaga K, Takagi M, Akao A, Kanematsu K, Kamei N, Okugawa Y, Yamada K. Cytotoxicity and actin‐depolymerizating activity of aplyronine A, a potent antitumor macrolide of marine origin, and its analogs. Tetrahedron 2002;58:1075–1102. [Google Scholar]
- 436. Ojika M, Kigoshi H, Yoshida Y, Ishigaki T, Nisiwaki M, Tsukada I, Arakawa M, Ekimoto H, Yamada K. Aplyronine A, a potent antitumor macrolide of marine origin, and the congeners aplyronines B and C: Isolation, structures and bioactivities. Tetrahedron 2007;63:3138–3167. [Google Scholar]
- 437. Ojika M, Kigoshi H, Suenaga K, Imamura Y, Yoshikawa K, Ishigaki T, Sakakura A, Mutou T, Yamada K. Aplyronines D‐H from the sea hare Aplysia kurodai: Isolation, structures and cytotoxicity. Tetrahedron 2012;68:982‒987. [Google Scholar]
- 438. Yamada K, Ojika M, Kigoshi H, Suenaga K. Aplyronine A, a potent antitumour macrolide of marine origin, and the congeners aplyronines B‐H: Chemistry and biology. Nat Prod Rep 2009;26:27–43. [DOI] [PubMed] [Google Scholar]
- 439. Kita M, Kigoshi H. Marine natural products that interfere with multiple cytoskeletal protein interaction. Nat Prod Rep 2015;32:534–542. [DOI] [PubMed] [Google Scholar]
- 440. Van den Mooter T, Teuwen LA, Rutten A, Dirix L. Trastuzumab emtansine in advanced human epidermal growth factor receptor 2‐positive breast cancer. Expert Opin Biol Ther 2015;15:749–760. [DOI] [PubMed] [Google Scholar]
- 441. Hirata K, Muraoka S, Suenaga K, Kuroda T, Kato K, Tanaka H, Yamamoto M, Takata M, Yamada K, Kigoshi H. Structure basis for antitumor effect of aplyronine A. J Mol Biol 2006;356:945–954. [DOI] [PubMed] [Google Scholar]
- 442. Kita M, Hirayama Y, Sugiyama M, Kigoshi H. Development of highly cytotoxic and actin‐depolymerizing biotin derivatives of aplyronine A. Angew Chem Int Ed 2011;50:9871‒9874. [DOI] [PubMed] [Google Scholar]
- 443. Kita M, Hirayama Y, Yoneda K, Yamagishi K, Chinen T, Usui T, Sumiya E, Uesugi M, Kigoshi H. Inhibition of microtubule assembly by a complex of actin and antitumor macrolide aplyronine A. J Am Chem Soc 2013;135:18089‒18095. [DOI] [PubMed] [Google Scholar]
- 444. Ohno O, Morita M, Kitamura K, Teruya T, Yoneda K, Kita M, Kigoshi H, Suenaga K. Apoptosis‐inducing activity of the actin‐depolymerizating agent aplyronine A and its side‐chain derivatives. Bioorg Med Chem Lett 2013;23:1467‒1471. [DOI] [PubMed] [Google Scholar]
- 445. Hong WP, Noshi MN, El‐Awa A, Fuchs PL. Synthesis of the C1‐C20 and C15‐C27 segments of aplyronine A. Org Lett 2011;13:6342–6345. [DOI] [PubMed] [Google Scholar]
- 446. Paterson I, Fink SJ, Lee LY, Atkinson SJ, Blakey SB. Total synthesis of aplyronine C. Org Lett 2013;15:3118–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Paterson I, Britton R, Ashton K, Knust H, Stafford J. Synthesis of antimicrofilament marine macrolides: Synthesis and configurational assignment of a C5‐C16 degradation fragment of reidispongiolide A. Proc Natl Acad Sci USA 2004;101:11986–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Yeung KS, Paterson I. Actin‐binding marine macrolides: Total synthesis and biological importance. Angew Chem Int Ed Engl 2002;41:4632–4653. [DOI] [PubMed] [Google Scholar]
- 449. Carbone M, Ciavatta ML, Wang JR, Cirillo I, Mathieu V, Kiss R, Mollo E, Guo YW, Gavagnin M. Extending the record of bis‐γ‐pyrone polypropionates from marine pulmonate mollusks. J Nat Prod 2013;76:2065–2073. [DOI] [PubMed] [Google Scholar]
- 450. Zhang X, Minale L, Zampella A, Smith CD. Microfilament depletion and circumvention of multiple drug resistance by sphinxolides. Cancer Res 1997;57:3751‒3758. [PubMed] [Google Scholar]
- 451. Guella G, Mancini I, Chiasera G, Pietra F. Sphinxolide, a 26‐membered antitumoral macrolide isolated from an unidentified Pacific nudibranch. Helv Chim Acta 1989;72:237‒246. [Google Scholar]
- 452. D'Auria MV, Gomez Paloma L, Minale L, Zampella A, Verbist JF, Roussakis C, Debitus C, Patissou J. Three new potent cytotoxic macrolides closely related to sphinxolide from the new Caledonian sponge Neosiphonia superstes . Tetrahedron 1993;49:8657–8664. [Google Scholar]
- 453. D'Auria MV, Gomez Paloma L, Minale L, Zampella A, Verbist JF, Roussakis C, Debitus C, Patissou J. Reidispongiolide A and B, two new potent cytotoxic macrolides from the new Caledonian sponge Reidispongia coerulea . Tetrahedron 1994;50:4829–4834. [Google Scholar]
- 454. Allingham JS, Zampella A, D'Auria MV, Rayment I. Structures of microfilament destabilizing toxins bound to actin provide insight into toxin design and activity. Proc Natl Acad Sci USA 2005;102:14527–14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. Mebs D. Chemical defense of a dorid nudibranch, Glossodoris quadricolor, from the Red sea. J Chem Ecol 1985;11:713‒716. [DOI] [PubMed] [Google Scholar]
- 456. Kakou Y, Crews P, Bakus GJ. Dendrolasin and latrunculin A from the Fijian sponge Spongia mycofijiensis and an associated nudibranch Chromodoris lochi . J Nat Prod 1987;50:482‒484. [Google Scholar]
- 457. Spector I, Shochet NR, Kashman Y, Groweiss A. Latrunculins: Novel marine toxins that disrupt microfilament organization in cultured cells. Science 1983;219:493‒495. [DOI] [PubMed] [Google Scholar]
- 458. Longley RE, McConnell OJ, Essich E, Harmody D. Evaluation of marine sponge metabolites for cytotoxicity and signal transduction activity. J Nat Prod 1993;56:915‒920. [DOI] [PubMed] [Google Scholar]
- 459. El Sayed KA, Youssef DTA, Marchetti M. Bioactive natural and semisynthetic latrunculins. J Nat Prod 2006;69:219‒223. [DOI] [PubMed] [Google Scholar]
- 460. Khanfar MA, Youssef DTA, El‐Sayed KA. 3D‐QSAR studies of latrunculin‐based actin polymerization inhibitors using CoMFA and CoMSIA approaches. Eur J Med Chem 2010;45:3662‒3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Konishi H, Kikuchi S, Ochiai T, Ikoma H, Kubota T, Ichikawa D, Fujiwara H, Okamoto K, Sakakura C, Sonoyama T, Kokuba Y, Sasaki H, Matsui T, Otsuji E. Latrunculin a has a strong anticancer effect in a peritoneal dissemination model of human gastric cancer in mice. Anticancer Res 2009;29:2091–2097. [PubMed] [Google Scholar]
- 462. Tymiak AA, Rinehart KL. Structures of Kelletinins I and II, antibacterial metabolites of the marine mollusk Kelletia kelletii . J Am Chem Soc 1983;105:7396‒7401. [Google Scholar]
- 463. Castiello D, Cimino G, De Rosa S, De Stefano S, Sodano G. High molecular weight polyacetylenes from the nudibranch Peltodoris atromaculata and the sponge Petrosia ficiformis . Tetrahedron Lett 1980;21:5047–5050. [Google Scholar]
- 464. Ueoka R, Ise Y, Matsunaga S. Cytotoxic polyacetylenes related to petroformyne‐1 from the marine sponge Petrosia sp. Tetrahedron 2009;65:5204‒5208. [Google Scholar]
- 465. Ortega MG, Zubia E, Carballo JL, Salvá J. Fulvinol, a new long‐chain diacetylenic metabolite from the sponge Reniera fulva . J Nat Prod 1996;59:1069‒1071. [Google Scholar]
- 466. Ciavatta ML, Nuzzo G, Takada K, Mathieu V, Kiss R, Villani G, Gavagnin M. Sequestered fulvinol‐related polyacetylenes in Peltodoris atromaculata . J Nat Prod 2014;77:1678‒1684. [DOI] [PubMed] [Google Scholar]
- 467. Kuroda T, Kigoshi H. Aplaminal: A novel cytotoxic aminal isolated from the sea hare Aplysia kurodai . Org Lett 2008;10:489‒491. [DOI] [PubMed] [Google Scholar]
- 468. Kigoshi H, lmamura Y, Yoshikawa K, Yamada K. Three new cytotoxic alkaloids, aplaminone, neoaplaminone and neoaplaminone sulfate from the marine mollusc Aplysia kurodai . Tetrahedron Lett 1990;31:4911‒4914. [Google Scholar]
- 469. Teeyapant R, Kreis P, Wray V, Witte L, Proksch P. Brominated secondary compounds from the marine sponge Verongia aerophoba and the sponge feeding gastropod Tylodina perversa . Z Naturforsch 1993;48c:640–644. [DOI] [PubMed] [Google Scholar]
- 470. Thoms C, Ebel R, Proksch P. Sequestration and possible role of dietary alkaloids in the sponge‐feeding mollusk Tylodina perversa In: Cimino G. & Gavagnin M, Eds. Mollusc: From Chemo‐Ecological Study to Biotechnological Application. Springer, Berlin Heidelberg, German: 2006. p 261–275. [DOI] [PubMed] [Google Scholar]
- 471. Gallimore WA, Scheuer PJ. Malyngamides O and P from the sea hare Stylocheilus longicauda . J Nat Prod 2000;63:1422‒1424. [DOI] [PubMed] [Google Scholar]
- 472. Appleton DR, Sewell MA, Berridge MV, Copp BR. A new biologically active malyngamide from a New Zealand collection of the sea hare Bursatella leachii . J Nat Prod 2002;65:630‒631. [DOI] [PubMed] [Google Scholar]
- 473. Suntornchashwej S, Suwanborirux K, Koga K, Isobe M. Malyngamide X: The first (7R)‐lyngbic acid that connects to a new tripeptide backbone from the Thai sea hare Bursatella leachii . Chem Asian J 2007;2:114‒122. [DOI] [PubMed] [Google Scholar]
- 474. Sone H, Kondo T, Kiryu M, Ishiwata H, Ojika M, Yamada K. Dolabellin, a cytotoxic bisthiazole metabolite from the sea hare Dolabella auricularia: Structural determination and synthesis. J Org Chem 1995;60:4774‒4781. [Google Scholar]
- 475. Suntornchashwej S, Chaichit N, Isobe M, Suwanborirux K. Hectochlorin and morpholine derivatives from the Thai sea hare, Bursatella leachii . J Nat Prod 2005;68:951‒955. [DOI] [PubMed] [Google Scholar]
- 476. Marquez BL, Watts KS, Yokochi A, Roberts MA, Verdier‐Pinard P, Jimenez JI, Hamel E, Scheuer PJ, Gerwick WH. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J Nat Prod 2002;65:866–871. [DOI] [PubMed] [Google Scholar]
- 477. Cetusic JRP, Green FR, III , Graupner PR, Oliver MP. Total synthesis of hectochlorin. Org Lett 2002;4:1307–1310. [DOI] [PubMed] [Google Scholar]
- 478. Andersen RJ, Faulkner DJ, Cun‐heng H, van Duyne GD, Clardy J. Metabolites of the marine prosobranch mollusc Lamellaria sp. J Am Chem Soc 1985;107:5492‒5495. [Google Scholar]
- 479. Imbri D, Tauber J, Opatz T. Synthetic approaches to the lamellarins—A comprehensive review. Mar Drugs 2014;12:6142–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Ballot C, Martoriati A, Jendoubi M, Buche S, Formstecher P, Mortier L, Kluza J, Marchetti P. Another facet to the anticancer response to lamellarin D: Induction of cellular senescence through inhibition of topoisomerase I and intracellular ROS production. Mar Drugs 2014;12:779‒798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Bailly C. Anticancer properties of lamellarins. Mar Drugs 2015;13:1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Marco E, Laine W, Tardy C, Lansiaux A, Iwao M, Ishibashi F, Bailly C, Gago F. Molecular determinants of topoisomerase I poisoning by lamellarins: Comparison with camptothecin and structure‐activity relationships. J Med Chem 2005;48:3796–3807. [DOI] [PubMed] [Google Scholar]
- 483. Gallego MA, Ballot C, Kluza J, Hajji N, Martoriati A, Castéra L, Cuevas C, Formstecher P, Joseph B, Kroemer G, Bailly C, Marchetti P. Overcoming chemoresistance of non‐small cell lung carcinoma through restoration of an AIF‐dependent apoptotic pathway. Oncogene 2008;27:1981–1992. [DOI] [PubMed] [Google Scholar]
- 484. Ballot C, Kluza J, Martoriati A, Nyman U, Formstecher P, Joseph B, Bailly C, Marchetti P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid lamellarin D. Mol Cancer Ther 2009;8:3307–3317. [DOI] [PubMed] [Google Scholar]
- 485. Khiati S, Seol Y, Agama K, Dalla Rosa I, Agrawal S, Fesen K, Zhang H, Neuman KC, Pommier Y. Poisoning of mitochondrial topoisomerase I by lamellarin D. Mol Pharmacol 2014;86:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010;17:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Vanhuyse M, Kluza J, Tardy C, Otero G, Cuevas C, Bailly C, Lansiaux A. Lamellarin D: A novel pro‐apoptotic agent from marine origin insensitive to P‐glycoprotein‐mediated drug efflux. Cancer Lett 2005;221:165–175. [DOI] [PubMed] [Google Scholar]
- 488. Hénon H, Conchon E, Hugon B, Messaoudi S, Golsteyn RM, Prudhomme M. Pyrrolocarbazoles as checkpoint 1 kinase inhibitors. Anticancer Agents Med Chem 2008;8:577–597. [PubMed] [Google Scholar]
- 489. Cantrell CL, Groweiss A, Gustafson KR, Boyd MR. A new staurosporine analog from the prosobranch mollusk Coriocella nigra . Nat Prod Lett 1999;14:39‒46. [Google Scholar]
- 490. Carté B, Faulkner DJ. Defensive metabolites from three nembrothid nudibranchs. J Org Chem 1983;48:2314‒2318. [Google Scholar]
- 491. Melvin MS, Ferguson DC, Lindquist N, Manderville RA. DNA binding by 4‐methoxypyrrolic natural products. Preference for intercalation at AT sites by tambjamine E and prodigiosin. J Org Chem 1999;64:6861–6869. [DOI] [PubMed] [Google Scholar]
- 492. Aldrich LN, Stoops SL, Crews BC, Marnett LJ, Lindsley CW. Total synthesis and biological evaluation of tambjamine K and a library of unnatural analogs. Bioorg Med Chem Lett 2010;20:5207‒5211. [DOI] [PubMed] [Google Scholar]
- 493. Carbone M, Irace C, Costagliola F, Castelluccio F, Villani G, Calado G, Padula V, Cimino G, Cervera JL, Santamaria R, Gavagnin M. A new cytotoxic tambjamine alkaloid from the Azorean nudibranch Tambja ceutae . Bioorg Med Chem Lett 2010;20:2668‒2670. [DOI] [PubMed] [Google Scholar]
- 494. Hernandez IP, Moreno D, Araujo Javier A, Torroba T, Perez‐Toma´ R, Quesada R. Tambjamine alkaloids and related synthetic analogs: Efficient transmembrane anion transporters. Chem Comm 2012;48:1556‒1558. [DOI] [PubMed] [Google Scholar]
- 495. Xu WQ, Song LJ, Liu Q, Zhao L, Zheng L, Yan ZW, Fu GH. Expression of anion exchanger 1 is associated with tumor progress in human gastric cancer. J Cancer Res Clin Oncol 2009;135:1323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Liedauer R, Svoboda M, Wlcek K, Arrich F, Jä W, Toma C, Thalhammer T. Different expression patterns of organic anion transporting polypeptides in osteosarcomas, bone metastases and aneurysmal bone cysts. Oncol Rep 2009;22:1485–1492. [DOI] [PubMed] [Google Scholar]
- 497. Buxhofer‐Ausch V, Secky L, Wlcek K, Svoboda M, Kounnis V, Briasoulis E, Tzakos AG, Jaeger W, Thalhammer T. Tumor‐specific expression of organic anion‐transporting polypeptides: Transporters as novel targets for cancer therapy. J Drug Deliv 2013;2013, Article ID 863539, 12 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. AbuAli G, Grimm S. Isolation and characterization of the anticancer gene organic cation transporter like‐3 (ORCTL3). Adv Exp Med Biol 2014;818:213–227. [DOI] [PubMed] [Google Scholar]
- 499. Liu T, Li Q. Organic anion‐transporting polypeptides: A novel approach for cancer therapy. J Drug Target 2014;22:14–22. [DOI] [PubMed] [Google Scholar]
- 500. Ullrich N, Caplanusi A, Brône B, Hermans D, Larivière E, Nilius B, Van Driessche W, Eggermont J. Stimulation by caveolin‐1 of the hypotonicity‐induced release of taurine and ATP at basolateral, but not apical, membrane of Caco‐2 cells. Am J Physiol Cell Physiol 2006;290:C1287–1296. [DOI] [PubMed] [Google Scholar]
- 501. van der Mark VA, de Waart DR, Ho‐Mok KS, Tabbers MM, Voogt HW, Oude Elferink RP, Knisely AS, Paulusma CC. The lipid flippase heterodimer ATP8B1‐CDC50A is essential for surface expression of the apical sodium‐dependent bile acid transporter (SLC10A2/ASBT) in intestinal Caco‐2 cells. Biochim Biophys Acta 2014;1842:2378–2386. [DOI] [PubMed] [Google Scholar]
- 502. Melvin MS, Wooton KE, Rich CC, Saluta GR, Kucera GL, Lindquist N, Manderville RA. Copper‐nuclease efficiency correlates with cytotoxicity for the 4‐methoxypyrrolic natural products. J Inorg Biochem 2001;87:129–135 [DOI] [PubMed] [Google Scholar]
- 503. Cavalcanti BC, Junior HVN, Seleghim MHR, Berlinck RGS, Cunha GMA, Moraes MO, Pessoa C. Cytotoxic and genotoxic effects of tambjamine D, an alkaloid isolated from the nudibranch Tambja eliora, on Chinese hamster lung fibroblasts. Chem Biol Interact 2008;174:155‒162. [DOI] [PubMed] [Google Scholar]
- 504. Pettit GR, Tang Y, Knight JC. Antineoplastic agents. 545. Isolation and structure of Turbostatins 1‐4 from the Asian marine mollusk Turbo stenogyrus . J Nat Prod 2005;68:974‒978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Andrianasolo EH, Haramaty L, MacPhail KL, White E, Vetriani C, Falkowski P, Lutz R. Bathymodiolamides A and B, ceramide derivatives from a deep‐sea hydrothermal vent invertebrate mussel, Bathymodiolus thermophilus . J Nat Prod 2011;74:842‒846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Esmaeelian B, Benckendorff K, Johnston MR, Abbott CA. Purified brominated indole derivatives from Dicathais orbita induce apoptosis and cell cycle arrest in colorectal cancer cell lines. Mar Drugs 2013;11:3802‒3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Carroll AR, Scheuer PJ. Kuanoniamines A, B, C, and D: Pentacyclic alkaloids from a tunicate and its prosobranch mollusk predator Chelynotus semperi . J Org Chem 1990;55:4426‒4431. [Google Scholar]
- 508. Carbone M, Li Y, Irace C, Mollo E, Castelluccio F, Di Pascale A, Cimino G, Santamaria R, Guo YW, Gavagnin M. Structure and cytotoxicity of phidianidines A and B: First finding of 1,2,4‐oxadiazole system in a marine natural product. Org Lett 2011;13:2516‒2519. [DOI] [PubMed] [Google Scholar]
- 509. Brogan JT, Stoops SL, Lindsley CW. Total synthesis and biological evaluation of Phidianidines A and B uncovers unique pharmacological profiles at CNS targets. ACS Chem Neurosci 2012;3:658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Singleton PA, Moss J, Karp DD, Atkins JT, Janku F. The mu opioid receptor: A new target for cancer therapy? Cancer 2015;121:2681–2688. [DOI] [PubMed] [Google Scholar]
- 511. Vitale RM, Gatti M, Carbone M, Barbieri F, Felicità V, Gavagnin M, Florio T, Amodeo P. Minimalist hybrid ligand/receptor‐based pharmacophore model for cxcr4 applied to a small‐library of marine natural products led to the identification of phidianidine a as a new cxcr4 ligand exhibiting antagonist activity. ACS Chem Biol 2013;8:2762‒2770. [DOI] [PubMed] [Google Scholar]
- 512. Lin HY, Snider BB. Synthesis of phidianidines A and B. J Org Chem 2012;77:4832–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Manzo E, Pagano D, Carbone M, Ciavatta ML, Gavagnin M. Synthesis of phidianidine B, a highly cytotoxic 1,2,4‐oxadiazole marine metabolite. Arkivoc 2012;ix:220–228. [Google Scholar]
- 514. Manzo E, Pagano D, Ciavatta ML, Carbone M, Gavagnin M. U.S. Patent No. 9.085.569 “New 1,2,4‐oxadiazol derivatives, process for their preparation and use thereof as intermediates in the preparation of indolic alkaloids”. PCT/IB2013/052163 US9085569 B2, 2015.
- 515. Fontana A, Cavaliere P, Wahidulla S, Naik CG, Cimino G. A new antitumor isoquinoline alkaloid from the marine nudibranch Jorunna funebris . Tetrahedron 2000;56:7305‒7308. [Google Scholar]
- 516. Petek BJ, Jones RL. PM00104 (Zalypsis®): A marine derived alkylating agent. Molecules 2014;19:12328–12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Charupant K, Suwanborirux K, Amnuoypol S, Saito E, Kubo A, Saito N. Jorunnamycins A—C, new stabilized renieramycin‐type bistetrahydroisoquinolines isolated from the Thai nudibranch Jorunna funebris . Chem Pharm Bull 2007;55:81‒86. [DOI] [PubMed] [Google Scholar]
- 518. Charupant K, Daikuhara N, Saito E, Amnuoypol S, Suwanborirux K, Owa T, Saito N. Chemistry of renieramycins. Part 8: Synthesis and cytotoxicity evaluation of renieramycin M‐jorunnamycin A analogues. Bioorg Med Chem 2009;17:4548‒4558. [DOI] [PubMed] [Google Scholar]
- 519. McPherson JR, Ong CK, Ng CC, Rajasegaran V, Heng HL, Yu WS, Tan BK, Madhukumar P, Teo MC, Ngeow J, Thike AA, Rozen SG, Tan PH, Lee AS, Teh BT, Yap YS. Whole‐exome sequencing of breast cancer, malignant peripheral nerve sheath tumor and neurofibroma from a patient with neurofibromatosis type 1. Cancer Med 2015;4:1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, Ye X, Chiu C, Pham T, Hongo JA, Bainbridge T, Firestein R, Blackwood E, Metcalfe C, Stawiski EW, Yauch RL, Wu Y, de Sauvage FJ. Targeting PTPRK‐RSPO3 colon tumours promotes differentiation and loss of stem‐cell function. Nature 2016;529:97–100. [DOI] [PubMed] [Google Scholar]
- 521. Rinehart KL, Fregeau NL, Warwick RA, Garcia Gravalos D, Avila J, Faircloth GT. Spisulosine compounds having antitumor activity. PCT WO0052521 A1 19991021, 1999.
- 522. Abad JL, Nieves I, Rayo P, Casas J, Fabriàs G, Delgado A. Straightforward access to spisulosine and 4,5‐dehydrospisulosine stereoisomers: Probes for profiling ceramide synthase activities in intact cells. J Org Chem 2013;78:5858–5866. [DOI] [PubMed] [Google Scholar]
- 523. Faircloth G, Cuevas C. Kahalalide F and ES285: Potent anticancer agents from marine molluscs In Cimino G, Gavagnin M, Eds. Molluscs: From Ecological Study to Biotechnological Application. Springer, New York: 2006. p 363‒379. [DOI] [PubMed] [Google Scholar]
- 524. Baird RD, Kitzen J, Clarke PA, Planting A, Reade S, Reid A, Welsh L, López Lázaro L, de las Heras B, Judson IR, Kaye SB, Eskens F, Workman P, deBono JS, Verweij J. Phase I safety, pharmacokinetic, and pharmacogenomic trial of ES‐285, a novel marine cytotoxic agent, administered to adult patients with advanced solid tumors. Mol Cancer Ther 2009;8:1430–1437. [DOI] [PubMed] [Google Scholar]
- 525. Salcedo M, Cuevas C, Alonso JL, Otero G, Faircloth G, Fernandez‐Sousa JM, Avila J, Wandosell F. The marine sphingolipid‐derived compound ES 285 triggers an atypical cell death pathway. Apoptosis 2007;12:395–409. [DOI] [PubMed] [Google Scholar]
- 526. Sánchez AM, Malagarie‐Cazenave S, Olea N, Vara D, Cuevas C, Díaz‐Laviada I. Spisulosine (ES‐285) induces prostate tumor PC‐3 and LNCaP cell death by de novo synthesis of ceramide and PKCzeta activation. Eur J Pharmacol 2008;584:237–245. [DOI] [PubMed] [Google Scholar]
- 527. Cuadros R, Wandosell F, Faircloth G, Fernandez‐Sousa JM, Avila J. The marine compound spisulosine, an inhibitor of cell proliferation, promotes the disassembly of actin stress fibers. Cancer Lett 2000;152:23–29. [DOI] [PubMed] [Google Scholar]
- 528. Schöffski P, Dumez H, Ruijter R, Miguel‐Lillo B, Soto‐Matos A, Alfaro V, Giaccone G. Spisulosine (ES‐285) given as a weekly three‐hour intravenous infusion: Results of a phase I dose‐escalating study in patients with advanced solid malignancies. Cancer Chemother Pharmacol 2011;68:1397–1403. [DOI] [PubMed] [Google Scholar]
- 529. Vilar E, Grünwald V, Schöffski P, Singer H, Salazar R, Iglesias JL, Casado E, Cullell‐Young M, Baselga J, Tabernero J. A phase I dose‐escalating study of ES‐285, a marine sphingolipid‐derived compound, with repeat dose administration in patients with advanced solid tumors. Invest New Drugs 2012;30:299–305. [DOI] [PubMed] [Google Scholar]
- 530. Massard C, Salazar R, Armand JP, Majem M, Deutsch E, García M, Oaknin A, Fernández‐García EM, Soto A, Soria JC. Phase I dose‐escalating study of ES‐285 given as a three‐hour intravenous infusion every three weeks in patients with advanced malignant solid tumors. Invest New Drugs 2012;30:2318–2326. [DOI] [PubMed] [Google Scholar]
- 531. Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev 2013;65:1866–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti‐cancer drug delivery. J Control Release 2010;148:135–146. [DOI] [PubMed] [Google Scholar]
- 533. Narang AS, Varia S. Role of tumor vascular architecture in drug delivery. Adv Drug Deliv Rev 2011;63:640–658. [DOI] [PubMed] [Google Scholar]
- 534. Byrne JD, Betancourt T, Brannon‐Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev 2008;60:1615–1626. [DOI] [PubMed] [Google Scholar]
- 535. Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012;2:3–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Kolate A, Baradia D, Patil S, Vhora I, Kore G, Misra A. PEG – A versatile conjugating ligand for drugs and delivery systems. J Control Rel 2014;192:67–81. [DOI] [PubMed] [Google Scholar]
- 537. Mura S, Nicolas J, Couvreur P. Stimuli‐responsive nanocarriers for drug delivery. Nat Mater 2013;12:991–1003. [DOI] [PubMed] [Google Scholar]
- 538. Pla D, Martí M, Farrera‐Sinfreu J, Pulido D, Francesch A, Calvo P, Cuevar C, Royo M, Aligué R, Albericio F, Álvarez M. Lamellarin D bioconjugates II: Synthesis and cellular internalization of dendrimer and nuclear location signal derivatives. Bioconj Chem 2009;20:1112–1121. [DOI] [PubMed] [Google Scholar]
- 539. Pungkham H, Swatdipakdi N, Theerasilp M, Karnkla S, Chittchang M, Ploypradith P, Nasongla N. PEG‐b‐PCL and PEG‐b‐PLA polymeric micelles as nanocarriers for Lamellarin N delivery. Conf Proc III Eng Med Biol Soc 2011;2011:3245–3248. [DOI] [PubMed] [Google Scholar]
- 540. Hosta L, Pla‐Roca M, Arbiol J, López‐Iglesias C, Samitier J, Cruz LJ, Kogan MJ, Albericio F. Conjugation of Kahalalide F with gold nanoparticles to enhance in vitro antitumor activity. Bioconj Chem 2009;20:138–146. [DOI] [PubMed] [Google Scholar]
- 541. Estella‐Hermoso de Mendoza A, Calvo P, Bishop A, Avilés P, Blanco‐Prieto MJ. Comparison of pharmacokinetic profiles of PM02734 loaded lipid nanoparticles and cyclodextrins: in vitro and in vivo characterization. J Biomed Nanotechnol 2012;8:703–708. [DOI] [PubMed] [Google Scholar]
- 542. Legigan T, Clarhaut J, Tranoy‐Opalinski I, Monvoisin A, Renoux B, Thomas M, Le Pape A, Lerondel S, Papot S. The first generation of β‐galactosidase‐responsive prodrugs designed for the selective treatment of solid tumors in prodrug monotherapy. Angewandte 2012;51:11606–11610. [DOI] [PubMed] [Google Scholar]
- 543. Burns KE, Robinson MK, Thévenin D. Inhibition of cancer cell proliferation and breast tumor targeting of pHLIP‐Monomethyl Aurisatin E Conjugates. Mol Pharm 2015;12:1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Batisse C, Dransart E, Sarkouh RA, Brulle L, Bai S‐K, Godefroy S, Johannes L, Schmidt F. A new delivery system for auristatin in STxB‐drug conjugate therapy. Eur J Med Chem 2015;95:483–491. [DOI] [PubMed] [Google Scholar]
- 545. Pan L‐Q, Wang H‐B, Xie Z‐M, Li Z‐H, Tang X‐J, Xu Y‐C, Zhang C, Naranmanduran H, Chen S‐Q. Novel conjugation of tumor‐necrosis‐factor‐related apoptosis‐inducing ligand (TRAIL) with monomethyl auristatin E for efficient antitumor drug delivery. Adv Mater 2013;25:4718–4722. [DOI] [PubMed] [Google Scholar]
- 546. Temming K, Meyer DL, Zabinski R, Dijkers ECF, Poelstra K, Molema G, Kok J. Evaluation of RGD‐targeted albumin carriers for specific delivery of auristatin E to tumor blood vessels. Bioconj Chem 2006;17:1385–1394. [DOI] [PubMed] [Google Scholar]
- 547. Temming K, Meyer DL, Zabinski R, Senter PD, Poelstra K, Molema G, Kok RJ. Improved efficacy of ανβ3‐targeted albumin conjugates by conjugation of a novel auristatin derivative. Mol Pharm 2007;4:686–694. [DOI] [PubMed] [Google Scholar]
- 548. Crisp JL, Savariar EN, Glasgow HL, Ellies LG, Whitney MA, Tsien RY. Dual targeting of integrin ανβ3 and matrix metalloproteinase‐2 for optical imaging of tumors and chemotherapeutic delivery. Mol Cancer Ther 2014;13:1514–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Buckel L, Savariar EN, Crisp JL, Jones KA, Hicks AM, Scanderbeg DJ, Nguyen QT, Sicklick JK, Lowy AM, Tsien RY, Advani SJ. Tumor radiosensitation by monomethyl auristatin E: Mechanism of action and targeted delivery. Cancer Res 2015;75:1376–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Newman DJ. Developing natural product drugs: Supply problems and how they have been overcome. Pharmacol Ther 2016;162:1–9. [DOI] [PubMed] [Google Scholar]
- 551. Gomes NG, Dasari R, Chandra S, Kiss R, Kornienko A. Marine invertebrate metabolites with anticancer activities: Solutions to the “supply problem.” Mar Drugs 2016;14:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Glaser KB, Mayer A. A renaissance in marine pharmacology: From preclinical curiosity to clinical reality. Biochem Pharmacol 2009;78:440–448. [DOI] [PubMed] [Google Scholar]
- 553. Leal MC, Nunes C, Alexandre D, Silva TL, Reis A, Dinis MT, Calado R. Parental diets determine the embryonic fatty acid profile of the tropical nudibranch Aeolidiella stephanieae: The effect of eating bleached anemones. Mar Biol 2012;159:1745–1751. [Google Scholar]
- 554. Cruz S, Calado R, Serôdio J, Cartaxana P. Crawling leaves: Photosynthesis in Sacoglossan sea slugs. J Exp Bot 2013;64:3999–4009. [DOI] [PubMed] [Google Scholar]
- 555. Carroll D, Kempf S. Laboratory culture of the aeolid nudibranch Berghia verrucicornis (Mollusca, Opisthobranchia): Some aspects of its development and life history. Biol Bull 1990;179:243–253. [DOI] [PubMed] [Google Scholar]
- 556. O'Brien FX, McKindsey CH, Landry T, Costa‐Pierce BA. Methods of sustainable shelfish aquaculture In: Christou P, Savin R, Costa‐Pierce BA, Misztal I, Whitelaw BA, Eds. Sustainable Food Production. Springer Science+Business Media, New York: 2013. p 1436–1548. [Google Scholar]
- 557. Shawl AL, Davis M. Captive breeding behaviour of four strombidae conch. J Shellfish Res 2004;23:157–164. [Google Scholar]
- 558. Dolorosa RG, Grant A, Gill JA, Avillanosa AL, Gonzales BJ. Indoor and deep sub‐tidal intermediate culture of Trochus niloticus for restocking. Rev Fish Sci 2013;21:414–423. [Google Scholar]
- 559. Capo T, Bardales A, Gillette P, Lara M, Schmale M, Serafy J. Larval growth, development, and survival of laboratory‐reared Aplysia californica: Effects of diet and veliger density. Comp Biochem Phys C 2008;149:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Wilson RM, Danishefsky SJ. Applications of total synthesis toward the discovery of clinically useful anticancer agents. Chem Soc Rev 2007;36:1207–1226. [DOI] [PubMed] [Google Scholar]
- 561. López‐Macià A, Jiménez JC, Royo M, Giralt E, Albericio F. Synthesis and structure determination of Kahalalide F. J Am Chem Soc 2001;23:11398–11401. [DOI] [PubMed] [Google Scholar]
- 562. Shilabin AG, Kasanah N, Wedge DE, Hamann MT. Lysosome and HER3 (ErbB3) selective anticancer agent Kahalalide F: Semisynthetic modifications and antifungal lead‐exploration studies. J Med Chem 2007;50:4340–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Jiménez JC, López‐Macià A, Gracia C, Varón S, Carrascal M, Caba JM, Royo M, Francesch AM, Cuevas C, Giralt E, Albericio F. Structure‐activity relationship of Kahalalide F synthetic analogues. J Med Chem 2008;51:4920‒4931. [DOI] [PubMed] [Google Scholar]
- 564. Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol 2012;30:631–637. [DOI] [PubMed] [Google Scholar]
- 565. Perez M, Fernandez C, Chicharro JL, Zarzuelo M, de la Calle F, Cuevas C, Francesch A, Gallego P, Manzanares I. Hemisynthetic methods and new compounds. WO 20000069862, 23 November 2000.
- 566. Nicolaou KC, Hale CRH, Nilewskia C, Ioannidou HA. Constructing molecular complexity and diversity: Total synthesis of natural products of biological and medicinal importance. Chem Soc Rev 2012;41:5185–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Kuttruff CA, Eastgate MD, Baran PS. Natural product synthesis in the age of scalability. Nat Prod Rep 2014;31:419–432. [DOI] [PubMed] [Google Scholar]
- 568. Yu MJ, Zheng W, Seletsky BM. From micrograms to grams: Scale‐up synthesis of eribulin mesylate. Nat Prod Rep 2013;30:1158–1164. [DOI] [PubMed] [Google Scholar]
- 569. Adams DJ. The Valley of Death in anticancer drug development: A re‐assessment. Trends Pharmacol Sci 2012;33:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Hammonds T. Academic‐Pharma drug discovery alliances: Seeking ways to eliminate the valley of death. Future Med Chem 2015;7:1891–1899. [DOI] [PubMed] [Google Scholar]
- 571. Tannock IF. Cell proliferation In: Tannock IF, Hill RP, Eds. The Basic Science of Oncology. New York, McGraww‐Hill, Inc, 1992. p 154–177. [Google Scholar]