

Abstract
The replacement of noble metal technologies and the realization of new reactivities with earth‐abundant metals is at the heart of sustainable synthesis. Alkene hydrogenations have so far been most effectively performed by noble metal catalysts. This study reports an iron‐catalyzed hydrogenation protocol for tri‐ and tetra‐substituted alkenes of unprecedented activity and scope under mild conditions (1–4 bar H2, 20 °C). Instructive snapshots at the interface of homogeneous and heterogeneous iron catalysis were recorded by the isolation of novel Fe nanocluster architectures that act as catalyst reservoirs and soluble seeds of particle growth.
Keywords: amides, hydrides, hydrogenations, iron, nanoclusters
Catalytic hydrogenations of unsaturated C=C bond systems are pivotal to modern chemical transformations and mostly performed with nickel or platinum group catalysts.1 While some of the largest technical processes are iron‐catalyzed hydrogenations (Haber–Bosch, Fischer–Tropsch), the potential of iron as abundant, non‐toxic, and cheap transition metal catalyst for C=C hydrogenations has only very recently been tapped.2 Significant progress in the design of molecular Fe catalysts was made by the introduction of tridentate bis(imino)pyridine ligands (PDI) by Budzelaar et al.3 and Chirik et al.4 The (PDI)Fe(N2)2 pre‐catalysts cleanly hydrogenate mono‐ and di‐substituted alkenes under mild conditions and exceed the productivity of some precious metal catalysts.4 Further improved activities were observed with the related bis(carbene)–pyridine iron(0) complexes (Scheme 1, top).4 On the other hand, ill‐defined or nanoparticulate Fe catalysts were prepared by decompositions of iron carbonyls or by reductions of iron salts with organometallic or hydride reagents but exhibited only moderate hydrogenation activities.5 While providing an operationally simple access to Fe‐based hydrogenation catalysts, the latter approaches provided limited mechanistic insight, often involved precipitation of heterogeneous species especially in the absence of suitable ligands, and generally displayed high catalyst sensitivity and limited scope. From our recent studies into the development of low‐valent iron catalysts for hydrogenations,6 we reasoned that an effective yet operationally simple protocol would fulfill the following criteria: 1) The active catalyst is prepared in situ by the reduction of iron(II) precursors with commercial reductants; 2) the catalyst contains bulky ligands that are cheap, easily available, coordinate iron in various low oxidation states, and prevent unwanted aggregation to larger, catalytically inactive particles; 3) the ligands create a lipophilic periphery that enhances solubilization under the non‐polar conditions of alkene hydrogenations; and iv) the catalytic hydrogenation operates under mild conditions without sophisticated additives in common organic solvents. With these framework conditions, we investigated combinations of iron(II) bis(1,1,1,3,3,3‐hexamethyl‐disilazan‐2‐ide), Fe(hmds)2,7 and various reductants. Documented herein are the benefits of using this simple catalytic system that presents tangible advances over the current state‐of‐the‐art that could not have been predicted: Clean hydrogenations of challenging alkenes (for exqmple, tetra‐substituted) proceed under very mild conditions. A most user‐friendly protocol can be adopted by simple mixing of the ferrous salt, reductant, and ligand. The isolation of novel soluble Fe nanocluster topologies provides new insight into reductive catalyst formation and cluster aggregation (Scheme 1, bottom).
Scheme 1.
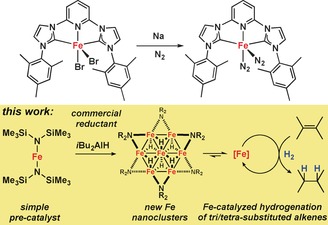
Soluble Fe catalysts for hydrogenations of alkenes.
There are several reports of the coordination chemistry of Fe(hmds)2 in the presence of various ligands, but only very few applications to catalytic reactions have been demonstrated.8 The displacement of hmds ligands from Fe(hmds)2 by formal hydride donors has not received significant attention despite its relevance to the preparation of simple hydridoiron species9 and hydrogenase model compounds.10 In the context of alkene hydrogenations, Chaudret et al. prepared catalytically active Fe nanoparticles by thermal decomposition of Fe(hmds)2 at 150 °C in the presence of H2.11 We studied the generation of active hydrogenation catalysts from Fe(hmds)2 and various simple and commercial hydride donors and reductants under mild conditions (Table 1). Ethylmagnesium chloride or zinc afforded poor hydrogenation catalysts (entries 1, 2). Similar low activity was observed when following Chaudret's protocol of thermal decomposition of Fe(hmds)2 to nanoparticles (entry 3).11 Extremely high hydrogenation activity was achieved in the presence of aluminium hydrides and organoaluminium reagents (entries 6–9).12 The most active catalyst was formed with diiso‐butylaluminium hydride (Dibal‐H) which afforded quantitative conversion of 1‐phenyl‐1‐cyclohexene at 1.3 bar H2 and 20 °C after 30 min. The operationally most convenient in situ catalyst formation from FeCl2, HN(SiMe3)2, and n‐butyl lithium gave nearly identical yields (entry 10). Complete inhibition was observed in the absence of Dibal‐H or the amido ligand N(TMS)2, respectively (entries 11, 12). Further tests of the catalyst mixtures revealed high chemoselectivity and robustness when employing Dibal‐H (Scheme 2, Table 2). This catalyst could be stored in solution for several days or dried in vacuum without significant loss of activity (entries 1–4, Table 2, turnover frequency (TOF) recorded after 7 min reaction at about 0 % conversion).
Table 1.
Selected optimization experiments.[a]

| Entry | Reductant (mol %) | Conditions | Yield [%][b] |
|---|---|---|---|
| 1 | EtMgCl (10) | 5 bar H2, 40 °C, 18 h | 5 (9) |
| 2 | Zn (10) | as entry 1 | <1 (1) |
| 3 | – | 5 bar H2, 150 °C, 18 h | 1 (1) |
| 4 | NaBH4 (5) | as entry 1 | 99 (99) |
| 5 | NaBH4 (5) | 1.3 bar H2, 20 °C, 3 h | 1 (2) |
| 6 | LiAlH4 (5) | as entry 4 | 99 (99) |
| 7 | Me3Al (10) | 1.3 bar H2, 20 °C, 0.5 h | 90 (98) |
| 8 | iBu3Al (10) | as entry 7 | 93 (99) |
| 9 | i Bu2AlH (10) | as entry 7 | 100 (100) |
| 10 | iBu2AlH (10) | FeCl2, HN(TMS)2, nBuLi[d] | 98 (99) |
| 11 | – | as entry 7 | <1 (1) |
| 12 | iBu2AlH (10) | as entry 7, FeCl2 [c] | <1 (1) |
[a] Conditions: 0.2 mmol alkene, 0.5 m in toluene, 5 mol % Fe[N(SiMe3)2]2, reductant, H2. [b] Yields determined by quantitative GC‐FID vs. internal n‐pentadecane. [c] 5 mol % FeCl2 instead of Fe(hmds)2. [d] 5 mol % FeCl2, 10 mol % HN(SiMe3)2, 10 mol % n‐butyl lithium (1.6 m in PhMe) instead of Fe(hmds)2.
Scheme 2.
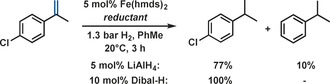
Chemoselectivity of the Fe(hmds)2/Dibal‐H catalyst.
Table 2.
Robustness of the Fe(hmds)2/Dibal‐H catalyst.

| Entry | Reductant | Catalyst treatment | TOF [h−1] |
|---|---|---|---|
| 1 | Dibal‐H | freshly prepared | 41 |
| 2 | Dibal‐H | storage for 5 d in solution | 37 |
| 3 | Dibal‐H | solvent removal, then dissolution | 30 |
| 4 | Dibal‐H | solvent removal, storage for 5 d, then dissolution | 27 |
| 5 | Me3Al | freshly prepared | 13 |
| 6 | Me3Al | storage for 1 d in solution | <1 |
| 7 | Dibal‐H | from FeCl2⋅1.5 thf, HN(TMS)2, n‐BuLi | 27 |
The optimized set of conditions was applied to the hydrogenation of various alkenes (Scheme 3). Mono‐, di‐, and tri‐substituted alkenes were cleanly reacted under 2 bar H2 pressure at room temperature.
Scheme 3.
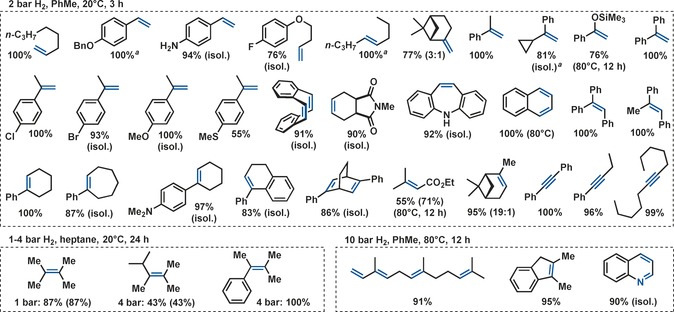
Substrate scope of iron‐catalyzed hydrogenations of alkenes and alkynes. Bonds in blue indicate the site of complete π‐bond hydrogenation. Standard conditions: 0.2 mmol alkene/alkyne, 0.5 m in toluene, 5 mol % Fe[N(SiMe3)2]2, 10 mol % Dibal‐H, 2 bar H2, 20 °C, 3 h. If not otherwise noted, yields were determined by quantitative GC‐FID vs. n‐pentadecane. Conversions are given in parentheses if <90 %. [a] 0.5 mol % Fe[N(SiMe3)2]2, 1 mol % Dibal‐H.
The mild conditions tolerated fluoride, chloride, bromide, silylenol ether, amine, imide, ester, thioether, and benzyl ether functions. The hydrogenations of some challenging substrates required elevated temperature and/or pressure. Remarkably mild conditions enabled the hydrogenation of tetra‐substituted alkenes (1–4 bar H2, 20 °C).4 The harsher conditions required for complete hydrogenation of 1,2‐dimethylindene might be a consequence of the low isomerization activity of the Fe(hmds)2/Dibal‐H catalyst.13 Notably, no ring‐opening of α‐cyclopropyl styrene was observed.14 With reduced catalyst loadings of 0.5 mol % Fe(hmds)2 and 1 mol % Dibal‐H, turnover frequencies (TOF in h−1) of 660 and 280 were recorded in the hydrogenations of 1‐octene and α‐methylstyrene, respectively (2 bar H2, PhMe, 20 °C, 5 min). Under the same conditions, conversion of 1‐phenyl‐1‐cyclohexene required 3 mol % catalyst loading which resulted in a TOF of 60 h−1. Alkynes were cleanly reacted to alkanes under identical conditions (Scheme 3).
Kinetic poisoning studies were performed to ascertain the topicity of the operating catalyst species.15 The addition of sub‐catalytic amounts of trimethylphosphine (PMe3) led to catalyst inhibition already at a catalyst/poison ratio of 10:1 (Scheme 4, top).16
Scheme 4.
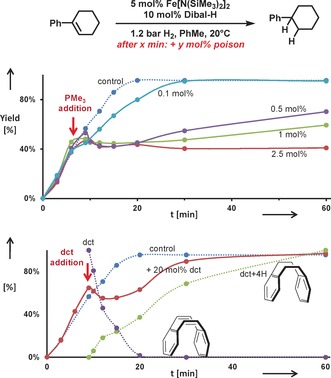
Poisoning studies with trimethylphosphine (PMe3, top) and dibenzo[a,e]cyclooctatetraene (dct, bottom).
Contrary to this, the selective homogeneous catalyst poison dibenzo‐[a,e]cyclooctatetraene17 (dct, 4 equiv per Fe) showed no significant inhibition but was merely a competing substrate for hydrogenation (Scheme 4, bottom). We thus postulate the operation of a heterotopic mechanism by polynuclear low‐valent Fe catalysts.
In an effort to identify potential catalytically active species, we investigated the reaction of Fe[N(SiMe3)2]2 with Dibal‐H under the conditions of the hydrogenation reactions (toluene or hexane, 20 °C). The reaction of Fe[N(SiMe3)2]2 and Dibal‐H in a toluene/hexane mixture underwent rapid color change from green to brown–black. Filtration, removal of the solvents, and crystallization from n‐hexane afforded the dark crystalline Fe4 nanocluster Fe3(hmds)4Fe(toluene) in 38 % yield (Scheme 5, Figure 1).18 Single crystal structure analysis showed a planar Fe4 core which is peripherally decorated with four hmds ligands of which two hmds adopt a bridging μ2‐coordination mode. One Fe atom bears an η6‐toluene. The paramagnetic complex had a melting point of 123 °C and exhibited an effective magnetic moment μ eff=2.0 μB (in C6D6). Two structurally related nanoclusters were isolated by slow solvent evaporation from the reaction of Fe[N(SiMe3)2]2 and Dibal‐H in n‐hexane. Crystal structure analysis established the dark‐red oligohydridoiron clusters Fe5(hmds)6FeH5 and Fe6(hmds)6FeH6 (35 % yield, 4/1, Scheme 5, Figure 1). The Fe6 cluster is a truncated derivative of the Fe7 cluster and bears one μ2‐H and four μ3‐H atoms coordinated to iron. The highly symmetrical Fe7 cluster, a low‐valent “Fe wheel”, contains six peripheral μ2‐hmds ligands and six μ3‐H ligands.19 The composition of the cluster mixture was further verified by X‐ray analysis, elemental analysis, and LIFDI‐MS (m/z 1301.2287, 1358.1793). The Fe4, Fe6, and Fe7 nanocluster architectures contain multiple iron centers in low oxidation states (formally Fe0, FeI, FeII) and constitute a distinct class of metallic cluster complexes20 that adopt rare planar Fen geometries and are void of the common carbonyl, nitrido, oxo and carbido ligands.21 Generally, discrete metallic clusters with direct interactions between the redox centers are considered as materials for optical, magnetic, and catalytic applications.22 Detailed studies of spectroscopic and coordination properties of the Fe nanoclusters are beyond the scope of this catalytic method development but will be reported soon. Preliminary studies proved that the Fe4 nanocluster is a competent hydrogenation pre‐catalyst in the presence of Dibal‐H and HN(TMS)2 (Scheme 6).
Scheme 5.
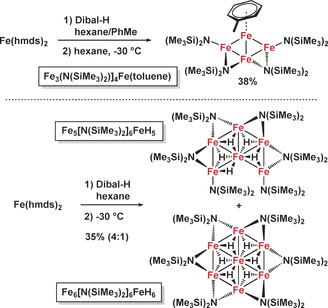
Synthesis of novel planar Fe4, Fe6, and Fe7 nanoclusters.
Figure 1.
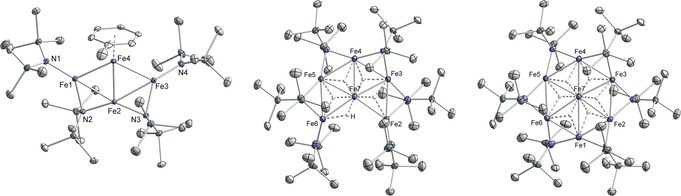
Crystal structures (50 % probability level, peripheral H atoms omitted) of Fe3(hmds)4Fe(toluene), Fe5(hmds)6FeH4, Fe6(hmds)6FeH6 (left to right).
Scheme 6.
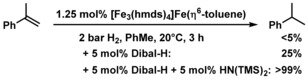
Catalytic hydrogenation with the isolated Fe4 nanocluster.
In summary, we have developed an iron‐catalyzed hydrogenation protocol that displays unprecedented activity for challenging tri‐ and tetra‐substituted alkenes under very mild reaction conditions. The catalyst is prepared by reaction of Fe[N(SiMe3)2]2 with diisobutylaluminum hydride or by a most user‐friendly in situ method from FeCl2. The isolation of novel low‐valent nanoclusters with planar Fe4, Fe6, and Fe7 geometries under such conditions provides new insight into the interface of homogeneous/heterogeneous catalysis and the growth of metallic nanoparticle materials. Further studies of the spectroscopic and chemical properties of these and related planar [(amido)Fe]n nanoclusters are currently being executed.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Evonik Foundation (T.N.G.) and the Fonds der Chemischen Industrie (M.V.) This work was funded by the European Research Council (ERC) through a Consolidator grant (683150).
T. N. Gieshoff, U. Chakraborty, M. Villa, A. Jacobi von Wangelin, Angew. Chem. Int. Ed. 2017, 56, 3585.
References
- 1.
- 1a. The Handbook of Homogeneous Hydrogenation (Eds.: J. G. de Vries, C. J. Elsevier), Wiley-VCH, Weinheim, 2007; [Google Scholar]
- 1b. Nishimura S., Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001. [Google Scholar]
- 2.
- 2a. Catalysis without Precious Metals (Ed.: R. M. Bullock), Wiley-VCH, Weinheim, 2010; [Google Scholar]
- 2b. Chirik P. J., Acc. Chem. Res. 2015, 48, 1687; [DOI] [PubMed] [Google Scholar]
- 2c. Junge K., Schröder K., Beller M., Chem. Commun. 2011, 47, 4849; [DOI] [PubMed] [Google Scholar]
- 2d. Le Bailly B. A. F., Thomas S. P., RSC Adv. 2011, 1, 1435. [Google Scholar]
- 3. Knijnenburg Q., Horton A. D., van der Heijden H., Gal A. W., Budzelaar P. H. M., WO2003042131 A1 20030522, 2003.
- 4.
- 4a. Bart S. C., Lobkovsky E., Chirik P. J., J. Am. Chem. Soc. 2004, 126, 13794; [DOI] [PubMed] [Google Scholar]
- 4b. Trovitch R. J., Lobkovsky E., Bill E., Chirik P. J., Organometallics 2008, 27, 1470; [Google Scholar]
- 4c. Hoyt J. M., Shevlin M., Margulieux G. W., Krska S. W., Tudge M. T., Chirik P. J., Organometallics 2014, 33, 5781; [Google Scholar]
- 4d. Yu R. P., Darmon J. M., Hoyt J. M., Margulieux G. W., Turner Z. R., Chirik P. J., ACS Catal. 2012, 2, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Phua P. H., Lefort L., Boogers J. A. F., Tristany M., de Vries J. G., Chem. Commun. 2009, 3747; [DOI] [PubMed] [Google Scholar]
- 5b. Rangheard C., de Julian Fernandez C., Phua P.-H., Hoorn J., Lefort L., de Vries J. G., Dalton Trans. 2010, 39, 8464; [DOI] [PubMed] [Google Scholar]
- 5c. Stein M., Wieland J., Steurer P., Tölle F., Mülhaupt R., Breit B., Adv. Synth. Catal. 2011, 353, 523; [Google Scholar]
- 5d. Hudson R., Rivière A., Cirtiu C. M., Luska K. L., Moores A., Chem. Commun. 2012, 48, 3360; [DOI] [PubMed] [Google Scholar]
- 5e. Welther A., Jacobi von Wangelin A., Curr. Org. Chem. 2013, 17, 326; [Google Scholar]
- 5f. Carter T. S., Guiet L., Frank D. J., West J., Thomas S. P., Adv. Synth. Catal. 2013, 355, 880; [Google Scholar]
- 5g. Frank D. J., Guiet L., Kaslin A., Murphy E., Thomas S. P., RSC Adv. 2013, 3, 25698; [Google Scholar]
- 5h. Hudson R., Hamasaka G., Osako T., Yamada Y. M. A., Li C.-J., Uozumi Y., Moores A., Green Chem. 2013, 15, 2141; [Google Scholar]
- 5i. MacNair A. J., Tran M.-M., Nelson J. E., Sloan G. U., Ironmonger A., Thomas S. P., Org. Biomol. Chem. 2014, 12, 5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Gärtner D., Welther A., Rad B. R., Wolf R., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2014, 53, 3722; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3796; [Google Scholar]
- 6b. Gieshoff T. N., Villa M., Welther A., Plois M., Chakraborty U., Wolf R., Jacobi von Wangelin A., Green Chem. 2015, 17, 1408; [Google Scholar]
- 6c. Welther A., Bauer M., Mayer M., Jacobi von Wangelin A., ChemCatChem 2012, 4, 1088. [Google Scholar]
- 7. Andersen R. A., Faegri K., Green J. C., Haaland A., Lappert M. F., Leung W.-P., Inorg. Chem. 1988, 27, 1782. [Google Scholar]
- 8.Selected recent examples:
- 8a. Lipschutz M. I., Chantarojsiri T., Dong Y., Tilley T. D., J. Am. Chem. Soc. 2015, 137, 6366; [DOI] [PubMed] [Google Scholar]
- 8b. Maddock L. C. H., Cadenbach T., Kennedy A. R., Borilovic I., Aromí G., Hevia E., Inorg. Chem. 2015, 54, 9201; [DOI] [PubMed] [Google Scholar]
- 8c. Hatakeyama T., Imayoshi R., Yoshimoto Y., Ghorai S. K., Jin M., Takaya H., Norisuye K., Sohrin Y., Nakamura M., J. Am. Chem. Soc. 2012, 134, 20262; [DOI] [PubMed] [Google Scholar]
- 8d. Yang J., Tilley T. D., Angew. Chem. Int. Ed. 2010, 49, 10186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10384; [Google Scholar]
- 8e. Yang J., Fasulo M., Tilley T. D., New J. Chem. 2010, 34, 2528. [Google Scholar]
- 9. Nakazawa H., Itazaki M., Top. Organomet. Chem. 2011, 33, 27. [Google Scholar]
- 10.
- 10a. Lubitz W., Ogata H., Rüdiger O., Reijerse E., Chem. Rev. 2014, 114, 4081; [DOI] [PubMed] [Google Scholar]
- 10b. Tschierlei S., Ott S., Lomoth R., Energy Environ. Sci. 2011, 4, 2340; [Google Scholar]
- 10c. Du P., Eisenberg R., Energy Environ. Sci. 2012, 5, 6012. [Google Scholar]
- 11. Kelsen V., Wendt B., Werkmeister S., Junge K., Beller M., Chaudret B., Chem. Commun. 2013, 49, 3416. [DOI] [PubMed] [Google Scholar]
- 12.Ziegler-type Fe/Al catalysts in hydrogenations:
- 12a. Sloan M. F., Matlack A. S., Breslow D. S., J. Am. Chem. Soc. 1963, 85, 4014; [Google Scholar]
- 12b. Takegami Y., Ueno T., Fujii T., Bull. Chem. Soc. Jpn. 1965, 38, 1279; [Google Scholar]
- 12c. Noskova N. F., Kazimova A. Zh., Kambarova K. K., Savelev S. R., Melamud N. L., Zh. Org. Khim. 1992, 28, 1352. [Google Scholar]
- 13.
- 13a.Isomerization of allylbenzene (to β-methylstyrene) and 1-octene (to 2-,3-,4-octenes) with 5 mol % Fe(hmds)2/10 mol % Dibal-H proceeded sluggishly under otherwise identical conditions (no H2).
- 13b. Mayer M., Welther A., Jacobi von Wangelin A., ChemCatChem 2011, 3, 1567. [Google Scholar]
- 14. Newcomb M. in Encyclopedia of Radicals in Chemistry, Biology and Materials (Eds.: C. Chatgilialoglu, A. Studer), Wiley, Chichester, 2012. [Google Scholar]
- 15.
- 15a. Widegren J. A., Finke R. G., J. Mol. Catal. A 2003, 198, 317; [Google Scholar]
- 15b. Astruc D., Lu F., Ruiz Aranzaes J., Angew. Chem. Int. Ed. 2005, 44, 7852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 8062; [Google Scholar]
- 15c. Crabtree R. H., Chem. Rev. 2012, 112, 1536. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Tseng K.-N. T., Kampf J. W., Szymczak N. K., ACS Catal. 2015, 5, 411; [Google Scholar]
- 16b. Li Y., Yu S., Wu X., Xiao J., Shen W., Dong Z., Gao J., J. Am. Chem. Soc. 2014, 136, 4031; [DOI] [PubMed] [Google Scholar]
- 16c. Alberico E., Sponholz P., Cordes C., Nielsen M., Drexler H.-J., Baumann W., Junge H., Beller M., Angew. Chem. Int. Ed. 2013, 52, 14162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14412. [Google Scholar]
- 17.
- 17a. Anton D. R., Crabtree R. H., Organometallics 1983, 2, 855; [Google Scholar]
- 17b. Franck G., Brill M., Helmchen G., J. Org. Chem. 2012, 89, 55; [Google Scholar]
- 17c. Sonnenberg J. F., Morris R. H., Catal. Sci. Technol. 2014, 4, 3426; [Google Scholar]
- 17d. Gärtner D., Stein A. L., Grupe S., Arp J., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2015, 54, 10545; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10691. [Google Scholar]
- 18.
- 18a.A structurally similar tetraborane motif: Maier A., Hofmann M., Pritzkow H., Siebert W., Angew. Chem. Int. Ed. 2002, 41, 1529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1600; [Google Scholar]
- 18b.A related synthesis of an (η6-toluene)FeI diamide complex: Janes T., Rawson J. M., Song D., Dalton Trans. 2013, 42, 10640. [DOI] [PubMed] [Google Scholar]
- 19. Lee Y., Anderton K. J., Sloane F. T., Ermert D. M., Abboud K. A., García-Serres R., Murray L. J., J. Am. Chem. Soc. 2015, 137, 10610. [DOI] [PubMed] [Google Scholar]
- 20.Non-planar nitrido/imido/amido Fe clusters:
- 20a. Hernández Sánchez R., Betley T. A., J. Am. Chem. Soc. 2015, 137, 13949; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Iovan D. A., Betley T. A., J. Am. Chem. Soc. 2016, 138, 1983; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Hernández Sánchez R., Bartholomew A. K., Powers T. M., Ménard G., Betley T. A., J. Am. Chem. Soc. 2016, 138, 2235; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Hernández Sánchez R., Zheng S.-L., Betley T. A., J. Am. Chem. Soc. 2015, 137, 11126; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20e. Hernández Sánchez R., Willis A. M., Zheng S.-L., Betley T. A., Angew. Chem. Int. Ed. 2015, 54, 12009; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12177; [Google Scholar]
- 20f. Powers T. M., Betley T. A., J. Am. Chem. Soc. 2013, 135, 12289; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20g. Hennessy E. T., Betley T. A., Science 2013, 340, 591; [DOI] [PubMed] [Google Scholar]
- 20h. Zhao Q., Betley T. A., Angew. Chem. Int. Ed. 2011, 50, 709; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 735; [Google Scholar]
- 20i. King E. R., Hennessy E. T., Betley T. A., J. Am. Chem. Soc. 2011, 133, 4917; [DOI] [PubMed] [Google Scholar]
- 20j.“Kochi-type” [Fe8Me12]− double decker: Muñoz S. B., Daifuku S. L., Brennessel W. W., Neidig M. L., J. Am. Chem. Soc. 2016, 138, 7492; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20k.Related Co6 and Co7 clusters: Ohki Y., Shimizu Y., Araake R., Tada M., Sameera W. M. C., Ito J.-I., Nishiyama H., Angew. Chem. Int. Ed. 2016, 55, 15821; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16053. [Google Scholar]
- 21.
- 21a. Akita M. in Comprehensive Organometallic Chemistry III, Vol. 6, 1st ed. (Eds.: R. H. Crabtree, D. M. P. Mingos), Elsevier, Amsterdam, 2007, p. 259; [Google Scholar]
- 21b. Whitmire K. H., Adv. Organomet. Chem. 1998, 42, 1 Anionic (CO)xFe3 triangles or non-planar (CO)yFe4 envelopes with bridging amido/imido/nitrido ligands: [Google Scholar]
- 21c. Zanello P., Laschi F., Cinquantini A., Della Pergola R., Garlaschelli L., Cucco M., Demartin F., Spalding T. R., Inorg. Chim. Acta 1994, 226, 1; [Google Scholar]
- 21d. Pergola R. D., Bandini C., Demartin F., Diana E., Garlaschelli L., Stanghellini P. L., Zanello P., J. Chem. Soc. Dalton Trans. 1996, 747; [Google Scholar]
- 21e. Bantel H., Suter P., Vahrenkamp H., Organometallics 1995, 14, 4424. [Google Scholar]
- 22.
- 22a. Metal Clusters in Chemistry (Eds.: P. Braunstein, L. A. Oro, P. R. Raithby), Wiley-VCH, Weinheim, 1999; [Google Scholar]
- 22b. Gatteschi D., Sessoli R., Villain J., Molecular Nanomagnets, Oxford University Press, Oxford, 2006; [Google Scholar]
- 22c. de Smit E., Weckhuysen B. M., Chem. Soc. Rev. 2008, 37, 2758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary