

Abstract
Resveratrol‐based natural products constitute a valuable source of unique compounds with diverse biological activities. In this report we investigate demethylation strategies to minimize formation of cyclized and dimerized products during the synthesis of viniferifuran and analogues. We found that boron trichloride/tetra‐n‐butylammonium iodide (BCl3/TBAI) is typically more effective than boron tribromide (BBr3). Based on these findings we carried out the first syntheses of dehydro‐δ‐viniferin, resveratrol‐piceatannol hybrid and anigopreissin A. In addition, we have developed a short and efficient route to viniferifuran that was obtained in 13% yield over six steps.
Keywords: demethylation, natural products, polyphenols, resveratrol oligomers, stilbenoids, total synthesis
Introduction
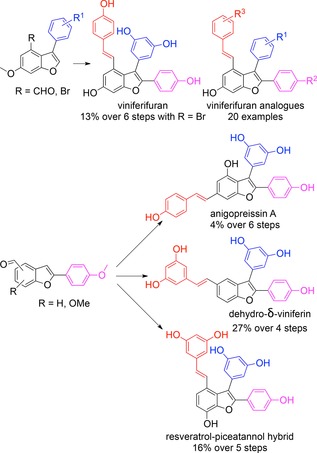
Stilbenoids of plant origin constitute a rich source of polyphenolic compounds with intriguing structures and diverse biological activities.1 To date the structures and absolute stereochemistry for several hundred higher order stilbenoids have been determined. Most of these often very complex natural products are formed by oligomerization of the key building block resveratrol. The large number of reports describing the positive effects of stilbenoids on human health have instigated wide interest in the chemistry and biology of resveratrol and its oligomers. The mode of action on the molecular level is, however, known for only a few stilbenoids, for example, diptoindonesin G (13, Figure 1) that was recently identified as an inhibitor of CHIP E3 ubiquitin ligase.2 Due to scarce quantity of many higher order stilbenoids in nature, intense efforts have been made to develop synthetic methods employing both biomimetic and de novo strategies. Biomimetic strategies usually involve metal‐ or enzyme‐catalyzed oxidative couplings of stilbenes but these protocols typically result in product mixtures and poor yields.1d,3 Recent improvements resulted in protocols for the preparation of racemic pallidol and quadrangularin by dimerization of protected resveratrol derivatives in excellent yields.4 However, biomimetic methods do not readily allow alteration of the core structures or substituent patterns, for example, to determine structure–activity relationships for bioactive stilbenoids. To address this Snyder and co‐workers developed de novo methods allowing the preparation of a large number of resveratrol dimers and higher order stilbenoids.5 These impressive studies spawned a number of syntheses applying innovative approaches for the preparation of racemic compounds.5,6 The first enantiototal synthesis of a stilbene dimer was the case of δ‐viniferin (3) where Shaw and co‐workers elegantly applied a carbene‐insertion chemistry using a rhodium catalyst to construct the dihydrobenzofuran core asymmetrically.7 Our interest in this intriguing family of natural products stems from the finding that the resveratrol tetramer (−)‐hopeaphenol blocks type III secretion, an indispensable virulence system, in the Gram‐negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa.8 While (−)‐hopeaphenol can be obtained in gram quantities from natural sources9 we focused on the de novo synthesis of partial structures and recently published total syntheses of (±)‐ampelopsin B (6)10 and (±)‐ϵ‐viniferin (1),10 the key intermediate to form almost, if not all, higher order of stilbene structures (Figure 1).1 To further advance our understanding of structural features that affect inhibition of type III secretion, we have continued to prepare resveratrol dimers. In this study we describe total syntheses of viniferifuran (2),11 its isomer anigopreissin A (11)12 and the recently reported resveratrol‐piceatannol hybrid (12)13 (Figure 1). Viniferifuran is a competitive inhibitor of syk kinase (IC50=62 nM)14 and this mechanism is believed to be key for the anti‐inflammatory activity of viniferifuran. Anigopreissin A was recently discovered as an inhibitor of HIV‐1 reverse transcriptase (IC50=8 μM) including two mutant enzymes resistant to the clinical drug nevirapine.12b Viniferifuran (2) is a highly substituted 7‐hydroxy‐5‐styryl‐2,3‐diarylbenzofuran structure that can be prepared in four steps starting with biomimetic dimerization of resveratrol to form (±)‐ϵ‐viniferin (1).15 Subsequent transformations including a key oxidative dehydrogenation by DDQ furnished viniferifuran (2).11 In our hands viniferifuran could be obtained in gram quantity in 11% yield over four steps (Supporting Information, SI‐1). In addition, we prepared a number of viniferifuran analogues and investigated conditions for demethylation of phenols, a critical transformation in the total synthesis of resveratrol oligomers.
Figure 1.
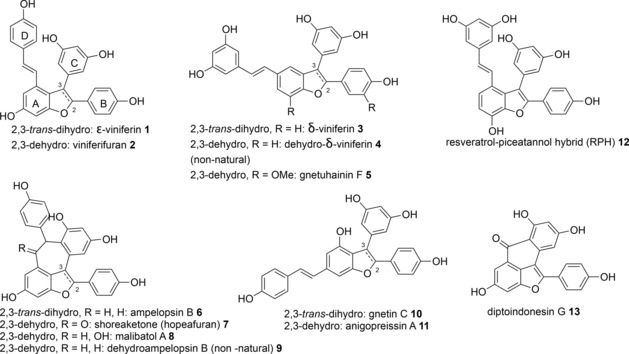
Selected natural and non‐natural resveratrol dimers and the stilbenoid diptoindonesin G.
Results and Discussion
Despite the number of methods available to construct the 2,3‐diarylbenzofuran core,16 only a few attempts have been made towards the total synthesis of viniferifuran. Kraus and Gupta17 and Chen et al.18 applied strategies relying on cyclization and subsequent dehydration of keto benzyl ethers by heating at high temperature (170 °C) with excess of the strong hindered phosphazene base P4‐t‐Bu17 or by using excess of the strong base LiTMP followed by dehydratation using pTsOH.18 Kim and Choi19 on the other hand employed a more versatile and milder procedure to construct a 3‐arylbenzofuran by cyclization of the corresponding β‐aryloxy ketone using Bi(OTf)3, followed by introduction of a 2‐aryl substituent by Pd(OAc)2‐catalyzed direct arylation.
After generation of an aldehyde, the styryl moiety was then introduced by a Wittig–Horner reaction.19 However, when reinvestigating the strategy by Kim and Choi (Supporting Information, SI‐2) the Wittig–Horner reaction in our hands only resulted in very low conversion of aldehyde 16a to the styryl moiety 17a. Using NaH instead of t‐BuOK20 under microwave irradiation at 120 °C during 30 min produced permethylated viniferifuran 17a in 80% yield. We then modified the route described by Kim and Choi19 to conveniently prepare permethylated viniferifuran 17a and analogues (Scheme 1). In this modified route, the ester moiety in 14a was transformed to the aldehyde 15a before harnessing the direct arylation10 to prepare intermediate 16a that is now ready for the Wittig–Horner olefination to give 17a. Interestingly, we could exploit the direct arylation of 15a with a low loading of 5% of PdCl(C3H5)(dppb),21 conditions that were found to be unsuccessful with the ester substrate 14a. Kraus and Gupta15 reported the final demethylation of 17a to form viniferifuran (2) in 67% yield using 1 M BBr3 in CH2Cl2 at 0 °C. However, when applying these conditions we obtained the cyclized product dehydroampelopsin B (9, Figure 1) as major product (46% yield after purification by HPLC) with only minor amounts of viniferifuran (not isolated) and mixtures of its dimers that were detected by LC‐MS (Supporting Information, SI‐2). Alternative protocols for demethylation were investigated and we found that BCl3/TBAI22 gave 2 and 9 in a 1:1 ratio without formation of dimerized products. HPLC purification furnished pure 2 and 9, each isolated in 23% yield (Supporting Information, SI‐2, and Table 1). We realized that the final demethylation would be a challenge when synthesizing analogues with variation of substituents on the A–D rings with the goal to allow elucidation of structure–activity relationships. This prompted us to investigate if BCl3/TBAI could be used as a general demethylation reagent. Interestingly, we also found that the Tf2O‐catalyzed cyclization of viniferifuran (2) gave racemic dehydroampelopsin B (9) in 50% yield (Supporting Information, SI‐1). We hypothesize that this compound might exist in nature since it is the core of shoreaketone (7) and malibatol A (8), both natural products (Figure 1).1
Scheme 1.
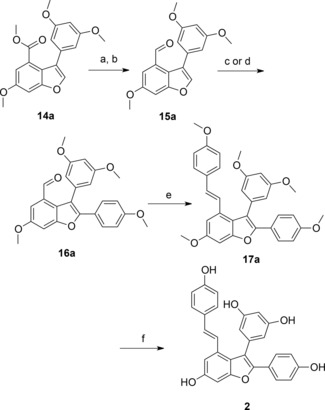
Modified synthetic route to viniferifuran. Reagents and conditions: a) LiAlH4 (3 equiv.), THF, 0 °C, 10 min. b) Dess–Martin periodinane (1.2 equiv.), DCM, 0 °C, 1 h, 80–90%, 2 steps. c) ArBr or ArI (1.5–2 equiv.), Pd(OAc)2 (0.1 equiv.), PCy3⋅HBF4 (0.2 equiv.), K2CO3 (1.5 equiv.), PivOH (3 equiv.), 100 °C, 20 h, 74%. d) ArBr (1.5–2 equiv.), PdCl(C3H5)dppb (0.05 equiv.), KOAc (2 equiv.), DMA, 150 °C, 20 h, 60%. e) Phosphonate derivatives (1.5 equiv.), NaH (3 equiv.), THF, 120 °C, MWI, 30 min, 80%. f) BCl3, TBAI, DCM, 0 °C to room temperature, 6 h, 23% (HPLC).
Table 1.
Demethylation with BCl3/TBAI to afford viniferifuran analogues.
| Substrate | R1 | R2 | R3 | Product | Product/cyclized form[d] | Yield [%][a] |
|---|---|---|---|---|---|---|
| 17a | 3,5‐diOMe | OMe | OMe | 2 | 1:1 | 23[b] |
| 17b | 3,5‐diOMe | F | OMe | 18a | <3:7 | (<10)[e] |
| 17c | 3,5‐diOMe | COOH | OMe | 18b | <3:7 | (<10)[e] |
| 17d | 3,5‐diOMe | OMe | H | 18c | >9:1 | 50 |
| 17e | 3,5‐diOMe | OMe | F | 18d | 1:0 | 51[b] (11)[c] |
| 17f | 3,5‐diOMe | OMe | NO2 | 18e | – | – |
| 17g | 3,5‐diOMe | NO2 | Cl | 18f | – | – |
| 17h | 3,5‐diOMe | NH2 | Cl | 18g | 1:0 | 18[b] |
| 17i | 2,5‐diOMe | OMe | OMe | 18h | 1:0 | 46 |
| 17j | 3‐OMe | OMe | OMe | 18i | 7:3 | 24[b] |
| 17k | 4‐OMe | OMe | OMe | 18j | 1:0 | 19[b] |
| 17l | 4‐CF3 | OMe | OMe | 18k | 1:0 | 54 |
| 17m | 4‐F | OMe | OMe | 18l | 1:0 | 53 (16)[c] |
| 17n | 3‐F | OMe | OMe | 18m | 1:0 | 55 |
| 17o | 3‐Cl | OMe | OMe | 18n | 1:0 | 55 |
| 17p | H | OMe | OMe | 18o | 1:0 | 53 |
[a] Isolated yields.
[b] Purified by HPLC.
[c] Yield with BBr3 as demethylation agent.
[d] Ratio based on 1H NMR of crude product.
[e] Estimated yield, not isolated.
We applied the modified route and prepared a number of permethylated compounds 17b–p with different substituents on rings B, C and D (Scheme 2, and Supporting Information, SI‐3). The carboxylic esters 14a–i were transformed to aldehydes 15a–i by a sequence of LiAlH4 reduction followed by a Dess–Martin oxidation. Direct arylation of 15a–i was then carried out with a set of aryl bromides carrying different electron‐withdrawing and electron‐donating substituents. The methylated products 16b–m were obtained in 35–74% yield using 10 mol% (PdOAc)2 or 47–60% using 5 mol% PdCl(C3H5)(dppb). Next, application of our improved conditions for the Wittig–Horner reaction to install the styryl moiety afforded the permethylated intermediates 17b–p in 52–82% yield. Finally, we investigated the impact of the different substituent patterns on demethylation and cyclization of 17b–p using BCl3/TBAI in DCM at 0 °C to room temperature for 6 h (Table 1).
Scheme 2.
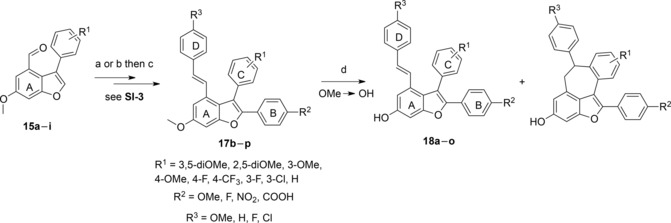
Modified synthetic route to viniferifuran. Reagents and conditions: a) ArBr or ArI (1.5–2 equiv.), Pd(OAc)2 (0.1 equiv.), PCy3⋅HBF4 (0.2 equiv.), K2CO3 (1.5 equiv.), PivOH (3 equiv.), 100 °C, 20 h, 35–74%. b) ArBr (1.5–2 equiv.), PdCl(C3H5)(dppb) (0.05 equiv.), KOAc (2 equiv.), DMA, 150 °C, 20 h, 47–60%. c) Phosphonate derivatives (1.5 equiv.), NaH (3 equiv.), THF, 120 °C, MWI, 30 min, 52–82%. d) BCl3, TBAI, DCM, 0 °C to room temperature, 6 h.
Under these conditions all the starting materials were consumed and we anticipated that cyclization would be suppressed in compounds with less nucleophilic C rings. Indeed, 18h (2,5‐diOH) and 18i (3‐OH) were obtained in higher yields than viniferifuran (2) and minor amounts of the cyclized product was observed only with starting material carrying the 3‐OMe group. This pattern was observed for other C‐ring substituents (3‐H, 3‐F, 3‐Cl, 4‐F, 4‐CF3, 4‐OMe) that furnished 18k–p in 19–55% yield. Modification of ring B with substituents such as 4‐F or 4‐COOH increased formation of the cyclized dehydroampelopsin B analogues 35 and 36, respectively (Supporting Information, SI‐4) and 18a and b was observed as minor products (not isolated). We also found that removal of the 4‐OMe group or replacement with a fluorine atom on ring D reduced the degree of cyclization and afforded 18c and 18d in 50 and 51% yields, respectively. Compounds 18e and 18f could not be isolated due to decomposition of the NO2 group during the demethylation. However, 18g could be obtained when NO2 was replaced by NH2. Several substrates were also demethylated with BBr3 that typically resulted in lower yields than BCl3/TBAI. In several cases, decomposition occurred or complex mixtures of dimerized products formed (Supporting Information, SI‐5). Acid‐catalyzed dimerization of stilbenes has been described using a variety of acids including BBr3.3,23
Our results indicate that BCl3/TBAI is superior to BBr3 for our purposes but for some compounds the yields are still low and in several cases HPLC was required to purify the compounds and remove excess TBAI. We therefore decided to explore alternative routes based on demethylation early in the synthetic sequence.
First we applied an alternative route where a set of D ring moieties were attached by applying the Wittig–Horner reaction on the early aldehyde intermediate 15a to afford 20a–c in 60–80% yield (Scheme 3). Interestingly, BBr3‐mediated demethylation could effectively occur in these cases and after acetylation the protected derivatives 21a–c were isolated in 38–62% yield. Bromination with NBS and subsequent deacetylation furnished the bromo polyphenolic substrates 22a–c. As a final step substituted B rings were introduced by Suzuki couplings to give the target compounds 18d and 18p‐‐s in 41–76% yield (Table 2). Next we exploited the strategy previously developed for the total synthesis of (±)‐ϵ‐viniferin.10 Direct arylation of the bromo derivative 19, prepared in three steps from 1‐bromo‐3,5‐dimethoxybenzene in 59% yield,10 installed ring B to give 23 in 36% yield and subsequent demethylation by BBr3 produced the bromo polyphenolic substrate 24 in 96% yield (Scheme 3). A set of D rings was introduced by Heck couplings of the corresponding styrene derivatives gave viniferifuran (2) in 64% yield and target compounds 18t–x in 25–64% yield (Table 2). This synthesis of viniferifuran is the shortest achieved to date and provides the target compound in 13% yield over 6 steps from 1‐bromo‐3,5‐dimethoxybenzene. Importantly this strategy avoids formation of dimers or cyclized products and also allows introduction of, for example, nitro and carboxylic acid substituents (cf. 18t–x) that are not compatible with a final demethylation.
Scheme 3.
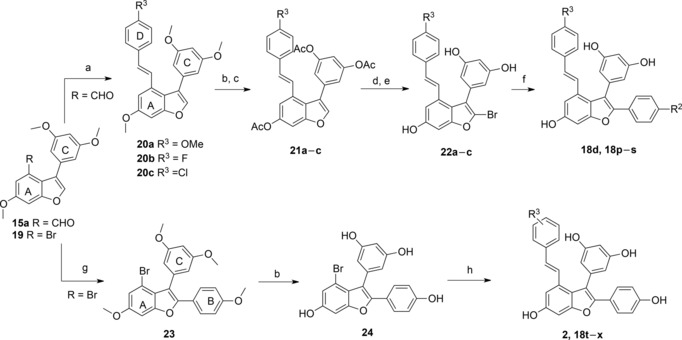
Alternative routes for the synthesis of viniferifuran and analogues. Reagents and conditions: a) Phosphonate derivatives (1.5 equiv.), NaH (3 equiv.), THF, 120 °C, MWI, 30 min, 60–80%. b) BBr3 (9–12 equiv.), DCM, −78 °C to room temperature, 6 h. c) Ac2O, THF, Et3N, room temperature, overnight, 38–62%, 2 steps. d) NBS (3 equiv.), pTsOH, room temperature, 24–48 h. e) KOH, MeOH, 0 °C, 30 min, 15–30%, 2 steps. f) PdCl2(dppf)⋅DCM (5% mol), boronic acid (1.5–2 equiv.), Na2CO3, DME/H2O (1/1), 70 °C, MWI, 30 min, 41–76%. g) 4‐Bromoanisole (2 equiv.), Pd(OAc)2 (0.1 equiv.), PCy3⋅HBF4 (0.2 equiv.), K2CO3 (1.5 equiv.), PivOH (3 equiv.), 100 °C, 20 h, 25–36%. h) Styrene derivatives (1.2 equiv.), Pd(OAc)2 (0.1 equiv.), dppp (0.1 equiv.), Et3N, DMF, 120 °C, 20 h, 25–64%.
Table 2.
Obtained viniferifuran analogues by final Suzuki or Heck couplings.
| Compound | R2 | R3 | Yield [%][a] |
|---|---|---|---|
| 18p | NHAc | OH | 76 |
| 18d | OH | F | 50 |
| 18q | F | F | 55 |
| 18r | OH | Cl | 41 |
| 18s | F | Cl | 41 |
| 18t | OH | 3‐NO2, 4‐OH | 25[b] |
| 18u | OH | 3‐OH, 4‐NO2 | 58 |
| 18v | OH | 4‐COOH | 60 |
| 18w | OH | 4‐NH2 | 64 |
| 18x | OH | 4‐EtOCOCH2O | 54 |
| viniferifuran 2 | OH | 4‐OH | 64 |
[a] Isolated yield.
[b] Purified by HPLC.
Having an understanding of the different reactivity of BBr3 and BCl3/TBAI, we turned our attention to the total synthesis of the benzofuryl stilbene dimers anigopreissin A (11),12 a resveratrol‐piceatannol hybrid (12)13 and dehydro‐δ‐viniferin (4)24 (Figure 1). The resveratrol‐piceatannol hybrid was isolated from a Vitis extract after a bioguided fractionation using a hepatitis C virus replication inhibition assay.13 Dehydro‐δ‐viniferin was recently synthesized by laccase‐biocatalyzed dimerization of resveratrol to form δ‐viniferin (3) followed by dehydrogenation, a strategy limited to 4‐hydroxystilbene starting materials.24
The final demethylations by BBr3 and BCl3/TBAI were studied on permethylated intermediates 29, 31 and 34 (Scheme 4). In order to prepare these intermediates, we constructed 2‐arylbenzofuran derivatives by a one pot two‐step Sonogashira cyclization applied to o‐iodophenol derivatives containing a free CHO group using a rapid and efficient protocol recently described by Markina et al.25 This procedure was applied to 25 and 26 to afford 28 and 30 in 74 and 90% yields, respectively. In the case of 27, addition of 1‐iodo‐3,5‐dimethoxybenzene during the cyclization step partially resulted in a direct coupling to introduce the C‐3 aryl on the benzofuran and 2,3‐diarylbenzofuran 33 and 2‐arylbenzofuran 32 were isolated in 25% and 60% yields, respectively. Intermediates 28 and 30 were then transformed by halogenation, Suzuki coupling to install the C‐3 aryl followed by the Wittig–Horner reaction to achieve the permethylated intermediates 29 and 31 in 31% (5 steps from 25) and 27% (4 steps from 26) yields, respectively. Interestingly, we could also apply direct arylation of 32 with 1‐bromo‐3,5‐dimethoxybenzene in the presence of 5 mol% PdCl(C3H5)dppb to give 33 in 60% yield. This is the first reported C‐3 arylation of a benzofuran when C‐2 is blocked by an aryl group containing an electron‐donating 4‐methoxy substituent. In a recent study this type of arylation was unsuccessful using Pd(OAc)2 and Sphos as ligand.26 A Wittig–Horner reaction was then applied to 33 to afford 34 in overall 43% yield (3 steps from 27). During demethylation, we again observed different reactivity between BBr3 and BCl3/TBAI. Surprisingly, BBr3‐mediated demethylation of stilbenes 31 and 34 which both contain a 3,5‐dimethoxystyryl moiety afforded the pure desired products 12 and 4 in 61% and 62% yields, respectively. The use of BCl3/TBAI was less efficient and in both cases HPLC was required for purification providing pure 12 and 4 in low yields. In contrast, BBr3 failed to produce anigopreissin A while BCl3/TBAI gave the target compound in 23% yield after purification by HPLC.
Scheme 4.
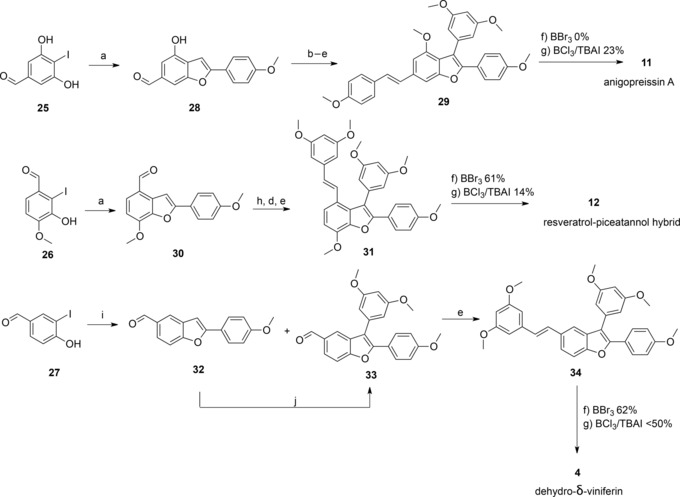
Total synthesis of anigopreissin A, resveratrol‐piceatannol hybrid and dehydro‐δ‐viniferin. Reagents and conditions: a) PdCl2(PPh3)2 (5% mol), CuI (3% mol), ethynylanisole (1.2–1.5 equiv.), Et3N/THF (2/1), MWI, 40 °C, 30 min then CH3CN, 100 °C, MWI, 30 min, 74–90%. b) MeI, K2CO3, DMF, room temperature, overnight, 77%. c) NIS (1 equiv.), pTsOH cat., CH3CN, room temperature, overnight, 59%. d) PdCl2(dppf)⋅DCM (5% mol), 3,5‐dimethoxyphenylboronic acid (1.5 equiv.), K2CO3, DME/H2O (4/1), 80–100 °C, MWI, 30 min, 83–91%. e) Phosphonate derivatives (1.5 equiv.), NaH (3 equiv.), THF, 120 °C, MWI, 30 min, 58–84%. f) BBr3, DCM, −78 °C to room temperature, 6 h. g) BCl3, TBAI, DCM, 0 °C to room temperature, 6 h. h) NBS (1.1 equiv.), DCM, room temperature, overnight, 50%. i) PdCl2(PPh3)2 (5% mol), CuI (3% mol), 4‐ethynylanisole (1.2–1.5 equiv.), Et3N/THF (2/1), MWI, 40 °C, 30 min then 1‐iodo‐3,5‐dimethoxybenzene (1.5 equiv.), CH3CN, 100 °C, MWI, 30 min, 25% 33+60% 32. j) 1‐Bromo‐3,5‐dimethoxybenzene (1.5 equiv.), PdCl(C3H5)dppb (5% mol), KOAc (2 equiv.), DMA, 150 °C, 20 h, 60%.
Conclusions
We have prepared a number of permethylated natural and non‐natural stilbenoid compounds and studied demethylations using BBr3 and BCl3/TBAI. The latter was found to be superior in demethylation of viniferifuran analogues and produced the product in higher yields and with less cyclized derivatives and higher order oligomers. In addition, strategies based on early demethylation were explored and viniferifuran (2) could be obtained in 13% yield over six steps, the shortest and most efficient synthesis reported so far. Finally, we achieved the first total syntheses of dehydro‐δ‐viniferin (4, 4 steps, 27%), resveratrol‐piceatannol hybrid (12, 5 steps, 16%) and anigopreissin A (11, 6 steps, 4%). The synthesis of dehydro‐δ‐viniferin also included a successful benzofuran C‐3 arylation with C‐2 blocked by an aryl group containing an electron‐donating substituent. The methods and strategies described have the potential be applicable in syntheses of other stilbenoid natural products and analogues to establish structure–activity relationships.
Experimental Section
General
LC‐MS analysis was carried out on a Waters LC system equipped with an Xterra MS C18 18.5 μm 4.6×50 mm column and an eluent system consisting of MeCN in water, both of which contained 0.2% formic acid. Detection was performed at 214 and 254 nm. Mass spectra were obtained by use of a Waters micromass ZG 2000, using both positive and negative electrospray ionization (ESI). 1H NMR and 13C NMR spectra were recorded on a Bruker DRX‐400 spectrometer in CDCl3 solution [residual CHCl3 (δ H=7.26 ppm, δ C=77.16 ppm) as internal standard] or in SO(CD3)2 solution [residual SO(CD3)(CD2H) (δ H=2.50 ppm, δ C=39.52 ppm) as internal standard] or in CO(CD3)2 solution [residual CO(CD3)(CD2H) (δ H=2.05 ppm, δ C=29.84 ppm) as internal standard] or in CD3OD solution [residual CD2HOH (δ H=3.31 ppm, δ C=49.00 ppm) as internal standard]. A Biotage Initiator 400W was used for microwave heating. TLC analysis was carried out using TLC aluminum sheets from EMD/Merck KGaA (mfr. no. Merck, 1.05554.0001). Product purification was done using Biotage flash chromatography (FC) with cartridge or ultra cartridge of 10 g, 25 g, 50 g or 100 g of silica gel. HPLC purification was carried out on a Gilson system equipped with a Macherey–Nagel Nucleodur C18 HTEC 5 μm 21×250 mm column. All eluents contained 0.005% formic acid and the flow rate was set to 20 mL/minute for CH3CN and 15 mL/minute for MeOH (method A: H2O/MeOH 80/20 to 0/100 over 30 min; method B: H2O/ CH3CN 80/20 to 0/100 over 30 min).
Procedure A: Demethylation with BBr3 [Exemplified with Dehydroampelopsin B (9)]
To a stirred solution of 17a (25 mg, 0.048 mmol, 1 equiv.) at 0 °C in DCM (3 mL) under a nitrogen atmosphere was added 1 M BBr3 solution in DCM (0.72 mL, 0.72 mmol, 15 equiv.). The mixture was allowed to warm up and stirred at room temperature for 6 h. The reaction was quenched with H2O (1 mL) at 0 °C. The mixture was diluted with EtOAc (25 mL), washed with H2O (5 mL) and brine (5 mL). The organic phase was dried with Na2SO4 and concentrated under reduced pressure. FC (DCM:MeOH 9:1) afforded a mixture that was purified using HPLC method A to give dehydroampelopsin B 9 as yellow solid; yield: 10 mg (46%).
Procedure B: Demethylation with BCl3/TBAI [Exemplified with Dehydroampelopsin B (9) and Viniferifuran (2)]
To a stirred solution of 17a (25 mg, 0.048 mmol, 1 equiv.), TBAI (265 mg, 0.72 mmol, 15 equiv.) at 0 °C in DCM (3 mL) under a nitrogen atmosphere was added 1 M BCl3 solution in DCM (0.72 mL, 0.72 mmol, 15 equiv.). The mixture was stirred at room temperature for 6 h. The reaction was quenched with H2O (1 mL) at 0 °C. The mixture was diluted with EtOAc (25 mL), washed with 10% Na2S2O3 solution (5 mL), H2O (5 mL) and brine (5 mL). The organic phase was dried with Na2SO4 and concentrated under reduced pressure. FC (DCM:MeOH 9:1) afforded a 1:1 mixture of 2 and 9 that was purified using HPLC method A to give viniferifuran 2 (yield: 5 mg, 23%) and dehydroampelopsin B 9 (yield: 5 mg, 23%) as yellow solids.
Anigopreissin A (11): Application of procedure B for demethylation of 29 (20 mg, 0.038 mmol FC (DCM:MeOH 9:1) followed by HPLC method A gave 11 as yellow solid; yield: 4 mg (23%). 1H NMR (400 MHz, CD3OD): δ=7.44–7.35 (m, 4 H), 7.15 (d, J=0.9 Hz, 1 H), 7.06 (d, J=16.2 Hz, 1 H), 6.98 (d, J=16.2 Hz, 1 H), 6.78 (d, J=8.6 Hz, 2 H), 6.76 (d, J=0.9 Hz, 1 H), 6.71 (d, J=8.8 Hz, 2 H), 6.42 (d, J=2.2 Hz, 2 H), 6.31 (t, J=2.2 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=159.5, 158.7, 158.4, 157.2, 153.0, 151.2, 137.2, 136.8, 130.7, 129.3, 128.9, 128.8, 127.3, 123.7, 119.1, 116.8, 116.6, 116.2, 110.3, 107.2, 102.8, 101.7;: ESI‐MS m/z=451.02, calcd. for [M−H]−: 451.12. Analytical data were in agreement with those reported in the literature.12
Resveratrol‐piceatannol hybrid (12): Application of procedure A for demethylation of 31 (50 mg, 0.09 mmol). FC (DCM:MeOH 9:1) gave 12 as yellow solid; yield: 26 mg (61%). Application of procedure B for demethylation of 31 (25 mg, 0.045 mmol). FC (DCM:MeOH 9:1) followed by HPLC method A gave 12 as yellow solid; yield: 3 mg (14%). 1H NMR (600 MHz, acetone‐d 6): δ=9.12–8.04 (br, OHs), 7.56 (d, J=8.9 Hz, 2 H), 7.43 (d, J=8.3 Hz, 1 H), 7.08 (d, J=16.2 Hz, 1 H), 6.83 (d, J=8.3 Hz, 1 H), 6.82 (d, J=8.9 Hz, 2 H), 6.76 (d, J=16.2 Hz, 1 H), 6.58 (t, J=2.2 Hz, 1 H), 6.49 (d, J=2.2 Hz, 2 H), 6.22 (d, J=2.1 Hz, 2 H).6.20 (t, J=2.1 Hz, 1 H); 13C NMR (150 MHz, acetone‐d 6): δ=161.3, 160.3, 159.6, 152.5, 143.8, 143.6, 142.1, 138.3, 131.6, 129.8, 128.2, 126.7, 125.0, 124.1, 121.8, 118.6, 117.2, 113.0, 110.9, 106.8, 104.7, 103.4; ESI.MS: m/z=467.04, calcd. for [M−H]: 467.11. Analytical data for the resveratrol‐piceatannol have not been reported in the literature.13
Dehydro‐δ‐viniferin (4): Application of procedure A for demethylation of 34 (15 mg, 0.029 mmol). FC (DCM:MeOH 9:1) gave 4 as yellow solid; yield: 8 mg (62%). Application of procedure B for demethylation of 34 (15 mg, 0.029 mmol). FC (DCM:MeOH 9:1) gave a mixture of 4 and partially deprotected products+excess of TBAI. 1H NMR (400 MHz, acetone‐d 6): δ=8.76 (s, 1 H, OH), 8.42 (s, 2 H, OHs), 8.22 (s, 2 H, OHs), 7.66–7.50 (m, 5 H), 7.23 (d, J=16.3 Hz, 1 H), 7.06 (d, J=16.3 Hz, 1 H), 6.60 (d, J=2.1 Hz, 2 H), 6.51 (d, J=2.2 Hz, 2 H), 6.45 (t, J=2.2 Hz, 1 H), 6.30 (t, J=2.1 Hz, 1 H); 13C NMR (100 MHz, acetone‐d 6): δ=161.1, 160.6, 159.9, 155.3, 153.4, 141.6, 136.4, 134.8, 132.6, 130.6, 130.5, 129.7, 124.9, 123.8, 119.5, 117.6, 117.3, 112.8, 109.9, 106.9, 104.1, 104.0; ESI‐MS: m/z=451.02, calcd, for [M−H]−: 451.12. Analytical data are in agreement with those reported in the literature.24
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Swedish Research Council (grant 2014‐4670). We are grateful to Dr. Anders Lindgren for helpful comments on the manuscript.
D. D. Vo, M. Elofsson, Adv. Synth. Catal. 2016, 358, 4085.
References
- 1.
- 1a. Shen T., Wang X.-N., Lou H.-X., Nat. Prod. Rep. 2009, 26, 916; [DOI] [PubMed] [Google Scholar]
- 1b. Quideau S., Deffieux D., Douart-Cassassus C., Pouysegu L., Angew. Chem. 2011, 123, 610; [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 586; [Google Scholar]
- 1c. Riviere C., Pawlus A. D., Merillon J.-M., Nat. Prod. Rep. 2012, 29, 1317; [DOI] [PubMed] [Google Scholar]
- 1d. Keylor M. H., Matsuura B. S., Stephenson C. R. J., Chem. Rev. 2015, 115, 8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao Z., Wang L., James T., Jung Y., Kim I., Tan R., Hoffmann F. M., Xu W., Chem. Biol. 2015, 22, 1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Velu S. S., Thomas N. F., Weber J.-F. F., Curr. Org. Chem. 2012, 16, 605, and references cited therein. [Google Scholar]
- 4.
- 4a. Li W., Li H., Li Y., Hou Z., Angew. Chem. 2006, 118, 7609; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006, 45, 7609; [Google Scholar]
- 4b. Matsuura B. S., Keylor M. H., Li B., Lin Y. X., Allison S., Pratt D. A., Stephenson C. R. J., Angew. Chem. 2015, 127, 3825; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2015, 54, 3754. [Google Scholar]
- 5.
- 5a. Snyder S. A., Zografos A. L., Lin Y., Angew. Chem. 2007, 119, 8334; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007, 46, 8186; [Google Scholar]
- 5b. Snyder S. A., Breazzano S. P., Ross A. G., Lin Y., Zografos A. L., J. Am. Chem. Soc. 2009, 131, 1753; [DOI] [PubMed] [Google Scholar]
- 5c. Snyder S. A., Wright N. E., Pflueger J. J., Breazzano S. P., Angew. Chem. 2011, 123, 8788; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 8629; [Google Scholar]
- 5d. Snyder S. A., Gollner A., Chiriac M. I., Nature 2011, 474, 461; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Wright N. E., Snyder S. A., Angew. Chem. 2014, 126, 3477; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 3409; [Google Scholar]
- 5f. Jepsen T. H., Thomas S. B., Lin Y., Stathakis C. I., de Miguel I., Snyder S. A., Angew. Chem. 2014, 126, 6865; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 6747. [Google Scholar]
- 6.For selected total syntheses of resveratrol-based oligomers from other groups, see:
- 6a. Nicolaou K. C., Wu T. R., Kang Q., Chen D. Y.-K., Angew. Chem. 2009, 121, 3492; [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 3440; [Google Scholar]
- 6b. Nicolaou K. C., Kang Q., Wu T. R., Lim C. S., Chen D. Y.-K., J. Am. Chem. Soc. 2010, 132, 7540; [DOI] [PubMed] [Google Scholar]
- 6c. Lee B. H., Choi Y. L., Shin S., Heo J.-N., J. Org. Chem. 2011, 76, 6611; [DOI] [PubMed] [Google Scholar]
- 6d. Klotter F., Studer A., Angew. Chem. 2014, 126, 2505; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 2473; [Google Scholar]
- 6e. Sudhakar G., Satish K., Chem. Eur. J. 2015, 21, 6475; and refs cited in ref.[1d] [DOI] [PubMed] [Google Scholar]
- 7. Soldi C., Lamb K. N., Squitieri R. A., Gonzalez-Lopez M., Di Maso M. J., Shaw J. T., J. Am. Chem. Soc. 2014, 136, 15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zetterström C. E., Hasselgren J., Salin O., Davis R. A., Quinn R. J., Sundin C., Elofsson M., PLoS ONE 2013, 8, e81969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davis R. A., Beattie K. D., Xu M., Yang X., Yin S., Holla H., Healy P. C., Sykes M., Shelper T., Avery V. M., Elofsson M., Sundin C., Quinn R. J., J. Nat. Prod. 2014, 77, 2633. [DOI] [PubMed] [Google Scholar]
- 10. Lindgren A. E. G., Öberg C. T., Hillgren J. M., Elofsson M., Eur. J. Org. Chem. 2016, 3, 426. [Google Scholar]
- 11.
- 11a. Ito J., Takaya Y., Oshima Y., Niwa M., Tetrahedron 1999, 55, 2529; [Google Scholar]
- 11b. Huang K. S., Lin M., Wang Y. H., Chin. Chem. Let. 1999, 10, 817. [Google Scholar]
- 12.
- 12a. Hoelscher D., Schneider B., Phytochemistry 1996, 43, 471; [Google Scholar]
- 12b.C. Tancharoen, Master Thesis, Kasetsart University, 2012;
- 12c. Brkljaca R., White J. M., Urban S., J. Nat. Prod. 2015, 78, 1600. [DOI] [PubMed] [Google Scholar]
- 13. Lee C. H., Yoon K. D., Heo T.-H., PCT Int. Appl PCT Int. Appl. WO 2015126129 A2 20150827, 2015.
- 14. Jiang M., Liu R., Chen Y., Zheng Q., Fan S., Liu P., Int. J. Mol. Sci. 2014, 15, 17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang G.-W., Wang H.-L., Capretto D. A., Han Q., Hu R.-B., Yang S.-D., Tetrahedron 2012, 68, 5216. [Google Scholar]
- 16. Jia Y., Li T., Yu C., Jiang B., Yao C., Org. Biomol. Chem. 2016, 14, 1982 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 17. Kraus G. A., Gupta V., Tetrahedron Lett. 2009, 50, 7180. [Google Scholar]
- 18. Chen D. Y.-K., Kang Q., Wu R. T., Molecules 2010, 15, 5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim I., Choi J., Org. Biomol. Chem. 2009, 7, 2788. [DOI] [PubMed] [Google Scholar]
- 20. Hwu J. R., Chuang K.-S., Chuang S. H., Tsay S.-C., Org. Lett. 2005, 7, 1545. [DOI] [PubMed] [Google Scholar]
- 21.For application of PdCl(C3H5)dppp in direct arylation of heterocycles, see:
- 21a. Loukotova L., Yuan K., Doucet H., ChemCatChem 2014, 6, 1303; [Google Scholar]
- 21b. Bheeter C. B., Chen L., Soule J.-F., Doucet H., Catal. Sci. Technol. 2016, 6, 2005, and references cited therein. [Google Scholar]
- 22. Brooks P. R., Wirtz M. C., Vetelino M. G., Rescek D. M., Woodworth G. F., Morgan B. P., Coe J. W., J. Org. Chem. 1999, 64, 9719. [Google Scholar]
- 23. Li X.-C., Ferrreira D., Tetrahedron 2003, 59, 1501. [Google Scholar]
- 24. Beneventi E., Conte S., Cramarossa M. R., Riva S., Forti L., Tetrahedron 2015, 71, 3052. [Google Scholar]
- 25. Markina N. A., Chen Y., Larock R. C., Tetrahedron 2013, 69, 2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dao-Huy T., Haider M., Glatz F., Schnurch M., Mihovilovic M. D., Eur. J. Org. Chem. 2014, 8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary