

Abstract
We have previously shown that mice inoculated intranasally with a wild-type baculovirus (Autographa californica nuclear polyhedrosis virus [AcNPV]) are protected from a lethal challenge by influenza virus. However, the precise mechanism of induction of this protective immune response by the AcNPV treatment remained unclear. Here we show that AcNPV activates immune cells via the Toll-like receptor 9 (TLR9)/MyD88-dependent signaling pathway. The production of inflammatory cytokines was severely reduced in peritoneal macrophages (PECs) and splenic CD11c+ dendritic cells (DCs) derived from mice deficient in MyD88 or TLR9 after cultivation with AcNPV. In contrast, a significant amount of alpha interferon (IFN-α) was still detectable in the PECs and DCs of these mice after stimulation with AcNPV, suggesting that a TLR9/MyD88-independent signaling pathway might also participate in the production of IFN-α by AcNPV. Since previous work showed that TLR9 ligands include bacterial DNA and certain oligonucleotides containing unmethylated CpG dinucleotides, we also examined the effect of baculoviral DNA on the induction of innate immunity. Transfection of the murine macrophage cell line RAW264.7 with baculoviral DNA resulted in the production of the inflammatory cytokine, while the removal of envelope glycoproteins from viral particles, UV irradiation of the virus, and pretreatment with purified baculovirus envelope proteins or endosomal maturation inhibitors diminished the induction of the immune response by AcNPV. Together, these results indicate that the internalization of viral DNA via membrane fusion mediated by the viral envelope glycoprotein, as well as endosomal maturation, which releases the viral genome into TLR9-expressing cellular compartments, is necessary for the induction of the innate immune response by AcNPV.
The baculovirus Autographa californica nuclear polyhedrosis virus (AcNPV) has long been used as a biopesticide and as an efficient tool for recombinant protein production in insect cells (39, 42). Subsequently, its efficacy for the delivery of high-level expression of foreign genes under the control of mammalian promoters in infected mammalian cells was also demonstrated (12, 26, 48). Since it causes no visible cytopathic effects, even at high titers, and does not replicate in mammalian cells (49), this baculovirus is now recognized as a useful viral vector, not only for the expression of foreign proteins in insect cells, but also for gene delivery to mammalian cells (4, 9, 12, 16, 26, 28, 37, 45, 48, 49, 53, 54).
AcNPV was also shown to be capable of stimulating interferon (IFN) production in mammalian cell lines and can confer protection from lethal encephalomyocarditis virus infections in mice (18). We demonstrated that intranasal inoculation with AcNPV induces a strong innate immune response and protects mice from a lethal challenge of influenza A and B viruses (1). Furthermore, inoculation with baculovirus induces the secretion of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and IL-12, in RAW264.7, a murine macrophage cell line. However, the precise mechanism of induction of the protective immune response by a pretreatment with AcNPV remained unclear.
Members of the IL-1 receptor/Toll-like receptor (TLR) superfamily are key mediators of innate and adaptive immunity (5). Toll, the first member of this superfamily to be identified, was initially discovered as a factor involved in dorsoventral axis formation in fly embryos and was later shown to participate in host defense mechanisms (38). A family of TLRs exists in mammals and has been shown to play an important role not only in the recognition of a wide variety of infectious pathogens and their products, but also in protection of the host from infections with pathogens. So far, 11 TLR family members and their corresponding ligands have been identified, with TLR1 being the only orphan receptor among them. Different TLRs have been shown to mediate immune responses to a variety of different pathogen-derived elements. For example, TLR4, TLR5, and TLR9 are essential for the recognition of lipopolysaccharides (LPS), bacterial flagellin, and bacterial DNA containing unmethylated CpG motifs, respectively (21, 24, 27, 46). TLR2 is implicated in the recognition of peptidoglycan (PGN) and lipopeptides (7, 13, 50, 57), while TLR6 can associate with TLR2 and recognize PGN and lipopeptides derived from mycoplasma (44). On the other hand, TLR3 has been shown to activate immune cells in response to virus-derived double-stranded RNA (6). Although synthetic imidazoquinoline compounds and guanosine analogs with antiviral activities have been shown to activate TLR7 and TLR8 (25, 36), it was recently demonstrated that single-stranded RNAs from RNA viruses are the natural ligands of these receptors (17, 23). The most recently identified TLR, termed TLR11, senses bacteria that cause infections of the bladder and kidney (60). In summary, TLRs recognize specific components derived from pathogens and activate a signaling cascade that causes proinflammatory cytokine production and subsequent immune responses.
TLRs share a common cytoplasmic Toll-IL-1 receptor (TIR) domain. MyD88, also a TIR domain-containing protein, associates with TLRs and acts as an adapter that recruits IL-1 receptor-associated kinase and TNF receptor-associated factor 6 (TRAF6) to TLRs. Macrophages isolated from MyD88-deficient mice fail to activate NF-κB and Jun N-terminal protein kinase or to produce inflammatory cytokines in response to microbial components such as lipopeptides, LPS, and CpG-rich bacterial DNA (20, 52), indicating that MyD88 is a critical component in the signaling pathway that leads to the production of inflammatory cytokines.
Viruses are obligate intracellular parasites; accordingly, viral proteins synthesized in host cells bear modifications that reflect the identity and characteristics of the host. Therefore, viral particles do not display exclusively pathogen-associated molecular patterns. Although the mechanisms by which the innate immune response is induced by viral infection are poorly understood, there is increasing evidence suggesting that TLRs function to detect viruses and trigger inflammatory responses. For instance, respiratory syncytial virus and mouse mammary tumor virus activate innate immunity through TLR4 (22, 34, 47), which is a signaling receptor for LPS. Similarly, hemagglutinin from wild-type measles virus was reported to activate TLR2 (10), which also recognizes certain elements of gram-positive bacteria and fungi. Herpes simplex virus type 1 (HSV-1) and human cytomegalovirus have also been shown to recognize TLR2 (15, 35), while vaccinia virus encodes proteins containing amino acid sequences similar to the Toll/IL-1 receptor domain and inhibits IL-1-, IL-18-, and TLR4-mediated signal transduction (11).
It was recently shown that HSV-1 and -2, whose genomes contain abundant CpG motifs, can induce angiogenesis and a variety of diseases, including herpes stromal keratitis, that produce chronic inflammatory responses via a TLR9/MyD88-dependent signaling pathway (33, 40, 61). HSV-1 and -2 are also able to trigger alpha interferon (IFN-α) secretion from plasmacytoid dendritic cells through TLR9/MyD88-dependent signaling (33, 40). The TLR9-mediated recognition of HSV by immunocompetent cells suggests that this recognition pathway may be important for the recognition of other DNA viruses.
For this study, we characterized the innate immune response induced by AcNPV. Peritoneal macrophages and splenic CD11c+ dendritic cells obtained from TLR9 or MyD88 knockout mice exhibited severe reductions in proinflammatory cytokine production following stimulation with AcNPV, whereas a significant amount of IFN-α was still detectable in these cells. In addition, the frequency of CpG motifs in the AcNPV genome was similar to that of bacterial DNA and significantly higher than that of mammalian DNA. Furthermore, stimulation by AcNPV was eliminated by a treatment with inhibitors of endosomal acidification. These results indicate that the internalization of viral AcNPV DNA via membrane fusion by envelope glycoproteins found in the endosome is required for the induction of a TLR9/MyD88-dependent innate immune response.
MATERIALS AND METHODS
Mice and cell culture.
C57BL/6 mice were purchased from Clea Japan, Inc., Tokyo, Japan. MyD88-deficient (MyD88−/−) mice were established as previously described (2) and backcrossed more than eight times with C57BL/6 mice. TLR9−/− mice were generated as previously described (24). The mice were injected intraperitoneally with 2 ml of 4% thioglycolate (Sigma-Aldrich Co., St. Louis, Mo.), and cells were harvested 3 days later by peritoneal lavage. The mouse macrophage cell line RAW264.7 was purchased from Riken Cell Bank (Tsukuba, Japan) and maintained in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (FCS), 1.5 mM l-glutamine, 100 U of penicillin/ml, and 100 μg of streptomycin/ml at 37°C in a 5% CO2 humidified incubator.
Viruses and reagents.
AcNPV was propagated in Spodoptera frugiperda (Sf-9) cells in Sf-900II insect medium supplemented with 10% (vol/vol) heat-inactivated FCS. A mutant baculovirus, AcNPVΔ64, which lacks the gp64 envelope protein and possesses the green fluorescent protein gene under the control of the polyhedrin promoter in the gp64 gene locus, was generated (Y. Kitagawa et al., unpublished data). AcNPV and AcNPVΔ64 were purified as previously described (1). The inactivation of AcNPV was performed with a Stratalinker 2400 (Stratagene, La Jolla, Calif.) using short-wavelength UV radiation (UVC, 254 nm) at a distance of 5 cm for 30 min on ice (1.6 × 104 mJ/cm2). The inactivation of infectivity was verified by a plaque assay with Sf-9 cells.
AcNPV DNA was isolated from the purified virions by a treatment with 10 mg of proteinase K (Sigma-Aldrich)/ml and 10% sodium dodecyl sulfate (SDS) in sterile phosphate-buffered saline (PBS) for 2 h at 55°C. The viral DNA was purified by phenol-chloroform-isoamyl alcohol extraction, precipitated at 12,000 × g, and resuspended in sterile endotoxin-free Tris-buffered saline. RNAs were removed by incubation with RNase A (10 mg/ml) (Wako Pure Chemical Industries, Osaka, Japan) for 1 h at 37°C, and the viral DNA was extracted as described above. The resultant DNA exhibited a single band by electrophoresis, and neither protein nor chromosomal DNA of insect cells was detected.
Phosphorothioate-stabilized mouse CpG (mCpG) oligodeoxynucleotides (ODN1668) (TCC-ATG-ACG-TTC-CTG-ATG-CT) and human CpG (hCpG) oligodeoxynucleotides (ODN2006) (TCG-TCG-TTT-TGT-CGT-TTT-GTC-GTT) were purchased from Invitrogen (Tokyo, Japan). Guanosine, 2′-deoxy-G, 8-bromo-G, 7-methyl-G, 7-allyl-8-oxo-G (loxoribine) was purchased from Invivogen (San Diego, Calif.). LPS derived from Salmonella enterica serovar Minnesota (Re-595), PGN derived from Staphylococcus aureus, monodansylcadaverine (MDC), and chloroquine were purchased from Sigma-Aldrich. Bafilomycin A1 and ammonium chloride were purchased from Wako Pure Chemical Industries. An anti-p39 mouse monoclonal antibody was kindly provided by G. F. Rohrmann. The virus stocks and the other TLR ligands were free of endotoxin (<0.01 endotoxin units/ml), as determined by use of a Pyrodick endotoxin measure kit (Seikagaku Co., Tokyo, Japan).
Production of authentic and truncated forms of gp64 proteins.
cDNAs encoding a deletion mutant of gp64 lacking the transmembrane region (gp64ΔTM) as well as a wild-type version of gp64 were obtained by PCRs with AcNPV DNA as a template. The same 5′ primer (5′-CATAAGCTTATGGTAAGCGCTATTGTTTTATAT-3′ ) was used to amplify the gp64 and gp64ΔTM cDNAs, and the 3′ primers were 5′-GATTCTAGAATATATTGTCTATTACGGTTTCT-3′ and 5′ GATTCTAGAATCGAAGTCAATTTAGCGGCCAA-3′, respectively. cDNAs were subcloned into HindIII and XbaI sites in pIB/V5-His (Invitrogen). The sequences of the recombinant plasmids, pIBgp64/V5-His and pIBgp64ΔTM/V5-His, were confirmed by DNA sequencing. These plasmids were transfected into Sf-9 cells by the use of Unifector (B-Bridge International, Inc., San Jose, Calif.). After 3 days of incubation, the recombinant gp64 proteins were purified from cell lysates or supernatants by use of a column of nickel-nitrilotriacetic acid beads (QIAGEN, Valencia, Calif.). The protein concentrations were determined by use of a Micro BCA protein assay kit (Pierce, Rockford, Ill.). The recombinant proteins were analyzed by SDS-12.5% polyacrylamide gel electrophoresis (SDS-12.5% PAGE) under reducing conditions, stained with GelCord Blue staining reagent (Pierce), and detected by immunoblotting analysis with an antihexahistidine monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, Calif.).
Isolation of peritoneal cells and cytokine production.
To evaluate cytokine production from macrophages in vitro, we seeded thioglycolate-elicited peritoneal cells (PECs) into 96-well plates at a concentration of 2 × 105 cells/well and stimulated them with various doses of AcNPV and loxoribine. After 24 h of incubation, the culture supernatants were collected and analyzed for cytokine production. The concentrations of IL-12 p40 and IFN-α in culture supernatants were determined by enzyme-linked immunosorbent assays (ELISAs). ELISA kits for OptEIA mouse IL-12 p40 Set and mouse IFN-α were purchased from BD PharMingen (San Diego, Calif.) and PBL Biomedical Laboratories (New Brunswick, N.J.), respectively. Total RNAs were isolated by the use of Sepazol-RNA I (Nacalai Tesque, Kyoto, Japan), electrophoresed, and transferred to nylon membranes. Hybridization was performed with the indicated cDNA probes as previously described (2). cDNA probes specific for IL-12 p40 were established as previously described (31). To determine the effects of infection with AcNPV on cytokine production, we seeded the mouse macrophage cell line RAW264.7 into six-well plates at a concentration of 106 cells/well and stimulated them with various TLR ligands, with or without endosomal inhibitors such as chloroquine, bafilomycin A1, MDC, and ammonium chloride. For cell stimulation, AcNPV (5 μg/ml), LPS (10 ng/ml), PGN (2.5 μg/ml), and mCpG (200 ng/ml) were used.
Preparation of splenic dendritic cells and cytokine secretion.
To prepare splenocytes containing dendritic cells (DCs), we cut spleen tissues into small fragments and incubated them with RPMI 1640 containing 400 U of collagenase (Wako)/ml and 15 μg of DNase (Sigma-Aldrich)/ml at 37°C for 20 min. For the last 5 min, 5 mM EDTA was added, and single-cell suspensions were prepared after red blood cell lysis. CD11c+ cells were purified by magnetic cell sorting with anti-CD11c microbeads (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according to the manufacturer's instructions and were used as splenic DCs. Enriched cells containing >90% CD11c+ cells were seeded into 96-well plates at a concentration of 105 cells/well and stimulated with various doses of AcNPV or loxoribine. Culture supernatants were collected, and the production of IL-12 p40 and IFN-α was determined by ELISAs.
Indirect immunofluorescence assay and flow cytometric analysis.
293T cells transfected with a plasmid encoding human TLR9 were dislodged with PBS containing 5 mM EDTA 48 h after transfection. The cells were incubated with PBS containing 2% FCS and an anti-Flag (M2) monoclonal antibody (1:1,000) (Santa Cruz Biotechnology) for 1 h at 4°C, washed twice with PBS containing 2% FCS, and further incubated with fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G (IgG) (Sigma-Aldrich) in PBS containing 2% FCS for 1 h at 4°C. The cells were then fixed with 4% paraformaldehyde for 20 min, and the surface expression of human TLR9 was observed by fluorescence microscopy (UFX-II microscope; Nikon, Tokyo, Japan). Intracellular staining was examined after permeabilization with 0.5% Triton X-100. Stained cells were also analyzed by flow cytometry with a FACSCalibur instrument (Becton Dickinson, San Jose, Calif.), and the data were analyzed with CellQuest software (Becton Dickinson).
NF-κB-luciferase reporter gene assays with 293T cells.
293T cells were transfected with an NF-κB-dependent luciferase reporter plasmid (pELAM-Luc) together with human TLR9 expression vectors by the use of Lipofectamine 2000 (Life Technologies, Grand Island, N.Y.). pELAM-Luc (kindly provided by D. T. Golenbock) contains a human E-selectin promoter introduced into the pGL3 reporter plasmid (Promega, Inc., Madison, Wis.). The human TLR9 expression vector (kindly provided by T. H. Chuang) consists of a preprotrypsin signal peptide and a Flag epitope tag followed by an in-frame human TLR9 cDNA sequence (14). At 24 h posttransfection, the cells were stimulated with hCpG DNA (10 μg/ml) or AcNPV DNA (10 μg/ml) for 24 h. The luciferase activity was determined as previously described (49) and calculated as the degree of induction compared with an untreated control.
Detection of AcNPV capsid protein in murine macrophage cells by Western blot analysis.
RAW264.7 murine macrophage cells (106 cells/well) infected with AcNPV at a dose of 40 μg/ml were washed extensively after 1 h of adsorption and harvested after 4 or 6 h of incubation. The cells were lysed in buffer containing 1% Triton X-100, 135 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1% glycerol, and protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Mannheim, Germany). The lysed sample was separated by SDS-12.5% PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Tokyo, Japan). An anti-p39 mouse monoclonal antibody was used to detect the AcNPV capsid protein, which was visualized with the SuperSignal West Femto chemiluminescent substrate (Pierce).
RESULTS
Immune system activation by AcNPV is not mediated by viral envelope glycoprotein.
It was previously reported that an IFN-stimulating preparation purified from Sf-9 cells infected with AcNPV exhibited IFN production both in vitro and in vivo and that induction was inhibited by monoclonal antibodies against the AcNPV envelope glycoprotein gp64 (18). To verify these observations, we constructed a mutant baculovirus lacking gp64, which we called AcNPVΔ64, and examined its ability to stimulate an immune response in RAW264.7 cells, which are highly sensitive to TLR stimulation and respond by producing inflammatory cytokines at a level comparable to that observed in primary macrophages (1). The absence of gp64 in purified particles of AcNPVΔ64 was confirmed by immunoblotting (Fig. 1A). The mutant virus lost the ability to induce TNF-α production in inoculated RAW264.7 cells (Fig. 1B), a result that is consistent with the previous observation that gp64 appears to play an important role in the induction of the immune response by AcNPV (18). Because some microbial products are known to induce cytokine production in macrophages, it was important to eliminate the possibility that contamination with microbial products contributed to the immune system activation by AcNPV. Although the stimulation of macrophages by AcNPV was completely eliminated by incubation at 70°C for 30 min (Fig. 1C), stimulation by the bacterial components PGN and LPS was resistant to heat treatment (Fig. 1C and data not shown). These data indicate that the activation of macrophages by AcNPV is mediated by heat-labile viral components rather than by LPS and PGN.
FIG. 1.
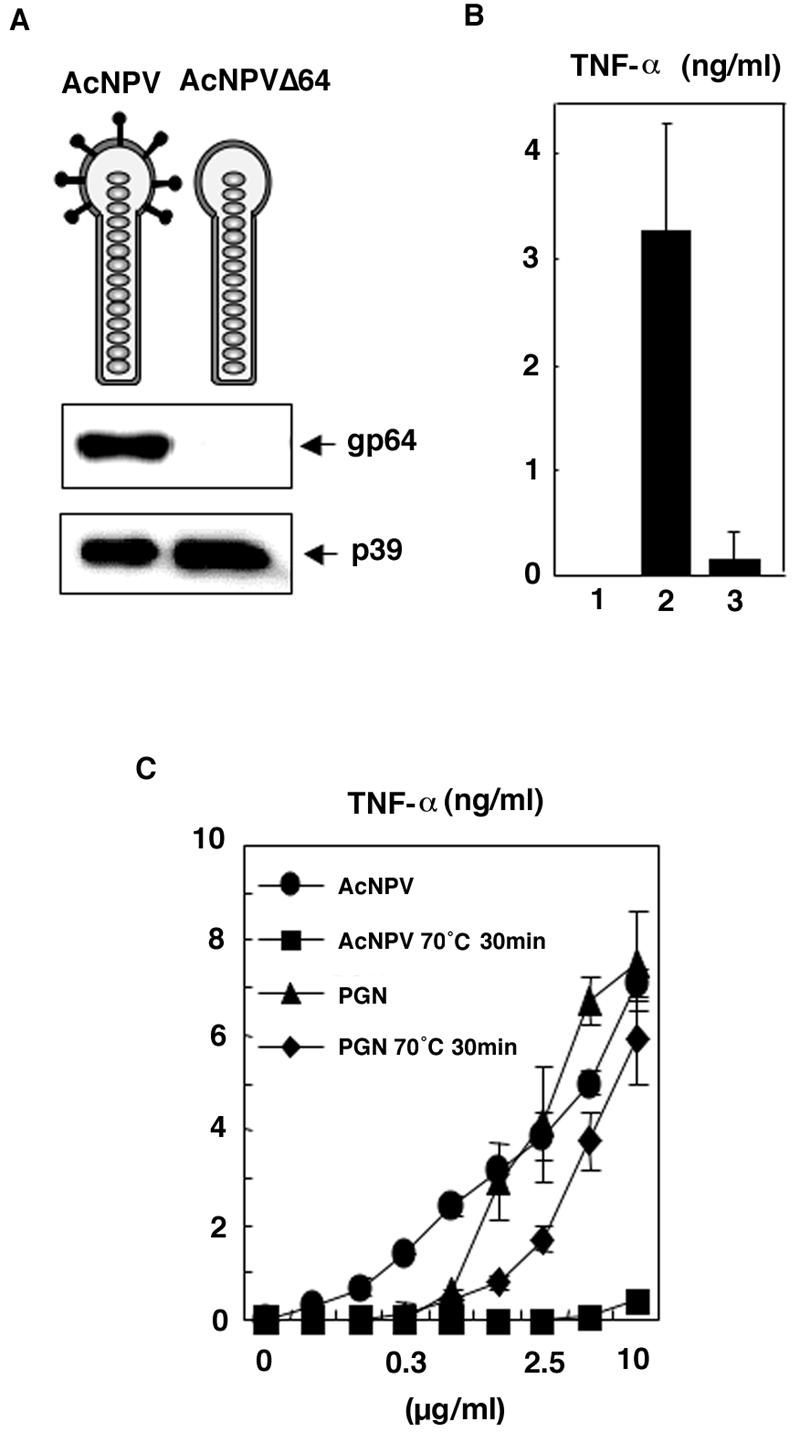
Immune system activation of macrophages by heat-denatured or gp64-deficient AcNPV. (A) Purified particles of the mutant virus, AcNPVΔ64, lack gp64, as assayed by immunoblotting. (B) The production of TNF-α in RAW264.7 cells (106 cells/well) inoculated with AcNPV (5 μg/ml) (bar 2) or AcNPVΔ64 (5 μg/ml) (bar 3) was determined 24 h after inoculation by a sandwich ELISA. 1 is an uninfected control. Data are shown as means ± SD. (C) AcNPV and PGN were incubated at 70°C for 30 min. Treated and untreated samples were inoculated into RAW264.7 cells (106 cells/well) and incubated for 24 h. The production of TNF-α was determined by a sandwich ELISA. Data are shown as means ± SD.
To further verify the involvement of gp64 in immune system stimulation by baculovirus, we prepared expression plasmids encoding both wild-type gp64 and a C-terminally truncated gp64 protein (gp64ΔTM) with a C-terminal His6 tag to allow for purification. Upon transfection of Sf9 cells, both recombinant proteins were detected, while gp64ΔTM was efficiently secreted into the culture supernatant (Fig. 2A). The protein from cells expressing gp64ΔTM was purified by column chromatography, producing a single band corresponding to gp64ΔTM and comparable to viral gp64 (Fig. 2B). We also tried to obtain the wild-type gp64 protein from the cell lysates but could not purify it to a homogeneous band (data not shown).
FIG. 2.

Immune system activation by AcNPV in macrophages is not mediated by gp64. (A) Wild-type gp64 and a deletion mutant lacking the transmembrane region of the gp64 envelope protein (gp64ΔTM) were expressed in Sf-9 cells. Whole-cell lysates and culture supernatants were subjected to SDS-PAGE under reducing conditions and visualized by immunoblotting with an antihexahistidine monoclonal antibody. Lane 1, cells transfected with pIB/V5-His; lanes 2 and 3, cells transfected with pIBgp64ΔTM/V5-His and pIBgp64/V5-His, respectively. The heavy chains of the antibody are indicated by asterisks. (B) Purified AcNPV virions (lane 2) and gp64ΔTM (lane 3) were analyzed by SDS-PAGE and Coomassie blue staining. Lane 1, molecular mass markers. (C) Activation of mouse macrophage RAW264.7 cells (106 cells/well) treated with the indicated amounts of AcNPV or gp64ΔTM. The production of TNF-α and IL-6 in culture supernatants after 24 h of incubation was determined by sandwich ELISAs. PGN was used as a positive control. Data are shown as means ± SD. (D) Production of IFN-α in RAW264.7 cells (106 cells/well) inoculated with AcNPV (5 μg/ml) or gp64ΔTM (5 μg/ml), as determined by a sandwich ELISA after 24 h of incubation. Data are shown as means ± SD. (E) Production of TNF-α in RAW264.7 cells (106 cells/well) inoculated with AcNPV (20 μg/ml) or PGN (2.5 μg/ml), with or without a pretreatment with the indicated amounts of gp64ΔTM for 2 h at 37°C. After 24 h of incubation, the production of TNF-α in culture supernatants was determined by a sandwich ELISA. Data are shown as means ± SD.
The activities of AcNPV, gp64ΔTM, and PGN on RAW264.7 cells were then examined. A dose-dependent induction of TNF-α and IL-6 was observed for RAW264.7 cells treated with AcNPV and PGN, whereas cytokine production was not observed for cells treated with gp64ΔTM (Fig. 2C). In addition, gp64ΔTM was not able to induce IFN-α production in RAW264.7 cells (Fig. 2D). Furthermore, the pretreatment of macrophage cells with gp64ΔTM inhibited immune system activation by AcNPV but had no effect on the activation by PGN (Fig. 2E), suggesting that the gp64ΔTM protein still retained some of the biological functions of the wild-type gp64 protein, at least in terms of its interaction with host cells. These results indicated that gp64 is an essential element of AcNPV-induced immune system activation in RAW264.7 cells but that it does not directly participate in the reaction. Viral components other than gp64 may be more directly involved in this process.
AcNPV induces inflammatory cytokine production through a MyD88/TLR9-dependent pathway.
Immune cells from MyD88- or TLR-deficient mice are unresponsive to TLR ligands, as assayed by their levels of cytokine production (5). Therefore, we used PECs and splenic CD11c+ DCs obtained from MyD88- and TLR-deficient mice to determine whether or not the TLR signaling pathway is responsible for the activation by AcNPV. Thioglycolate-elicited PECs were isolated from wild-type, MyD88−/−, TLR2−/−, TLR4−/−, and TLR9−/− mice and examined by ELISA and Northern blot analysis for the induction of IL-12 following exposure to AcNPV. Wild-type macrophages inoculated with AcNPV produced large amounts of IL-12 in a dose-dependent manner, whereas MyD88- or TLR9-deficient macrophages had severely reduced IL-12 production (Fig. 3A). PECs from TLR2−/− and TLR4−/− mice produced IL-12 at wild-type levels in response to AcNPV (Fig. 3A).
FIG. 3.

AcNPV activates PECs and DCs in a MyD88/TLR9-dependent manner. (A) PECs (2 × 105 cells/well) from wild-type (C57BL/6) or MyD88-, TLR2-, TLR4-, or TLR9-deficient mice were stimulated with the indicated amounts of AcNPV or loxoribine. The production of IL-12 p40 in culture supernatants was measured by a sandwich ELISA. Data are shown as means ± SD. (B) Northern blot analysis of murine macrophage cells stimulated with AcNPV. PECs (6 × 106 cells/well) from wild-type or MyD88- or TLR9-deficient mice were stimulated with AcNPV (10 μg/ml) for the indicated times. Total RNAs were extracted and subjected to Northern blot analysis. (C) Splenic CD11c+ DCs were prepared from wild-type or MyD88-, TLR4-, or TLR9-deficient mice and enriched by magnetic cell sorting. Splenic DCs (105 cells/well) were stimulated with the indicated amounts of AcNPV or loxoribine for 24 h. The production of IL-12 p40 in supernatants was measured by a sandwich ELISA. Data are shown as means ± SD.
Loxoribine is a potent inducer of cytokine production in macrophages and functions through a TLR7-dependent pathway (36). PECs from wild-type, TLR2−/−, TLR4−/−, and TLR9−/− mice all produced IL-12 in response to loxoribine, whereas no IL-12 production was observed in PECs from MyD88−/− mice (Fig. 3A). The transcription of IL-12 p40 mRNA was also impaired in MyD88- and TLR9-deficient macrophages stimulated with AcNPV (Fig. 3B). We further examined the response of splenic CD11c+ DCs to AcNPV and loxoribine. Wild-type and TLR4−/− splenic CD11c+ DCs produced IL-12 in response to AcNPV in a dose-dependent manner, whereas the production of IL-12 was severely impaired in MyD88−/− and TLR9−/− mice (Fig. 3C). In response to loxoribine, splenic CD11c+ DCs from TLR4−/− and TLR9−/− mice exhibited higher IL-12 production levels than wild-type cells, whereas the production of IL-12 was completely inhibited in MyD88−/− mice (Fig. 3C). These results indicate that AcNPV induces the production of inflammatory cytokines in immunocompetent cells through a MyD88/TLR9-dependent pathway.
AcNPV produces IFN-α through a MyD88/TLR9-independent pathway.
IFNs are important mediators of the early host defense against various viral infections. Since AcNPV has also been shown to be a potent inducer of IFN-α (Fig. 2D) (18), we investigated whether IFN-α production induced by AcNPV is dependent on the MyD88 and TLR9 signaling pathways. Although IFN-α induction by the TLR9 ligand, CpG oligonucleotides, was completely abolished in PECs and splenic CD11c+ DCs derived from MyD88−/− or TLR9−/− mice (data not shown), IFN-α production in response to AcNPV was less impaired (Fig. 4A). This contrasted sharply with the complete loss of IL-12 production observed for these cells (Fig. 3). Macrophages from MyD88−/− and TLR9−/− mice exhibited a slight reduction in IFN-α and IFN-β mRNA transcription in response to AcNPV (Fig. 4B). These results indicate that AcNPV induces the production of inflammatory cytokines in immunocompetent cells through a MyD88/TLR9-dependent pathway, while other MyD88/TLR9-independent pathways are also involved in the production of IFNs.
FIG. 4.

IFN production by AcNPV is mediated by a MyD88/TLR9-independent process. (A) PECs (2 × 105 cells/well) and splenic CD11c+ DCs (1 × 105 cells/well) were prepared from wild-type or MyD88- or TLR9-deficient mice and stimulated with the indicated amounts of AcNPV or loxoribine for 24 h. The production of IFN-α in culture supernatants was measured by a sandwich ELISA. Data are shown as means ± SD. (B) Northern blot analysis of murine macrophage cells stimulated with AcNPV. PECs (6 × 106 cells/well) from wild-type or MyD88- or TLR9-deficient mice were stimulated with AcNPV (10 μg/ml) for the indicated times. Total RNAs were then extracted and subjected to Northern blot analysis.
AcNPV DNA stimulates immune system activation in macrophage cell lines.
CpG motifs present in the genomes of many bacteria are unmethylated, whereas eukaryotic genomes are much more likely to undergo methylation. Previous work demonstrated that bacterial DNAs and certain oligonucleotides containing unmethylated CpG dinucleotides can stimulate PECs and DCs (19, 32). In addition, TLR9 is essential for the immune response to CpG-rich DNA, since TLR9-deficient mice are refractory to such stimulation (24). The frequency of bioactive CpG motifs in the AcNPV genome was similar to that observed for Escherichia coli and HSV DNAs (61) and significantly higher than that in murine and entomopoxvirus DNAs (Table 1).
TABLE 1.
CpG motif frequencies in AcNPV and other genomesa
| Motif | Frequency of appearance
|
||||
|---|---|---|---|---|---|
| E. coli | Mouse | HSV-1 | AcNPV | AmEPV | |
| CACGTT | 1.30 | 0.11 | 0.76 | 0.90 | 0.17 |
| AGCGTT | 1.70 | 0.17 | 0.42 | 1.12 | 0.15 |
| AACGTC | 0.60 | 0.11 | 0.73 | 0.98 | 0.17 |
| AGCGTC | 1.30 | 0.15 | 0.85 | 0.85 | 0.15 |
| GGCGTC | 1.40 | 0.15 | 4.0 | 1.10 | 0.02 |
| GGCGTT | 2.50 | 0.15 | 1.51 | 1.37 | 0.10 |
| Average | 1.53 | 0.14 | 1.38 | 1.05 | 0.13 |
The frequency at which each CpG hexamer appeared in the E. coli, mouse, HSV-1, AcNPV, and Amsacta moorei entomopoxvirus genomes was determined by using published sequence data. The GenBank accession numbers for the complete genomes of AcNPV and AmEPV are NC001623 and NC002520, respectively. The complete genomes of E. coli K-12 and HSV-1 and mouse chromosome sequences were described previously (61).
To determine the methylation status of the AcNPV genome, we digested DNAs isolated from AcNPV, Sf-9 cells, E. coli, and 293T cells with the restriction enzyme HpaII, which cannot cleave when the cytosine adjacent to the cleavage site (CC↓GG) is methylated. While DNA isolated from 293T cells was refractory to HpaII digestion, DNAs from AcNPV, Sf-9 cells, and E. coli were sensitive to HpaII digestion, indicating that most of the CpG dinucleotides in AcNPV were unmethylated (Fig. 5A).
FIG. 5.
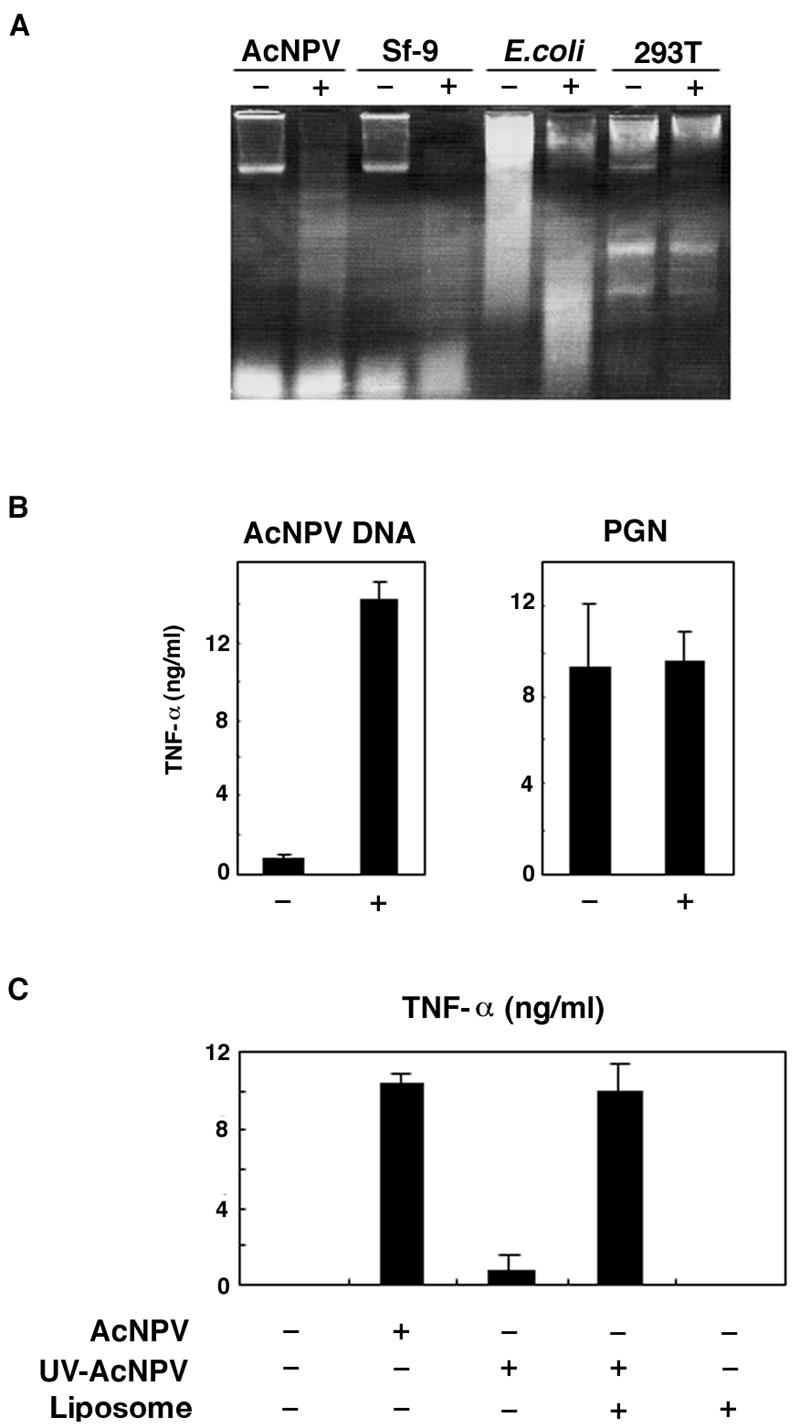
Activation of mouse macrophage cell line by AcNPV DNA. (A) Methylation status of genomic DNA. Genomic DNAs obtained from AcNPV, Sf-9 cells, E. coli, and 293T cells were digested with the methylation-sensitive restriction enzyme HpaII. Undigested (−) and digested (+) samples were analyzed by agarose gel electrophoresis. (B) RAW264.7 cells (106 cells/well) were treated with AcNPV DNA (5 μg/ml) or PGN (2.5 μg/ml) in the absence (−) or presence (+) of liposomes for 24 h, and the production of TNF-α in culture supernatants was determined by a sandwich ELISA. Data are shown as means ± SD. (C) Activation of RAW264.7 cells (106 cells/well) inoculated with untreated or UV-inactivated AcNPV (5 μg/ml) in the presence or absence of liposomes was assessed by the production of TNF-α in culture supernatants. Data are shown as means ± SD.
To determine the ability of AcNPV DNA to stimulate an immune response in vitro, we purified the viral DNA from virions. RAW264.7 cells were then treated with purified viral DNA or PGN with or without liposomes (Fig. 5B). The transfection of viral DNA with liposomes resulted in the production of TNF-α, but this effect was not observed in the absence of liposomes. The enhancement of TNF-α production by liposomes was not observed in cells treated with PGN, and the addition of liposomes alone did not elicit TNF-α production (Fig. 5C). These results indicate that the internalization of viral DNA is necessary for the activation of the AcNPV-mediated TLR9 signaling pathway. Thus, the impaired immune system activation by AcNPVΔ64 in macrophages may result from a failure to internalize viral DNA via gp64-mediated membrane fusion.
To further confirm that viral DNA activates the signaling pathway following internalization via gp64, we inactivated AcNPV by UV irradiation and examined the production of TNF-α in RAW264.7 cells. UV irradiation diminished the AcNPV-mediated induction of TNF-α, but the addition of liposomes restored the activation (Fig. 5C). These results suggest that the denaturation of gp64 by UV irradiation impaired the fusion capability of the envelope protein, thus inhibiting the internalization of viral DNA into the cell via membrane fusion.
AcNPV DNA induces NF-κB activation through human TLR9.
Signaling via TLRs occurs through the sequential recruitment of the adapter molecule MyD88 and the serine-threonine kinase IL-1 receptor-associated kinase, which leads to the activation of mitogen-activated protein kinases and the nuclear factor NF-κB (51). To assess whether or not the expression of human TLR9 confers cellular responsiveness to AcNPV DNA, we transfected 293T cells with a human TLR9 expression plasmid and a pELAM luciferase reporter plasmid together with AcNPV or hCpG, which was used as a positive control (Fig. 6A). Although NF-κB activation was not observed for cells transfected with undigested AcNPV DNA, HindIII-digested viral DNA and hCpG exhibited significant NF-κB activation, suggesting that undigested viral DNA is incapable of penetrating cells by transfection. No activation of NF-κB was observed in 293T cells cotransfected with a human TLR2 or TLR4 expression plasmid when stimulated with digested AcNPV DNA (data not shown).
FIG. 6.

AcNPV DNA induces NF-κB activation through human TLR9. (A) 293T cells were transfected with an empty or human TLR9 expression vector together with a pELAM luciferase reporter plasmid. Twenty-four hours after transfection, the cells were stimulated with digested or undigested AcNPV DNA (10 μg/ml). hCpG (10 μg/ml) was used as a positive control. The luciferase activity was determined at 24 h posttransfection and expressed as the level of induction compared with that detected in cells transfected with the human TLR9 expression vector alone. Data are shown as means ± SD. (B) Immunofluorescence micrographs of 293T cells transfected with an N-terminal Flag-tagged human TLR9 expression vector and stained with an anti-Flag (M2) monoclonal antibody. The intracellular (left) and cell surface (right) expression of TLR9 is shown. Nuclei were stained with propidium iodide (PI). Samples were observed by confocal microscopy. (C) The surface and intracellular expression of human TLR9 in 293T cells transfected with an N-terminal Flag-tagged human TLR9 expression vector (+) or an empty vector (−) and stained with an anti-Flag monoclonal antibody was examined by fluorescence-activated cell sorting.
Recent work demonstrated that the endogenous expression of TLR3, TLR7, TLR8, and TLR9 was mainly detected in the cytoplasmic vesicles of macrophages (58). To examine the localization of transiently expressed TLR9, we transfected 293T cells with a TLR9 expression plasmid and examined TLR9 expression by immunofluorescence microscopy and cell sorting. The expression of TLR9 in the cytoplasm was three times higher than that at the cell surface (Fig. 6B and C). These results indicate that the introduction of AcNPV DNA into the cytoplasm is specifically detected by human TLR9 and results in the activation of NF-κB.
AcNPV requires endosomal maturation to induce immune system activation in macrophages.
To further explore the role of endocytosis in the signal transduction pathway triggered by AcNPV DNA, we examined the effect of endosomal maturation or acidification inhibitors. As shown in Fig. 7A, chloroquine was able to inhibit immune system activation of RAW264.7 cells treated with AcNPV and mCpG oligonucleotides in a dose-dependent manner, but no inhibition of LPS or PGN activation was observed. Other inhibitors of endosomal maturation, such as ammonium chloride, bafilomycin A1, and MDC, inhibited AcNPV-induced, but not LPS-induced, immune system activation (Fig. 7B). Together with our other data, these results indicate that endosomal acidification and/or maturation is a key step in AcNPV-induced immune system activation via TLR9, a process that requires the release of the viral genome into TLR9-expressing cytoplasmic vesicles following the internalization of viral DNA by endocytosis through gp64-mediated membrane fusion.
FIG. 7.

AcNPV requires endosomal maturation to induce immune system activation in macrophages. (A) RAW264.7 cells (106 cells/well) were stimulated with AcNPV (5 μg/ml), mCpG (200 ng/ml), LPS (10 ng/ml), or PGN (2.5 μg/ml) at the indicated concentrations of chloroquine. After 24 h of incubation, the production of TNF-α in culture supernatants was determined by a sandwich ELISA. Chloroquine was added to the cells 2 h before stimulation. Data are shown as means ± SD. (B) RAW264.7 cells (106 cells/well) were treated with AcNPV (5 μg/ml) or LPS (10 ng/ml) and with the indicated concentrations of endosomal maturation inhibitors. After 24 h of incubation, the production of TNF-α in culture supernatants was determined by a sandwich ELISA. The inhibitors were added to the cells 2 h before stimulation. Data are shown as means ± SD.
AcNPV penetrates macrophages via the phagocytic pathway.
To further confirm that baculovirus was internalized into macrophages, we inoculated RAW264.7 cells with a recombinant baculovirus carrying a luciferase gene under the control of a mammalian promoter, AcCAGluc (49). As shown in Fig. 8A, the expression of luciferase was observed in 293T cells, but not RAW264.7 cells, that were infected with AcCAGluc. The viral capsid protein was clearly detected by immunoblotting for both 293T and RAW264.7 cells infected with AcNPV, but the protein level was greatly diminished in RAW264.7 cells by 6 h postinoculation, probably as a result of degradation (Fig. 8B). These results suggest that baculovirus can penetrate into different cells via gp64-mediated endocytosis but that it translocates into different subcellular compartments in different cells. In 293T cells, the nucleocapsid was apparently able to reach the nucleus, where the reporter gene was efficiently transcribed following uncoating. However, in the immunocompetent RAW264.7 cells, the nucleocapsid appeared to have been trapped by the phagocytic pathway, and degraded viral DNA was then translocated into TLR9-expressing intracellular compartments (58).
FIG. 8.

AcNPV penetrates macrophages through the phagocytic pathway. (A) 293T and RAW264.7 cells (106 cells/well) were inoculated with a recombinant baculovirus possessing the luciferase gene under the control of the CAG promoter, AcCAGluc (49) (10 and 20 μg/ml). Cells were harvested 24 h after infection, and relative luciferase activities were determined. (B) 293T and RAW264.7 cells (106 cells/well) were inoculated with AcCAGluc (40 μg/ml), washed extensively after 1 h of adsorption, and harvested after 4 or 6 h of incubation. The presence of the p39 capsid protein in cells inoculated with AcNPV was determined by immunoblotting with an anti-p39 monoclonal antibody.
DISCUSSION
We have previously demonstrated that intranasal inoculation with AcNPV induces a strong innate immune response that protects mice from a lethal challenge with influenza virus (1). The lungs of mice inoculated with AcNPV exhibited a marked infiltration of macrophages, which presumably inhibit the growth of influenza virus in the lung tissues. The baculovirus envelope glycoprotein gp64 contains mannose, fucose, and N-acetyl-glucosamine modifications but no detectable galactose or terminal sialic acid residues (29). The mannose receptor (MR) recognizes a range of carbohydrates present on the surfaces and cell walls of microorganisms. MR is primarily expressed on macrophages and DCs and is involved in MR-mediated endocytosis and phagocytosis. In addition, MR plays a key role in host defense and the induction of innate immunity (8). Therefore, it is tempting to speculate that gp64 interacts with MR through its mannose modifications in macrophages and DCs of mice inoculated with AcNPV. However, our data contradict such a model; instead, we show that it is AcNPV DNA, not the gp64 glycoprotein, that induces immune system activation in a MyD88/TLR9-dependent manner.
Recently, it was shown that plasmacytoid DCs (pDCs) naturally produce IFN-α in response to viruses (30). HSV-1 and -2, whose genomes contain abundant CpG motifs, are able to induce the production of IFN-α in pDCs. The HSV-induced production of IFN-α in pDCs derived from MyD88- and TLR9-deficient mice was completely eliminated (33, 40). The recognition of the HSV genome by TLR9 was shown to be mediated by an endocytic pathway that can be inhibited by chloroquine or bafilomycin A1. In this study, we demonstrated that AcNPV induces proinflammatory cytokines through a MyD88/TLR9-dependent signaling pathway, whereas signaling molecules other than MyD88 may participate in IFN-α production in response to AcNPV. Recently, MyD88-independent TLR signaling events involving TIR domain-containing adaptor inducing IFN-β (TRIF) were described (59). Therefore, it is possible that the TRIF pathway is one means by which AcNPV induces MyD88-independent IFN production. However, future studies are needed to clarify the precise mechanisms of this induction.
While UV irradiation of AcNPV abolishes its ability to stimulate an immune response, the addition of liposomes is able to restore this activity. UV-inactivated HSV is capable of inducing the production of IFN-α in pDCs (40), indicating that viral replication is not required for the HSV-induced immune response. In contrast, UV irradiation of AcNPV abolishes immune stimulation in macrophages, while internalization of the inactivated virus by liposomes restores the activity. These results, in conjunction with our data for AcNPVΔ64, indicate that the AcNPV-induced production of cytokines in immunocompetent cells requires a fusion process mediated by gp64 that leads to internalization of the viral genome into the cells.
Recently, several viral envelope glycoproteins were shown to induce immune system activation through TLRs (10, 22, 34, 47). However, gp64 does not directly participate in a TLR-mediated immune response. TLR family members are expressed differentially at very low levels on the surfaces of different immune cells and appear to respond to different stimuli (43). A recent study indicated that LPS and CpG-rich DNA activate TLRs in distinct cellular compartments (3). Internalization and endosomal maturation are required for CpG-rich DNA to activate TLR9, but not for LPS to activate TLR4 on the plasma membrane. We showed here that the inhibition of endosomal maturation by a treatment with chloroquine abolishes the immune system activation of AcNPV in a dose-dependent manner. These results imply that immune system activation by AcNPV through TLR9 requires membrane fusion via gp64 as well as the liberation of the viral genome into cytoplasmic vesicles expressing TLR9.
Interestingly, Lund et al. demonstrated that the TLR7-mediated immune recognition of single-stranded RNAs from vesicular stomatitis virus and influenza virus requires endosomal acidification (41). The recognition of HSV-1 and HSV-2 viral DNAs through a TLR9/MyD88-dependent pathway in pDCs also requires endosomal acidification (40). These data indicate that TLR7 and TLR9 expressed in the endosomal or lysosomal compartments of immunocompetent cells recognize the viral genome entering the cell through receptor-mediated endocytosis or phagocytosis, leading to the secretion of inflammatory cytokines and IFNs. However, the precise mechanisms by which viral genomes translocate to TLR-expressing compartments are still unknown.
Since the first report on the immunostimulatory potential of bacterial DNA, which found that the main immunogenic fraction of mycobacterial lysates consists of genomic DNA (55, 56), substantial progress has been made towards understanding the immunostimulatory potency of CpG-rich DNA motifs, which are more common in bacteria than in vertebrates. For instance, TLR9 was shown to be responsible in vivo for immune system stimulation by oligodeoxynucleotides containing unmethylated CpG motifs (24). Like bacteria, AcNPV contains a significant number of potentially bioactive CpG motifs. Interestingly, the frequency of CpG motifs in HSV DNA, which has been shown to be involved in the induction of angiogenesis in stromal keratitis (61), was similar to that in E. coli DNA. In contrast, the frequency of CpG motifs in the genome of an insect poxvirus was much lower than that for AcNPV (Table 1).
In conclusion, we have demonstrated that AcNPV has the ability to induce innate immune system activation through a MyD88/TLR9-dependent pathway. The molecular mechanisms of viral uptake, intracellular processing, and the induction of potent antiviral activity in immune cells require further investigation. However, the strong immune response induced by AcNPV makes it a promising candidate for a novel, adjuvant-containing vaccine vehicle against infectious diseases. In particular, our findings raise the possibility that AcNPV may be harnessed therapeutically to induce a host immune response against various infectious diseases caused by pathogens invading the respiratory tract.
Acknowledgments
We are grateful to T. H. Chuang for providing us with the TLR9 expression plasmid, D. T. Golenbock for the pELAM Luc plasmid, and G. F. Rohrmann for the p39 monoclonal antibody. We also thank I. Yanase and H. Murase for secretarial work. T.A. and H.H. are Research Fellows of the Japan Society for the Promotion of Science.
This work was supported by grants-in-aid from the Ministry of Health, Labor and Welfare in Japan to Y.M. and from the 21st Century Center of Excellence Program of Japan to Y.M. and S.A.
Footnotes
This study is dedicated to the memory of Ikuko Yanase.
REFERENCES
- 1.Abe, T., H. Takahashi, H. Hamazaki, N. Miyano-Kurosaki, Y. Matsuura, and H. Takaku. 2003. Baculovirus induces an innate immune response and confers protection from lethal influenza virus infection in mice. J. Immunol. 171:1133-1139. [DOI] [PubMed] [Google Scholar]
- 2.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143-150. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad-Nejad, P., H. Hacker, M. Rutz, S. Bauer, R. M. Vabulas, and H. Wagner. 2002. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptor at distinct cellular compartments. Eur. J. Immunol. 32:1958-1968. [DOI] [PubMed] [Google Scholar]
- 4.Airenne, K. J., M. O. Hiltunen, M. P. Turunen, A. M. Turunen, O. H. Laitinen, M. S. Kulomaa, and S. Y. Herttuala. 2000. Baculovirus-mediated periadventitial gene transfer to rabbit carotid artery. Gene Ther. 7:1499-1504. [DOI] [PubMed] [Google Scholar]
- 5.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675-680. [DOI] [PubMed] [Google Scholar]
- 6.Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413:732-738. [DOI] [PubMed] [Google Scholar]
- 7.Aliprantis, A. O., R. B. Yang, M. R. Mark, S. Suggett, B. Devaux, J. D. Radolf, G. R. Klimpel, P. Godowski, and A. Zychlinsky. 1999. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285:736-739. [DOI] [PubMed] [Google Scholar]
- 8.Apostolopoulos, V., and I. F. McKenzie. 2001. Role of the mannose receptor in the immune response. Curr. Mol. Med. 1:469-474. [DOI] [PubMed] [Google Scholar]
- 9.Barsoum, J., R. Brown, M. Mckee, and F. M. Boyce. 1997. Efficient transduction of mammalian cells by a recombinant baculovirus having the vesicular stomatitis virus G glycoprotein. Hum. Gene Ther. 8:2011-2018. [DOI] [PubMed] [Google Scholar]
- 10.Bieback, K., E. Lien, I. M. Klagge, E. Avota, J. Schneider-Schaulies, W. P. Duprex, H. Wagner, C. J. Kirschning, V. Ter Meulen, and S. Schneider-Schaulies. 2002. Hemagglutinin protein of wild-type measles virus activates Toll-like receptor 2 signaling. J. Virol. 76:8729-8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowie, A., E. Kiss-Toth, J. A. Symons, G. L. Smith, S. K Dower, and L. A. O. Neill. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 97:10162-10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyce, F. M., and N. L. R. Bucher. 1996. Baculovirus mediated gene transfer into mammalian cells. Proc. Natl. Acad. Sci. USA 93:2348-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brightbill, H. D., D. H. Libraty, S. R. Krutzik, R. B. Yang, J. T. Belisle, J. R. Bleharski, M. Maitland, M. V. Norgard, S. E. Plevy, S. T. Smale, P. J. Brennan, B. R. Bloom, P. J. Godowski, and R. L. Modlin. 1999. Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science 285:732-736. [DOI] [PubMed] [Google Scholar]
- 14.Chuang, T. H., J. Lee, L. Kline, J. C. Mathison, and R. J. Ulevitch. 2002. Toll-like receptor 9 mediates CpG-DNA signaling. J. Leukoc. Biol. 71:538-544. [PubMed] [Google Scholar]
- 15.Compton, T., E. A. Kurt-Jones, K. W. Boehme, J. Belko, E. Latz, D. T. Golenbock, and R. W. Finberg. 2003. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 77:4588-4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Condreay, J. P., S. M. Witherspoon, W. C. Clay, and T. A. Kost. 1999. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc. Natl. Acad. Sci. USA 96:127-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, E. Reis, and C. Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529-1531. [DOI] [PubMed] [Google Scholar]
- 18.Gronowski, A. M., D. M. Hilbert, K. C. F. Sheehan, G. Garotta, and R. D. Schreiber. 1999. Baculovirus stimulates antiviral effect in mammalian cells. J. Virol. 73:9944-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hacker, H., H. Mischak, T. Miethke, S. Liptay, R. Schmid, T. Sparwasser, K. Heeg, G. B. Lipford, and H. Wagner. 1998. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 17:6230-6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hacker, H., R. M. Vabulas, O. Takeuchi, K. Hoshino, S. Akira, and H. Wagner. 2000. Immune cell activation by bacterial CpG-DNA through myeloid differential marker 88 and tumor necrosis factor receptor-associated factor (TRAF) 6. J. Exp. Med. 192:595-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng, S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099-1103. [DOI] [PubMed] [Google Scholar]
- 22.Haynes, L. M., D. D. Moore, E. A. Kurt-Jones, R. W. Finberg, L. J. Anderson, and R. A. Tripp. 2001. Involvement of Toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 75:10730-10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303:1526-1529. [DOI] [PubMed] [Google Scholar]
- 24.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740-745. [DOI] [PubMed] [Google Scholar]
- 25.Hemmi, H., T. Kaisho, O. Takeuchi, S. Sato, H. Sanjo, K. Hoshino, T. Horiuchi, H. Tomizawa, K. Takeda, and S. Akira. 2002. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3:196-200. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann, C., V. Sandig, G. Jennings, M. Rudolph, P. Schlag, and M. Strauss. 1995. Efficient gene transfer into human hepatocytes by baculovirus vectors. Proc. Natl. Acad. Sci. USA 92:10099-10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the LPS gene product. J. Immunol. 162:3749-3752. [PubMed] [Google Scholar]
- 28.Huser, A., M. Rudolph, and C. Hofmann. 2001. Incorporation of decay-accelerating factor into the baculovirus envelope generates complement resistant gene transfer vectors. Nat. Biotechnol. 19:451-455. [DOI] [PubMed] [Google Scholar]
- 29.Jarvis, D. L., and E. E. Finn. 1995. Biochemical analysis of the N-glycosylation pathway in baculovirus-infected lepidopteran insect cells. Virology 212:500-511. [DOI] [PubMed] [Google Scholar]
- 30.Kadowaki, N., S. Antonenko, J. Y. Lau, and Y. J. Liu. 2000. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 192:219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P. F. Muhlradt, S. Sato, K. Hoshino, and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167:5887-5894. [DOI] [PubMed] [Google Scholar]
- 32.Krieg, A. M., and H. Wagner. 2000. Causing a commotion in the blood: immunotherapy progresses from bacteria to bacterial DNA. Immunol. Today 21:521-526. [DOI] [PubMed] [Google Scholar]
- 33.Krug, A., G. D. Luker, W. Barchet, D. A. Leib, S. Akira, and M. Colonna. 2004. Herpes simplex virus type 1 (HSV-1) activates murine natural interferon-producing cells (IPC) through Toll-like receptor 9. Blood 103:1433-1437. [DOI] [PubMed] [Google Scholar]
- 34.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 5:398-401. [DOI] [PubMed] [Google Scholar]
- 35.Kurt-Jones, E. A., M. Chan, S. Zhou, J. Wang, G. Reed, R. Bronson, M. M. Arnold, D. M. Knipe, and R. W. Finberg. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 101:1315-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee, J., T. H. Chuang, V. Redecke, L. She, P. M. Pitha, D. A. Carson, E. Raz, and H. B. Cottam. 2003. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 100:6646-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehtolainen, P., K. Tyynela, J. Kannasto, K. J. Airenne, and S. Y. Herttuala. 2002. Baculoviruses exhibit restricted cell type specificity in rat brain: a comparison of baculovirus- and adenovirus-mediated intracerebral gene transfer in vivo. Gene Ther. 9:1693-1699. [DOI] [PubMed] [Google Scholar]
- 38.Lematitre, B., E. Nicolas, L. Michaut, J. M. Reichhart, and J. A. Hoffmann. 1996. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973-983. [DOI] [PubMed] [Google Scholar]
- 39.Luckow, V. A., and M. D. Summers. 1988. Signals important for high-level expression of foreign genes in Autographa californica nuclear polyhedrosis virus expression vectors. Bio/Technology 6:47-55. [DOI] [PubMed] [Google Scholar]
- 40.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lund, J., L. Alexopoulou, A. Sato, M. Karow, N. C. Adams, N. W. Gale, A. Iwasaki, and R. A. Flavell. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 101:5598-5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuura, Y., R. D. Possee, H. A. Overton, and D. H. L. Bishop. 1987. Baculovirus expression vector. The requirement for high level expression of proteins, including glycoproteins. J. Gen. Virol. 68:1233-1250. [DOI] [PubMed] [Google Scholar]
- 43.Muzio, M., D. Bosisio, N. Polentarutti, G. Damico, A. Stoppacciaro, R. Mancinelli, C. vant Veer, G. Penton-Rol, L. P. Ruco, P. Allavena, and A. Mantovani. 2000. Differential expression and regulation of Toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 164:5998-6004. [DOI] [PubMed] [Google Scholar]
- 44.Ozinsky, A., D. M. Underhill, J. D. Fontenot, A. M. Hajjar, K. D. Smith, C. B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. USA 97:13766-13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pieroni, L., D. Maione, and N. L. Monica. 2001. In vivo gene transfer in mouse skeletal muscle mediated by baculovirus vectors. Hum. Gene Ther. 12:871-881. [DOI] [PubMed] [Google Scholar]
- 46.Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085-2088. [DOI] [PubMed] [Google Scholar]
- 47.Rassa, J. C., J. L. Meyers, Y. Zhang, R. Kudaravalli, and S. R. Ross. 2002. Murine retroviruses activate B cells via interaction with Toll-like receptor 4. Proc. Natl. Acad. Sci. USA 99:2281-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sandig, V., C. Hofmann, S. Steinert, G. Jennings, P. Schlag, and M. Strauss. 1996. Gene transfer into hepatocytes and human liver tissue by baculovirus vectors. Hum. Gene Ther. 7:1937-1945. [DOI] [PubMed] [Google Scholar]
- 49.Shoji, I., H. Aizaki, H. Tani, K. Ishii, T. Chiba, I. Saito, T. Miyamura, and Y. Matsuura. 1997. Efficient gene transfer into various mammalian cells, including non-hepatic cells, by baculovirus vectors. J. Gen. Virol. 78:2657-2664. [DOI] [PubMed] [Google Scholar]
- 50.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takeda, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443-451. [DOI] [PubMed] [Google Scholar]
- 51.Takeuchi, O., and S. Akira. 2001. Toll-like receptors: their physiological role and signal transduction system. Int. Immunopharmacol. 1:625-635. [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi, O., A. Kaufmann, K. Grote, T. Kawai, K. Hoshino, M. Morr, P. F. Muhlradt, and S. Akira. 2000. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 164:554-557. [DOI] [PubMed] [Google Scholar]
- 53.Tani, H., M. Nishijima, H. Ushijima, T. Miyamura, and Y. Matsuura. 2001. Characterization of cell-surface determinants important for baculovirus infection. Virology 279:343-353. [DOI] [PubMed] [Google Scholar]
- 54.Tani, H., C. K. Limn, C. C. Yap, M. Onishi, M. Nozaki, Y. Nishimune, N. Okahashi, Y. Kitagawa, R. Watanabe, R. Mochizuki, K. Moriishi, and Y. Matsuura. 2003. In vitro and in vivo gene delivery by recombinant baculoviruses. J. Virol. 77:9799-9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tokunaga, T., H. Yamamoto, S. Shimada, H. Abe, T. Fukuda, Y. Fujisawa, Y. Furutani, O. Yano, T. Kataoka, T. Sudo, N. Makiguchi, and T. Suganuma. 1984. Antitumor activity of deoxyribonucleic acid fraction from Mycobacterium bovis BCG. I. Isolation, physicochemical characterization and antitumor activity. JNCI 72:955-962. [PubMed] [Google Scholar]
- 56.Tokunaga, T., T. Yamamoto, and S. Yamamoto. 1999. How BCG led to the discovery of immunostimulatory DNA. Jpn. J. Infect. Dis. 52:1-11. [PubMed] [Google Scholar]
- 57.Underhill, D. M., A. Ozinsky, A. M. Hajjar, A. Stevens, C. B. Wilson, M. Bassetti, and A. Aderem. 1999. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401:811-815. [DOI] [PubMed] [Google Scholar]
- 58.Wagner, H. 2004. The immunobiology of the TLR9 subfamily. Trends Immunol. 25:381-386. [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640-643. [DOI] [PubMed] [Google Scholar]
- 60.Zhang, D., G. Zhang, M. S. Hayden, M. B. Greenblatt, C. Bussey, R. A. Flavell, and S. Ghosh. 2004. A Toll-like receptor that prevents infection by uropathogenic bacteria. Science 303:1522-1526. [DOI] [PubMed] [Google Scholar]
- 61.Zheng, M., D. M. Klinman, M. Gierynska, and B. T. Rouse. 2002. DNA containing CpG motifs induces angiogenesis. Proc. Natl. Acad. Sci. USA 99:8944-8949. [DOI] [PMC free article] [PubMed] [Google Scholar]