

Abstract
Objective
A disintegrin and metalloproteinase ADAM17 (TNF-α converting enzyme) regulates soluble TNF levels. We tested the hypothesis that aging-induced activation in adipose tissue (AT)-expressed ADAM17 contributes to the development of remote coronary microvascular dysfunction (CMD) in obesity.
Approach and Results
Coronary arterioles (CA,~90 μm) from right atrial appendages and mediastinal AT were examined in patients (age:69±11yrs.,BMI:30.2±5.6) who underwent open-heart surgery. CA and AT were also studied in 6-month and 24-month lean and obese mice fed a normal or high-fat diet (HFD). We found that obesity elicited impaired endothelium-dependent CA dilations only in older patients and in aged HFD mice. Transplantation of AT from aged obese, but not from young or aged mice increased serum cytokine levels, including TNF, and impaired CA dilation in the young recipient mice. In patients and mice obesity was accompanied by age-related activation of ADAM17, which was attributed to vascular endothelium-expressed ADAM17. Excess, ADAM17-shed TNF from AT arteries in older obese patients was sufficient to impair CA dilation in a bioassay in which the AT artery was serially connected to a CA. Moreover, we found that the increased activity of endothelial ADAM17 is mediated by a diminished inhibitory interaction with caveolin-1, owing to age-related decline in caveolin-1 expression in obese patients and mice or to genetic deletion of caveolin-1.
Conclusions
The present study indicates that aging and obesity cooperatively reduce Cav1 expression, increase vascular endothelial ADAM17 activity and soluble TNF release in AT, which may contribute to the development of remote CMD in older obese patients.
Keywords: human, aging, obesity, adipose tissue, endothelium, coronary arterioles, caveolin-1
Subject Codes: Vascular Biology, Endothelium/Vascular Type/Nitric Oxide, Coronary Circulation, Inflammation
Graphical abstract
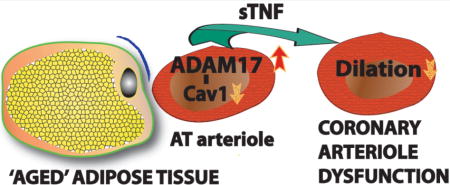
INTRODUCTION
Dysfunction of coronary resistance arteries develops in older patients, often in the absence of occlusive atherosclerotic lesions in the large coronary artery, which contributes to poor myocardial perfusion.1 Cardiovascular complications arising specifically from coronary microvascular dysfunction (CMD) are less defined2 and often viewed as an inevitable consequence of aging. CMD has recently emerged as the primary underlying pathology of a subset of cardiovascular diseases, such as heart failure with preserved ejection fraction (HFpEF).3 HFpEF is increasingly prevalent in older patients, including both men and women, who often suffer from several comorbidities.4 There is general agreement that HFpEF is uniquely driven by a chronic systemic inflammatory state, which selectively targets and adversely affects the coronary microcirculation.5, 6 In 732 elderly Framingham study subjects inflammatory mediators, such as TNF, interleukin-6 and C-reactive protein were associated with increased risk for heart failure in people without prior myocardial infarction.7 The nature of the underlying mechanisms of inflammatory mediator-induced CMD remains poorly understood. It is known that comorbid conditions such as obesity can dramatically accelerate the timeline to many cardiovascular complications and particularly so with advanced age.8 Therefore, in this study we sought to investigate the mechanisms by which pro-inflammatory changes in ‘aged’ adipose tissue contribute to the development of CMD in older obese patients with cardiovascular diseases.
It has been long recognized that expansion of visceral adipose tissue (abdominal and intrathoracal) adversely affects metabolic homeostasis.9 It is also known that pathological adipose tissue expansion is associated with excess production of adipokines10, 11 and pro-inflammatory cytokines, including tumor necrosis factor-α (TNF).12–14 TNF plays a well-established role in the development of insulin resistance in type 2 diabetes.15, 16 In animal models of obesity-associated metabolic syndrome, increased plasma TNF was shown to be associated with coronary artery vasodilator dysfunction, due to TNF-mediated increases in reactive oxygen species (ROS) production and impaired endothelial nitric oxide (NO) synthase (eNOS) function.17, 18 We recently provided evidence that increased ROS and peroxynitrite formation impairs NO-mediated coronary arteriole dilation in patients with type 2 diabetes.19 Previous clinical studies have revealed that elevated plasma and adipose tissue TNF levels strongly correlate with the severity of coronary artery disease in elderly.12, 20–22 In this context, we23 and others24 have found that aging exacerbates obesity-associated vascular dysfunction in mouse models and that adipose tissue-derived cytokines, including TNF, play a pivotal role in the vascular impairment. While these aforementioned studies provide evidence for a pathological role of adipose tissue-derived TNF in age-related vascular dysfunction, the underlying mechanisms of increased TNF production and its causative relationship with CMD remain largely unknown.
A disintegrin and metalloproteinase, ADAM17, also known as TNF-α converting enzyme or TACE, regulates soluble TNF levels.25 ADAM17 mRNA expression has been detected in mouse adipose tissue (both in visceral and subcutaneous fat pads), which is readily up-regulated after high fat feeding.26 ADAM17 is sequestered and localized within cell membrane caveolae, where its activity is regulated through its interaction with caveolin-1 (Cav1), the main structural protein of membrane caveolae.27–29 Our recent studies have shown that diabetic patients exhibit ROS-dependent disruption of vascular endothelial caveolae19 and that mice with genetic deletion of Cav1 develop coronary artery dilator dysfunction19 and high blood pressure, when fed a high fat diet.30 The nature of mechanisms by which adipose tissue ADAM17 contributes to excess TNF release and the role of endothelial caveolae dysfunction in this process remain unknown in aging. These observations and considerations led us to hypothesize that aging of the adipose tissue is associated with transition to pro-inflammatory phenotype, which is characterized by ADAM17 activation and excess TNF production thereby contributing to the development of CMD in older obese patients with cardiovascular diseases.
MATERIAL AND METHODS
“Materials and Methods are available in the online-only Data Supplement.”
RESULTS
Reduced dilation in coronary arterioles in older obese patients
Coronary arteriole dilator function was examined in isolated coronary arterioles obtained from consecutive patients who underwent open-heart surgery. The active (spontaneously developed or pre-constricted by U46619, 10 nM, mean arteriolar tone: 28.4±1.7%) and passive (in the absence of extracellular Ca2+) baseline diameters of isolated coronary arterioles were not significantly different in the four groups (diameters in μm, Younger-BMI<30, active: 93±9, passive: 126±12; Younger-BMI≥30, active: 86±5, passive: 136±6; Older-BMI<30, active: 80±12, passive: 108±11; Older-BMI≥30, active: 92±13, passive: 120±14). We found that the magnitude of endothelium-dependent coronary arteriole dilation in response to bradykinin was significantly decreased in older (≥69 yrs.) patients, which was even further reduced in older obese (BMI≥30) individuals (Fig. 1a). When examined by regression analyses the age-related decline was found to be significant only in obese patients (BMI≥30), whereas a non-significant negative trend was observed in the non-obese (BMI<30) (Fig. 1c). The age-related decline in bradykinin-induced coronary arteriole dilation was similar in men and women (r= −0.486 vs. r= −0.629, P=0.131, Supplementary Fig. I). In addition, no significant correlation was observed between age, obesity and SNP (NO donor)-induced coronary arteriole dilation in this patient population (Fig. 1b & 1d).
Figure 1. Reduced dilation in isolated coronary arterioles in older and obese patients.
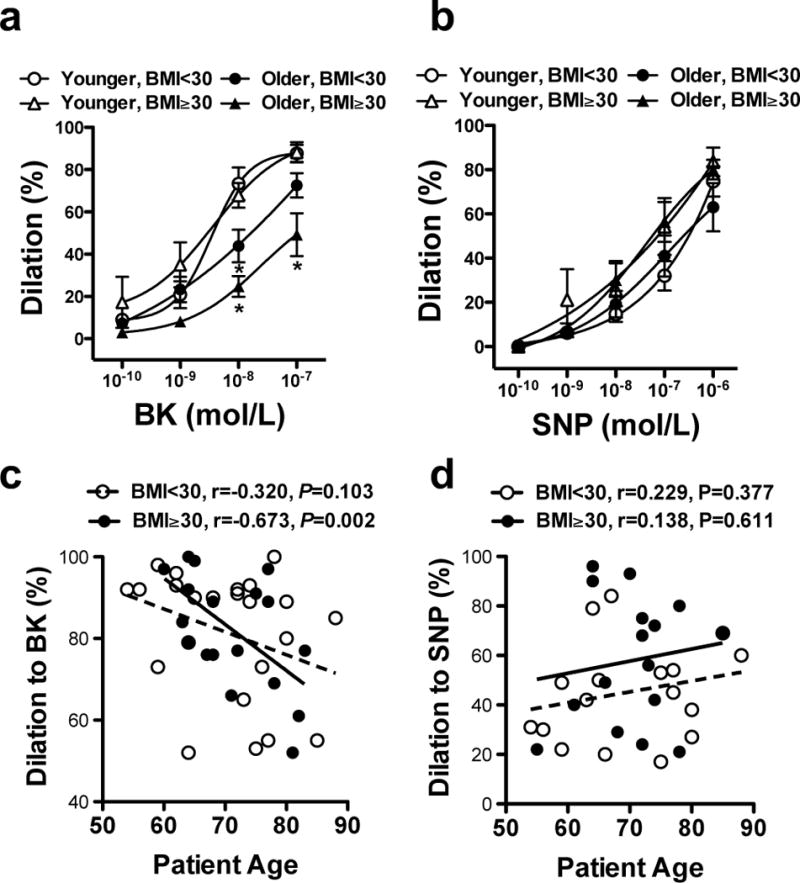
Changes in diameter of coronary arterioles isolated from non-obese (BMI<30) or obese (BMI≥30), younger (< 69 yrs, n = 26) or older (≥ 69, yrs., n = 23) patients (note that age group segregation was based on the mean age of this cohort) are shown in response to cumulative concentrations of bradykinin (BK, panel a) or sodium nitroprusside (SNP, panel b). Pearson correlations between the magnitude of BK- (to 10−7 M, panel c) and SNP-induced (to 10−6 M, panel d) dilations and age in obese patients and non-obese patients are shown. Regression lines are also shown in obese (solid line) and non-obese (dashed line) patient groups on panels c & d. Data are shown either as scatter dot plot or as means ± SEM. *P<0.05.
Reduced coronary artery dilation is associated with increased serum levels of pro-inflammatory cytokines in aged obese mice
We examined mechanisms that could underlie obesity and/or aging-induced development of coronary artery dilator dysfunction in a mouse model of HFD-induced obesity. We assessed coronary artery dilations in young (6-month) and aged (24-month) obese mice fed a 3-month HFD, similar as described previously.24 Young and aged mice on a normal chow diet served as controls. Both young and aged mice had a similar weight gain after 3-month of HFD (gain in young: 10.7±2.6 g vs. aged: 14.4±4.6 g, non significant), although the final body weights were significantly higher in the aged HFD fed mice (young: 29.4±2.6 g vs. young HFD 45.1±2.6 g and aged: 39.4±5.2 g vs. aged HFD 63.1±6.2 g, P<0.05). The active (spontaneously developed or pre-constricted by U46619, 10 nM, mean arteriolar tone: 38.8±9.9%) and passive (in the absence of extracellular Ca2+) baseline diameters of isolated coronary arterioles were not significantly different in the four groups (diameters in μm, Young, active: 97±6, passive: 133±4; Young-HFD, active: 83±7, passive: 128±15; Aged, active: 77±9, passive: 155±12; Aged-HFD, active: 79±6, passive: 140±16). We found that coronary artery dilation in response to the endothelium-dependent agonist, acetylcholine (ACh), but not to SNP, was significantly reduced in the aged, with a further decline in aged HFD mice, when compared to young mice with normal or with a HFD (Fig. 2a & 2b). These changes in the coronary artery dilator function were associated with significantly increased serum levels of cytokines, including TNF and IL-1β in the aged mice, and even a more prominent increase in TNF and IL-6 levels was observed in aged HFD mice, when compared to the young groups (Fig. 3a).
Figure 2. Reduced coronary artery dilation in aged obese mice is recapitulated in the young after transplantation of aged obese adipose tissue.
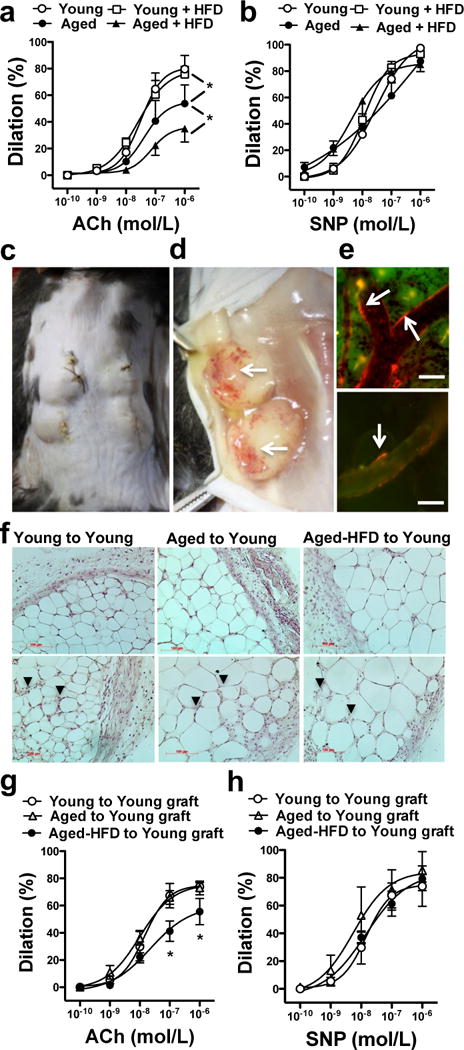
Changes in diameter of coronary arterioles isolated from Young or Aged mice fed a normal or HFD in response to cumulative concentrations of ACh (panel a) or SNP (panel b) are shown (n = 5−7, in each group). Panel c-e: Illustration of a 7-day adipose tissue allotransplantation in mice. Panel d shows vascularized adipose tissue grafts after 7-day of transplantation. Panel e depicts images of rhodamine-dextran fluorescence (injected through the tail vein of young recipient mice and shown in red, white arrows) in ear vessel (upper) and in the adipose tissue graft’s vessel lumens (lower). Bar: 20 μm. Representative images of hematoxylin stained adipose tissue allografts harvested from young recipients of Young to Young, Aged to Young or Aged-HFD to Young transplants (f panels, black arrowheads on lower panels indicate sites of infiltrated polymorphonuclear leukocytes). Panel g-h: Changes in diameter of coronary arterioles isolated from the young recipient mice after adipose tissue transplantation from young mice (Young to Young graft), from aged (Aged to Young graft) or from aged-HFD mice (Aged-HFD to Young graft) in response to ACh (panel g) or SNP (panel h, n=6 in each group). Data are means ± SEM. *P < 0.05.
Figure 3. Inflammatory mediators in the serum of young and aged mice on normal or HFD and also in adipose tissue transplanted mice.
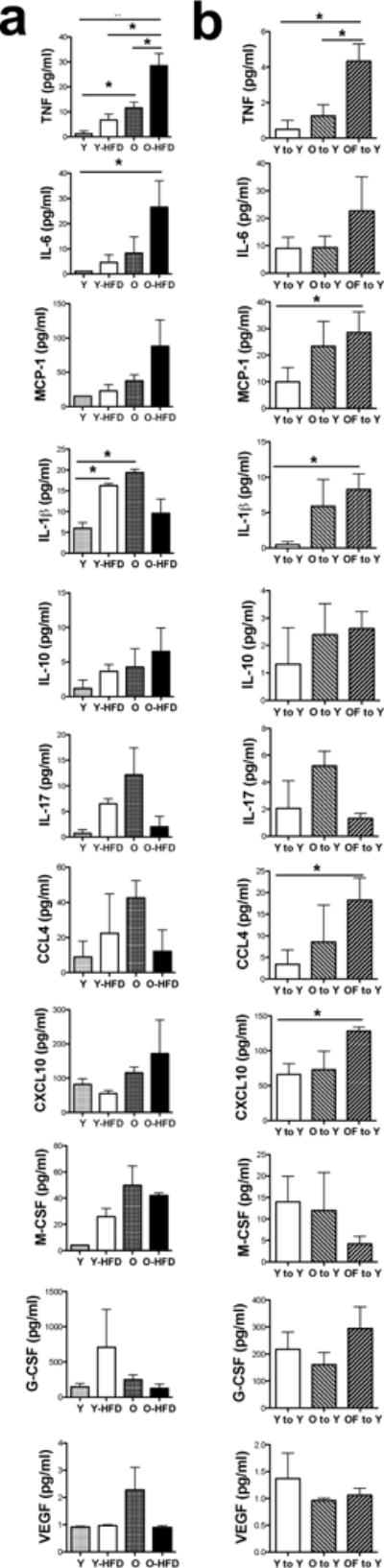
Serum levels of inflammatory cytokines and mediators were measured in young (Y) and aged (O) mice fed a normal or HFD (Y-HFD and O-HFD), as well as in young mice after 7-day adipose tissue transplantation either from young (Y to Y), aged to young (O to Y) or aged HFD mice (OF to Y) using a high sensitivity magnetic beads based multiplex ELISA (n=5–6 in each group). Data are shown as mean ± SEM. *P < 0.05
Adipose tissue transplantation from aged obese mice increases TNF levels and impairs coronary artery dilation in the young recipient
To test whether adipose tissue (AT) from the aged mice is a significant source of pro-inflammatory cytokines and whether it contributes to the development of coronary artery dilator dysfunction we performed AT transplantation between aged and aged-HFD fed mice to normal young recipient mice (Fig 2c & 2d), similarly as described previously.31 For controls we transplanted AT from normal young mice into normal young recipients. To assure engrafting and connection with the host circulation we injected rhodamine-dextran through the femoral vein of the recipient mice and detected the rhodamine dye in the vessel lumen within the transplanted fat pads (Fig. 2e). Allografts showed no sign of necrosis (Fig. 2f, upper panels), but were infiltrated by polymorphonuclear leukocytes (PMNLs, Fig. 2f lower panels) with a similar extent among the experimental groups as assessed by a semi-quantitative histology analysis (14±4 vs. 10±3 vs. 14±3 PMNLs/field of observation, in young-to-young vs. aged-to-young vs. aged HFD-to-young allografts, respectively). Importantly, we found that after a 7-day AT transplantation from aged HFD, but not from aged, to young mice coronary artery dilation to ACh became significantly reduced in the young recipients (Fig. 2g & 2h). Among the inflammatory cytokines and chemokines we had found elevated in the aged HFD mice, TNF and MCP-1 levels were significantly increased in the serum of young recipients (Fig. 3b). We also observed elevated IL-1β, CCL4 and CXCL10 serum levels after AT transplantation, but these cytokine levels were not increased in the aged HFD mice, and some of these changes may be attributed to transplantation procedures (Fig. 3b).
Obesity and aging cooperatively activates ADAM17 in mouse and human adipose tissue
Given the consistently and prominently elevated serum TNF levels in aged HFD fed mice, as well as in young mice receiving AT transplants from the aged HFD mice, we next examined obesity and age-related changes in the expression and activity of the TNF converting enzyme, ADAM17 in AT. Immunohistology analysis revealed that ADAM17 is abundantly expressed in adipocytes and even more prominently in blood vessels in the aged AT (Fig. 4a). We found that ADAM17 protein expression (pro and matured forms, Fig. 4b–d) was significantly increased in the AT of young-HFD and aged-HFD mice, when compared to controls. On the other hand, ADAM17 enzyme activity was found to be significantly increased only in the aged and aged-HFD mice (Fig. 4e) Two-way ANOVA revealed that the increased ADAM17 protein expression was related to HFD, but not to aging (HFD: P=0.021, aging: P=0.496, interaction: P=0.159), whereas increased ADAM17 enzyme activity was related to both HFD and aging (HFD: P=0.028, aging: P=0.002, interaction: P=0.919).
Figure 4. ADAM17 expression and activity is elevated in adipose tissue of aged HFD mice.
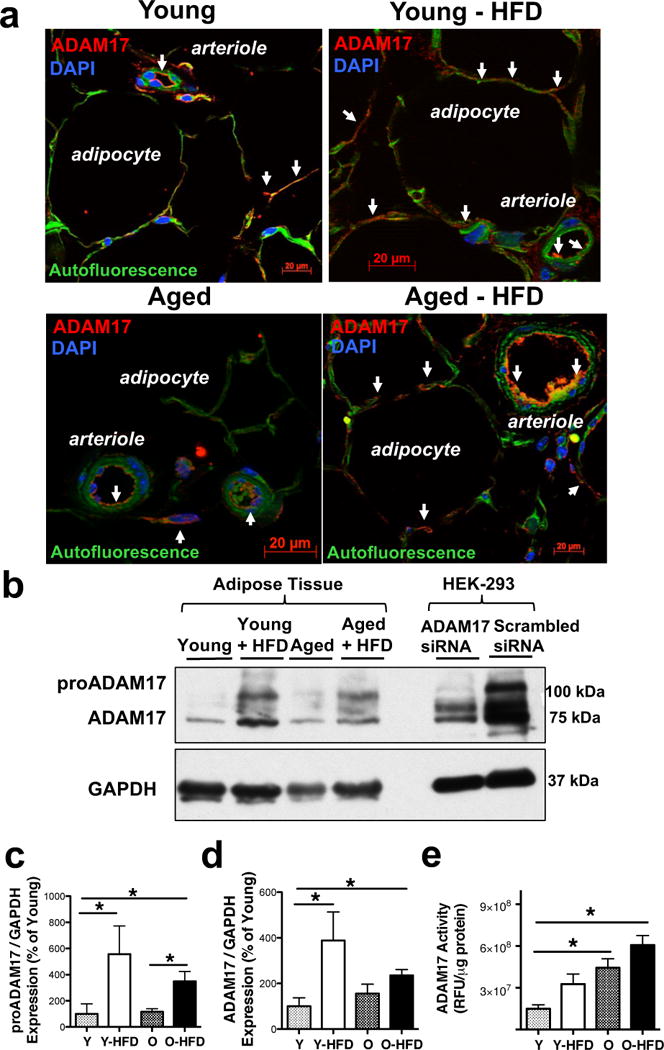
Representative images of ADAM17 immunofluorescence staining (shown in red, white arrows, panels a) of adipose tissue in Young and Aged mice with normal or HFD are shown. DAPI: nuclei staining in blue, autofluorescence: green. Representative images of Western immunoblot (panel b) and summary data (panel c & d, n=4 in each group) show proADAM17, mature ADAM17 and GAPDH (used for loading control) expression in adipose tissue in Young and Aged mice with normal or HFD. The specificity of ADAM17 antibody is shown in HEK-293 cells by using siRNA knockdown of ADAM17. Summary data of ADAM17 enzyme activity (normalized to protein concentration) in adipose tissue homogenates in Young and Aged mice with or without HFD (panel e, n=4 in each group). Data are shown as mean ± SEM. *P < 0.05.
We then examined age-related changes in ADAM17 expression and activity in AT in obese patients. We found no changes in adipocyte size in AT between younger and older obese patients (Fig. 5a–c) with a similar BMI (BMI in younger: 32±3 vs. older: 31±2, not significantly different), indicating a similar level of increased adiposity in this subset of patients. Similar to mice we observed a prominent microvascular endothelial expression of ADAM17 in AT (Fig. 5d & 5e and Supplementary Fig. II), which was also confirmed with immunoelectron microscopy (Fig. 5f–h). Importantly, we found a significant, age-related increase in ADAM17 enzyme activity in freshly dissected AT blood vessels, whereas only a positive trend was observed in isolated adipocytes (Fig. 5i & 5j).
Figure 5. Vascular expression and activity of ADAM17 in human adipose tissue.
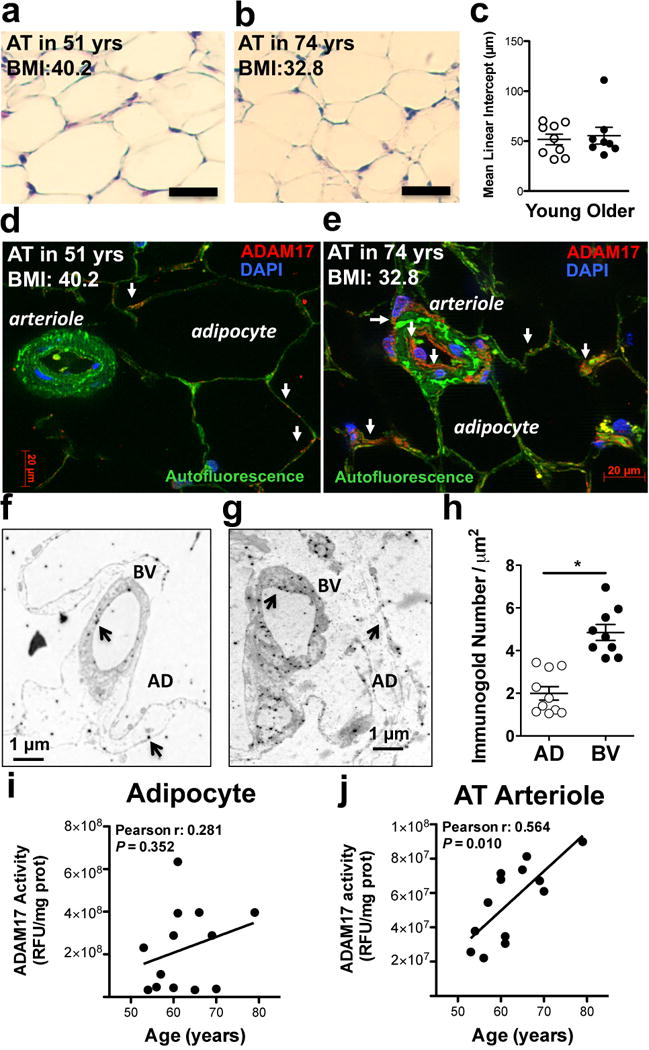
Representative images of hematoxylin staining (panels a & b, bar=50 μm) with mean linear intercept analysis (panel c), ADAM17 immunofluorescence (shown in red, white arrows, DAPI: nuclei staining in blue, autofluorescence: green, panels d & e) and ADAM17 immuno-electronmicroscopy (panels f & g) with immunogold (black arrows) distribution analysis (random areas from the total of 106.6 μm2 blood vessels and 132.5 μm2 adipocytes) in adipocytes (AD) versus blood vessels (BV) (panel h), in adipose tissue (AT) surgical samples in younger (< 69 yrs.) and older (≥ 69, yrs.) obese patients are shown (BMI in younger: 32±3 vs. older: 31±2, not significantly different). Individual measurements of ADAM17 enzyme activity (normalized to protein concentration, measured in triplicates in each patient) in adipocytes and isolated AT arteries are shown as a function of patients’ age (n=13, Pearson correlations with linear regression lines).
Bioactive, soluble TNF is shed by ADAM17 in arteries of adipose tissue in older obese patients
Next, we examined whether vascular endothelium-expressed ADAM17 itself is able to generate the soluble form of biologically active TNF. First, we directly measured soluble TNF in superfusates from freshly dissected AT arteries and found that the level of TNF was significantly elevated in AT artery superfusates in older obese patients (Fig. 6a). To test if TNF is bioactive, we performed a bioassay experiment in which AT artery was serially connected to a coronary arteriole, which served as a biosensor (Fig. 6b). This set-up permitted us to test the impact of soluble mediators produced by the upstream AT artery on endothelium-dependent dilation in the distal coronary arteriole. When the coronary arteriole was serially connected to the AT artery from an older obese patient, bradykinin-induced dilation in the coronary arteriole was significantly reduced, compared to AT arteries from younger obese individuals (Fig. 6c). The impaired coronary arteriole dilation imposed by ‘aged-obese’ AT vessels was restored to level of young AT vessels by the ADAM17 inhibitor, TIMP3, PEGylated soluble TNF receptor or by a TNF blocking antibody (Fig. 6d–f). Selective removal of the endothelium from AT arteries also prevented the impaired distal coronary arteriole dilation imposed by AT vessels from older obese subjects (Supplementary Fig. IV). Moreover, when isolated coronary arterioles were exposed directly to TNF, dilation to bradykinin, but not to SNP, was significantly reduced (Fig. 6g & 6h). These data provide evidence that in AT, membrane-bound TNF in the vascular endothelium is efficiently cleaved by ADAM17 into a bioactive, soluble form, which is then able to impair coronary arteriole dilator function in older obese patients.
Figure 6. Bioactive, soluble TNF is shed by ADAM17 in arteries of adipose tissue in older obese patients.
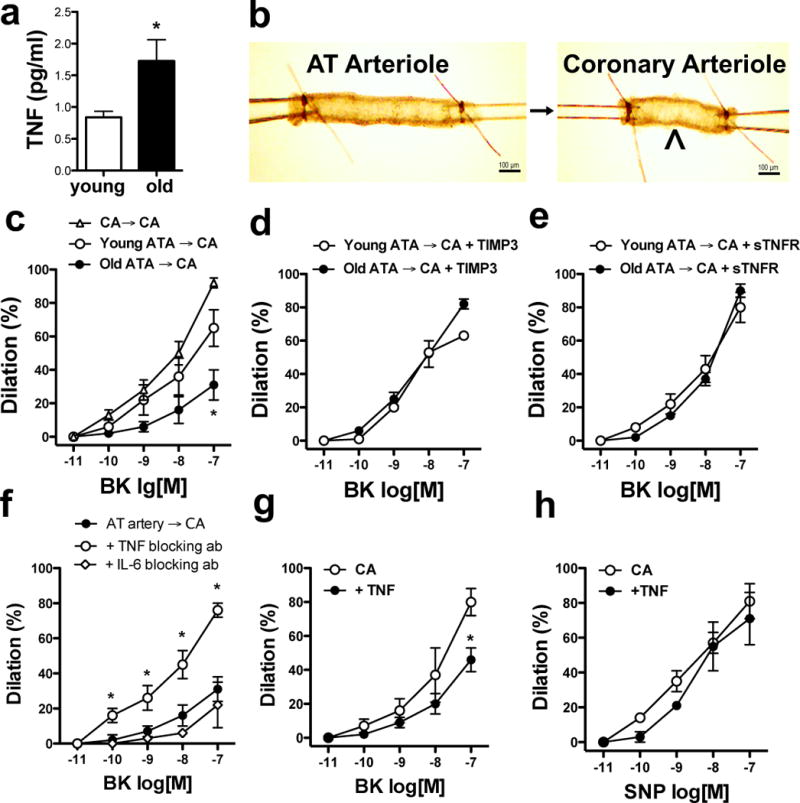
Summary data of TNF concentrations in adipose tissue arteriole superfusates in younger (< 69 yrs., n=4) and older (≥ 69, yrs., n=4) obese patients are shown, as measured by high sensitivity multiplex ELISA (panel a). Images depict the design of a bioassay experiment, in which adipose tissue (AT) arteries (diameter: 100–250 μm) were directly connected to a biosensor artery (coronary arteriole, CA, diameter: ~ 100 μm) with a small glass capillary (panel b). Connecting two CA with a glass capillary served as control. Diameter changes in the CA (biosensor) were measured in response to cumulative concentrations of endothelium-dependent agonist, bradykinin (BK, 10−11 – 10−7 M) in younger (< 69 yrs., n=6) and older (≥ 69, yrs., n=5) obese patients. In panels d-f, the CA (biosensor) was superfused by the AT artery and responses to BK were determined following selective treatment of the AT artery as indicated in the figure legends. BK and SNP-induced dilations in CA were also obtained in the absence or presence of exposure to exogenous TNF (Panel g & h). Data are shown as means ± SEM. *P<0.05.
Diminished interaction with caveolin-1 augments ADAM17 activity in endothelial cells
We next examined the mechanisms that underlie age-related increases in vascular ADAM17 activity. We measured protein expression of ADAM17 in AT arteries but found no significant differences between younger and older obese patents (Fig. 7a & 7b). Data from our mouse studies also suggest that the protein expression level of ADAM17 does not simply correlate with ADAM17 enzyme activity. Recent evidence indicates that, caveolin 1 (Cav1, the main structural protein of endothelial cell membrane caveolae) physically interacts with and regulates ADAM17 activity.27, 29 The possible interaction between ADAM17 and Cav1 was also confirmed in this study by dual immunogold labeling of ADAM17 and Cav1 (Fig. 7c). Interestingly, we found that the expression of Cav1 in AT arteries prominently declines with age (Fig. 7a & 7c) and this corresponds with a significantly lower degree of co-localization of ADAM17 and Cav1 in AT arteries in older obese patients (Fig. 7d & 7e).
Figure 7. Reduced interaction between caveolin-1 and ADAM17 in adipose tissue vascular endothelium.
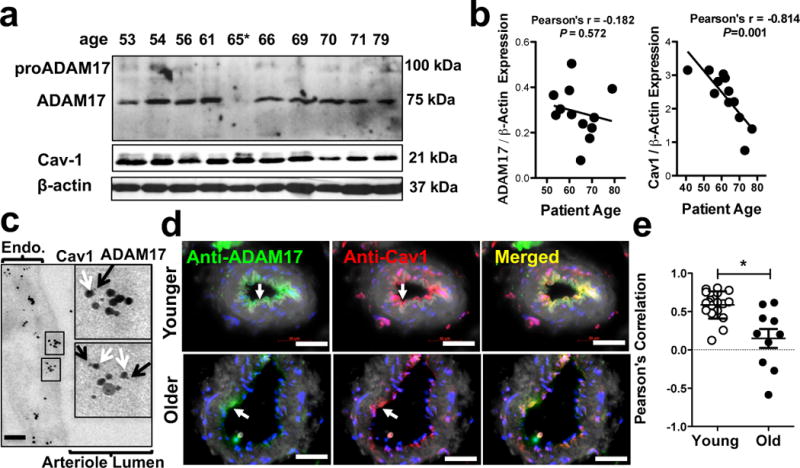
Representative Western immunoblots (panel a) and summary data (b panels) show ADAM17 and Cav1 expression (normalized to β-actin) in homogenates of adipose tissue arterioles obtained from individual obese patients in relation to their age (Pearson’s correlations, linear regression lines results from 13 patients are shown). Representative micrographs of double label immuno-electron microscopy (panel c) show endothelial distribution of anti-ADAM17 (5 nm, inset, black arrows) and anti-Cav1 (15 nm, inset white arrows) immunogold particles in adipose tissue vascular endothelium (representative images from 3 patients). Representative immunohistochemistry images for colocalization of ADAM17 and Cav1 in adipose tissue arteries in younger (< 69 yrs.) and older (≥ 69, yrs.) obese patients are shown (panel d). Green: ADAM17; Red: Cav-1; Blue, DAPI (nuclei); Merged: yellow indicates overlap of red and green signal; white, autofluorescence (at 488 nm). Scale bar, 50 μm. Summary data of Pearson correlation analysis for determining the degree of co-localization of ADAM17 and Cav-1. Multiple random regions (from 5 younger and 5 older obese patients) along the endothelial cells were selected manually, and a scatterplot of ADAM17 and Cav-1 fluorescent intensities (normalized to the background adjacent to the region of interest) was generated. ImageJ was used to determine pixel signal intensities for each region of interest (panel e). Data are shown either as scatter dot plot or as means ± SEM. *P<0.05.
In line with these human data we found a significantly reduced expression of Cav1 in AT in aged HFD mice, however we observed increased Cav1 expression in young HFD animals (Fig. 8a). Along with our finding showing that aged mice exhibit a significantly elevated ADAM17 activity we raised the possibility that Cav1 is a negative regulator of ADAM17 activity in AT. In support of this hypothesis we found that the serum level of TNF (Fig. 8c) and the activity of ADAM17 in AT (Fig. 8d) are both significantly elevated in mice with genetic deletion of Cav1, when compared to that of wild type mice. We found no significant changes in the protein expression of ADAM17 in AT (Fig. 8b) and in the aorta in the Cav1 knockout mice (Supplementary Fig. V).
Figure 8. Loss of caveolin-1 augments ADAM17 activity in vascular endothelial cells.
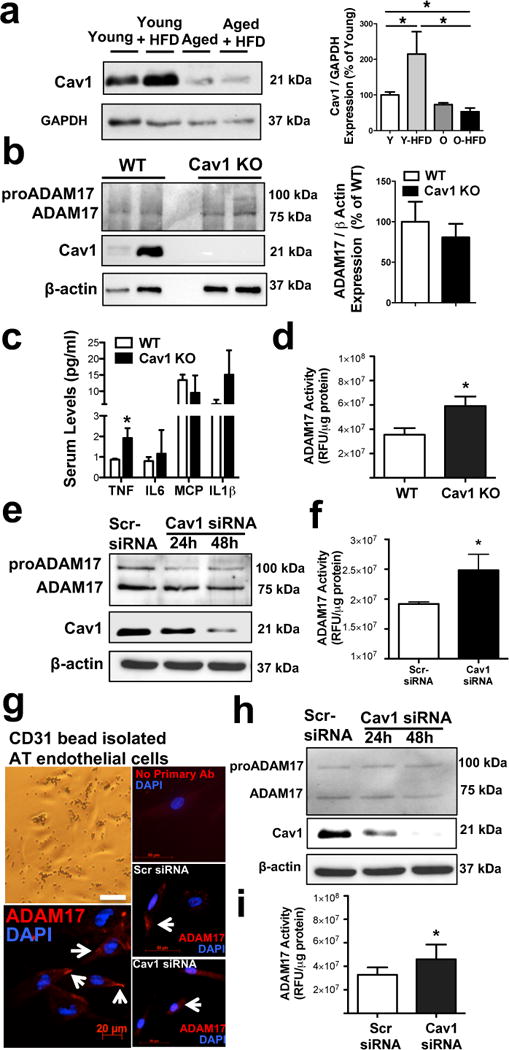
Representative Western immunoblots and summary data (N=4 in each group) show Cav1 expression (normalized to GAPDH) in homogenates of adipose tissue obtained from young and aged mice with normal or HFD (panel a). Representative Western immunoblots and summary data (N=4 in each group) show Cav1 and ADAM17 expression (normalized to β-actin) in adipose tissue obtained from wild-type (WT) or Cav1 knockout mice (panel b). Serum levels of TNF, IL-6, MCP-1 and IL1β as well as adipose tissue ADAM17 enzyme activities were measured in WT and Cav1 knockout mice (panel c & d, n=4 in each group). ADAM17 and Cav1 protein expression as well as ADAM17 enzyme activities were measured in cultured human coronary artery endothelial cells (Panel e & f) or CD31 micro bead isolated and cultured human adipose tissue endothelial cell (panel g, h & i) that were transfected with scrambled siRNA control (Scr-siRNA) or with Cav1-targeted siRNAs (Santa Cruz) for 24 hours or 48 hours (ADAM17 immunocytochemistry and ADAM17 enzyme activity was assessed in cells after 48 hours of transfection, N=3 in each group). Data are shown as means ± SEM. *P<0.05.
Moreover, we genetically silenced Cav1 and measured ADAM17 activity in cultured human coronary endothelial cells (HCAEC) and also in primary isolated and cultured AT-derived vascular endothelial cells. We have found that depletion of Cav1 resulted in a significantly increased ADAM17 enzyme activity without affecting ADAM17 protein expression, in both HCAEC and in AT-derived endothelial cells (Fig. 8e–i).
DISCUSSION
The present study demonstrates that coronary microvascular dysfunction (CMD) is more prevalent in older patients who are obese. We identify prominent expression and increased biological activity of ADAM17 in the vascular endothelium of adipose tissue as a significant source of the excess soluble TNF production observed in these patients and a primary mechanism underlying impaired coronary arteriole dilation. We also found that the increased activity of ADAM17 arises from diminished inhibitory interactions with caveolin-1, owing to an age-related decline in caveolin-1 expression. Collectively, our findings have important implications for pro-inflammatory changes in ‘aged-obese’ adipose tissue, which contributes to the development of CMD and related cardiac complications in older, obese patients.
Cardiovascular complications arising from CMD frequently develops in the absence of occlusive atherosclerotic lesions or may occur after the successful reopening the large occluded coronary arteries.1 As such, CMD has recently been linked to HFpEF, which develops most often in older patients with various comorbidities, including obesity, hypertension and type 2 diabetes but without compromised large coronary arteries.3 In HFpEF the underlying pathology seems to be driven by a chronic systemic inflammation state,5, 6 but the pathological mediators of chronic inflammation and the underlying mechanisms of CMD remain poorly characterized. Obesity is considered one of the most potent risk factors for many cardiovascular diseases, and is growing in prevalence in older adults at an alarming rate.8 Our earlier studies showed that in mice and rats with HFD-induced obesity32, 33 and also in obese patients34, 35 coronary arteriole dilator function is surprisingly preserved, in spite of significant adiposity and weight gain and elevated glucose and serum lipid levels. In the present study we also found that younger obese patients as well as younger HFD-fed obese mice have maintained endothelium-dependent coronary arteriole dilations. Such maintained or even enhanced vasodilator capacity of coronary microvessels may imply vascular adaptations to hemodynamic and/or metabolic changes in the heart early on in obesity, a debated phenomenon, which is often implicated in obesity paradox.36, 37 We23 and others24, 38 reported that aging of obese mice or rats, however, induces a prominent impairment in vasodilator function in mesenteric and skeletal muscle arteries. Similarly, in this study we found a significant age-related decline in human coronary arteriole dilator function in both obese men and women, but not in lean patients. These results were mirrored in mice, in which aging and obesity cooperatively induced impaired coronary artery dilations. Based on these observations we reasoned that adipose tissue-specific mediator(s) may change and contribute to the development of aging-related deleterious vascular effects. A previous study by Bailey-Downs et al. indicated that higher levels of circulating as well as adipose tissue-derived pro-inflammatory cytokines coincide with age-related development of vascular dysfunction in obese mice.24 In the present study we examined a direct, causative interrelationship between altered adipose tissue-derived inflammatory mediators and the development of CMD in aging and obesity. To this end we performed adipose tissue transplantation across aged, aged obese and young mice.31 Importantly, we noted that coronary arteriole vasodilatory capacity in young recipients became significantly impaired following transplantation of aged-obese, but not young or aged adipose tissue. We found that among the measured cytokines and inflammatory mediators TNF was markedly elevated in the young recipients of aged-obese fat transplants, suggesting an important and selective role for TNF in the development of coronary arteriole dilator dysfunction.
Adipose tissue, including the mediastinal fat pad, has been recognized as a significant source of the pro-inflammatory cytokine, TNF.12, 21, 22, 39 Adipocytes and especially adipose tissue-infiltrated macrophages have been implicated as being the source of excess TNF, which promotes obesity-associated systemic complications, such as type 2 diabetes.21, 40 While our study does not exclude the involvement of macrophages and adipocytes, we identify an important role of the vascular endothelium as a prominent source of soluble TNF in adipose tissue. It has been shown that vascular endothelial cells produce TNF in response to inflammatory stimulation by LPS or IL-1, in vitro.41–43 Overexpression of TNF in cultured human umbilical vein endothelial cells (HUVECs) results in the production of soluble TNF, which can be reduced by inhibition of TNF-converting enzyme, ADAM17.41 The pathophysiological relevance of TNF production in the vascular endothelium and the regulated release of soluble TNF by ADAM17 have not yet been determined. In the present study we have demonstrated for the first time that ADAM17 is functionally present in vascular endothelial cells in both rodent and human adipose tissue. We found that adipose tissue ADAM17 expression is induced by obesity, and observed that ADAM17 becomes selectively activated in the aged adipose tissue. We also found that the release of soluble TNF by endothelial ADAM17 in adipose tissue arteries from older obese patients exhibited potent biological activity and was able to impair endothelium-dependent dilator function in remote coronary arterioles. In view of the elevated systemic and adipose tissue TNF levels in older obese patients,21, 40, 44 it is likely that endothelial cells in adipose tissue contribute to systemic TNF levels. Unfortunately, serum samples were not available in our patient cohort and this hypothesis can only be supported by our data from aged-obese mice, including that of ‘aged-obese’ adipose tissue transplantation experiments. Whether increased activity of ADAM17 in adipose tissue endothelium correlates with elevated serum TNF levels in older obese patients has yet to be determined.
In this study we further examined the potential mechanisms by which vascular endothelial ADAM17 becomes activated in the aged-obese adipose tissue. It has been shown that ADAM17 is expressed in mouse adipose tissue (both in visceral and subcutaneous fat pads) and that mRNA expression level is increased in HFD-fed mice.26 In this study we found that both young and aged HFD mice had significantly increased protein expression of ADAM17 (young mice even higher), but only the aged mice exhibited a significantly increased ADAM17 enzyme activity, which was accompanied by elevated TNF levels in the serum of aged obese animals. Results from our human study showed that age-related increases in ADAM17 activity cannot be simply attributed to increased ADAM17 protein expression. Among other important regulatory mechanisms of ADAM17 activation, such as achieved by phosphorylation of the enzyme,25 recent studies have shown that ADAM17 is localized within cell membrane caveolae, where its activity is regulated through caveolae compartmentalization and a direct interaction with caveolin-1 (Cav1), the main structural protein of membrane caveolae.27–29 In the present study we confirmed that ADAM17 is located in a close proximity of Cav1 in endothelium cell membrane in the adipose tissue. Of note that Cav1 expression in adipose tissue arteries was significantly reduced with age in the obese patients, and this was associated with a significantly lower degree of co-localization between Cav1 and ADAM17. Consistent with these observations aging of mice was accompanied by a significantly reduced Cav1 levels in adipose tissue, which led to a significantly elevated ADAM17 enzyme activity. By contrast, young HFD mice had increased adipose tissue Cav1 and ADAM17 expression, which was however associated with only a modest increase in ADAM17 enzyme activity.
In line with our results earlier in vitro studies have shown that Cav1 negatively regulates the enzyme activity of ADAM17 (i.e. loss of Cav1 expression and diminished interaction increases ADAM17 activity) in cultured vascular smooth muscle cells27 and neuronal cells.29 In contrast, a study by D’Alessio et al found that ADAM17 co-immunoprecipitates with Cav1 and its shedding activity is reduced after disruption of caveolae with methyl-β-cyclodextrin or by Cav1 silencing in HUVECs (EA.hy926).28 Similarly, a study by Takayanagi et al showed that in Cav1 knockout mice angiotensin II-induced increase in ADAM17 expression and activity is diminished in the aortic vascular smooth muscle cells, which supports a positive regulatory role by Cav1 on ADAM17, in vivo.45 These studies indicated that Cav1 plays an important regulatory role by repressing or promoting ADAM17 enzyme activity, but also highlighted the complexity and ambiguity of mechanisms through which Cav1 regulate ADAM17, both under normal or pathological conditions, in vitro and in vivo.
Our recent studies have shown that mice lacking Cav1 exhibit impaired NO-mediated coronary arteriole dilation19 and that they are highly susceptible to the development of microvascular dysfunction and hypertension upon commencing a HFD.30 It is known that Cav1 plays an important role in the regulation and caveolae compartmentalization of eNOS,46 where eNOS activity is inhibited by Cav1.47–49 However, in Cav1 knockout mice lacking caveolae or under pathological conditions that accompanied by disrupted endothelial caveolae function/structure the activation of eNOS becomes compromised and this may explain diminished NO-mediated dilation in coronary arterioles.19 The nature of pathological mechanisms by which loss of Cav1/caveolae function, directly or indirectly initiates coronary arteriole dysfunction remains incompletely understood. Interestingly, it has been shown that Cav1 knockout mice exhibit elevated TNF levels in the lung (even at baseline), which is further enhanced in response to bacteria-induced inflammatory stimuli.50, 51 In the present study we found that serum level of TNF is significantly elevated in Cav1 knockout mice, and thus we have raised the possibility that increased ADAM17 activity may play a role in this process. In support of this results from the present study show that adipose tissue in Cav1 knockout mice exhibit an increased ADAM17 activity. In addition, we found that genetic silencing of Cav1 in human adipose tissue-derived and also coronary artery endothelial cells lead to increased activity of ADAM17. These findings are in line with previous studies indicating an inhibitory effect by Cav1 on several vascular endothelial signaling pathways.19, 30, 33, 52 Remarkably, recent studies revealed that impairments in Cav1-regulated fundamental homeostatic pathways are among the most important mediators of premature aging (reviewed in).53 In this process, a diminished Cav1-mediated internalization and thus sustained activation of various plasma membrane localized proteins/receptors promote cell senescence.53 As such, an age-related decline in Cav1 expression and/or caveolae dysfunction would be a prime example whereby aging enhances ADAM17 activition and promotes excess soluble TNF production, via a diminished Cav1-mediated ADAM17 internalization. Further studies are needed to examine the molecular base of interactions between ADAM17 and Cav1, Cav1-dependent ADAM17 internalization and its impact on ADAM17 enzyme activity and soluble TNF production in more details.
There are several limitations in our study. Our human study was conducted in consecutive patients with heart diseases who underwent open-heart surgery. This observational study was designed to determine the prevalence of CMD and also the impact of obesity and aging in this patient cohort. The majority (~70%) of these patients also had large coronary artery disease and almost all patients had multiple comorbidities, including hypertension and diabetes. These comorbid conditions are highly prevalent in heart failure patients. Approximately 18% of these patients had heart failure and 5 had HFpEF diagnosis. Unfortunately these limitations along with the limited number of HFpEF patients and the lack of follow up do not allow us to draw any firm conclusions regarding a mechanistic connection between CMD and the current or future incidence of HFpEF, especially in relation to increased ADAM17 activity and excess soluble TNF production by ‘aged-obese’ adipose tissue. In addition, various biochemical measurements, such as ADAM17 protein expression and activity as well as immunohistochemistry and immunoelectron microscopic assessment of ADAM17 tissue distribution were performed in a subset of consecutive patients, which patient cohort was overlapping with those with functional assessments of coronary arterioles, but may differ in the occurrence of comorbidities and medications. We did not have permission and did not collect blood from these patients who underwent heart surgery and therefore assessment of serum inflammatory mediators, such as TNF levels was not possible. Experiments in mouse models of aging, obesity and adipose tissue transplantation were designed to overcome these and other limitations of our human study, which largely yielded supportive, complementary results regarding the role of obesity- and aging-induced pro-inflammatory changes in adipose tissue vascular endothelium. In order to provide further insights into the molecular mechanism through which vascular endothelium expressed ADAM17 contributes to soluble TNF production by the ‘aged and obese’ adipose tissue employing transgenic mouse models may be considered in future studies. As such, endothelium selective ADAM17 knockout mice were recently developed.54 These transgenic mice show impaired collateral blood vessel growth, which indicates a pivotal role of endothelial ADAM17 in angiogenesis. It is known that adequate adipose tissue vascularization is critical for the maintenance of metabolism and function of the expanded adipose tissue, in which TNF-mediated inflammatory mechanisms, likely through ADAM17 activation, play an important role.55, 56 Therefore, a rather complex autocrine/paracrine (within the adipose tissue) and remote cardiovascular and other systemic effects of vascular endothelial ADAM17 activation and soluble TNF production can be envisioned in obesity and aging, which has yet to be investigated in future studies.
In conclusion, this study is the first to identify vascular endothelium-expressed ADAM17 in rodent and human adipose tissue as a significant source of bioactive, soluble TNF in obesity and aging. Our results indicate an important role for vascular endothelial ADAM17 in contributing to the excess production of TNF in older obese patients. Aging-related reduction in Cav1 expression and subsequently increased activity of endothelial ADAM17 may lead to excess TNF production. We propose that the augmented ADAM17/TNF signaling pathway in the vascular endothelium of adipose tissue acts remotely to promote coronary arteriole dysfunction and thereby contribute to the development of CMD and its related cardiac complications in older obese patients.
Supplementary Material
Table 1.
Patient demographics, diseases, and medications
| n | 74 |
| Female/Male | 30/44 |
| Age (years) | 69±11 |
| Body weight (Kg) | 87±18 |
| BMI (Kg/m2) | 30.2±5.6 |
| SBP (mmHg) | 133±24 |
| DBP (mmHg) | 73±13 |
| Serum glucose (mg/dL) | 128±49 |
| Underlying disease, n | |
| Coronary artery disease | 54 |
| Peripheral vascular disease | 6 |
| HFrEF (EF=30±8) | 9 |
| HFpEF (EF=58±5) | 6 |
| Prior AMI | 25 |
| Hypertension | 68 |
| Type 1 diabetes | 2 |
| Type 2 diabetes | 25 |
| COPD | 11 |
| Stroke | 9 |
| Hyperlipidemia | 50 |
| Medications, n | |
| Aspirin | 44 |
| Lipid lowering | 49 |
| Insulin | 8 |
| Oral antidiabetic drug | 17 |
| Beta blocker | 44 |
| ACEI/ARB | 40 |
| Diuretic | 33 |
| Anticoagulant | 25 |
| Calcium channel blocker | 24 |
|
| |
| Surgical procedure, % | |
| CABG | 54 |
| Valve Replacement | 21 |
n, number of patients studied; for continues variables mean ± SD are shown.
BMI- body mass index, SBP- systolic blood pressure, DBP- diastolic blood pressure, HFrEF- heart failure with reduced ejection fraction, HFpEF- heart failure with preserved ejection fraction, EF- ejection fraction, AMI- previous acute myocardial infarction, COPD, chronic obstructive pulmonary disease, ACEI/ARB- angiotensin converting enzyme inhibitors/angiotensin receptor blockers, CABG- coronary artery bypass graft.
HIGHLIGHTS.
The present study demonstrates that coronary microvascular dysfunction is more prevalent in older patients who are obese.
We identify vascular endothelium-expressed ADAM17 in rodent and human obese adipose tissue as a significant source of bioactive, soluble TNF in aging.
Our results indicate an important role for vascular endothelial ADAM17 in contributing to the excess production of TNF in older obese subjects.
Aging-related reduction in Cav1 expression and subsequently increased activity of endothelial ADAM17 leads to excess TNF production in obesity.
The augmented ADAM17/TNF signaling pathway in the vascular endothelium of adipose tissue acts remotely to promote coronary arteriole dysfunction in older obese patients.
Acknowledgments
The authors thank Penny Roon, and Perry Libby (Electron Microscopy and Histology Core Laboratory) for their assistance in immunoelectron microscopy study.
Source of Funding. This study was supported by Grants HL104126 (ZB), HL126949, HL112640, AR070029, and HL134354 (NW) from the National Institutes of Health, 16PRE27550006 (H.D.) from the American Heart Association, and by the Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313) to MBG.
Abbreviations
- ADAM17
A disintegrin and metalloproteinase 17
- TNF
tumor necrosis factor-α
- AT
adipose tissue
- CMD
coronary microvascular disease
- CA
coronary arterioles
- HFD
high fat diet
- Cav1
caveolin-1
- HFpEF
heart failure with preserved ejection fraction
- ROS
reactive oxygen species
- ACh
acetylcholine
- SNP
sodium nitroprusside
Footnotes
Disclosures. No potential conflicts of interest relevant to this article are reported.
References
- 1.Nemes A, Forster T, Csanady M. Diminished coronary flow velocity reserve and aortic distensibility in elderly patients with chest pain and negative coronary angiograms. Aging clinical and experimental research. 2008;20:297–301. doi: 10.1007/BF03324859. [DOI] [PubMed] [Google Scholar]
- 2.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 3.Udelson JE. Heart failure with preserved ejection fraction. Circulation. 2011;124:e540–543. doi: 10.1161/CIRCULATIONAHA.111.071696. [DOI] [PubMed] [Google Scholar]
- 4.Silverman MG, Patel B, Blankstein R, Lima JA, Blumenthal RS, Nasir K, Blaha MJ. Impact of race, ethnicity, and multimodality biomarkers on the incidence of new-onset heart failure with preserved ejection fraction (from the multi-ethnic study of atherosclerosis) Am J Cardiol. 2016;117:1474–1481. doi: 10.1016/j.amjcard.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 6.Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016;4:312–324. doi: 10.1016/j.jchf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D’Agostino RB, Framingham Heart S Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: The framingham heart study. Circulation. 2003;107:1486–1491. doi: 10.1161/01.cir.0000057810.48709.f6. [DOI] [PubMed] [Google Scholar]
- 8.Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: A report from the american heart association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol. 2015;208:501–512. doi: 10.1083/jcb.201409063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen MK, Noblet JN, Sassoon DJ, Conteh AM, Goodwill AG, Tune JD. Perivascular adipose tissue and coronary vascular disease. Arterioscler Thromb Vasc Biol. 2014;34:1643–1649. doi: 10.1161/ATVBAHA.114.303033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Payne GA, Borbouse L, Kumar S, Neeb Z, Alloosh M, Sturek M, Tune JD. Epicardial perivascular adipose-derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase c-beta pathway. Arterioscler Thromb Vasc Biol. 2010;30:1711–1717. doi: 10.1161/ATVBAHA.110.210070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, Sarov-Blat L, O’Brien S, Keiper EA, Johnson AG, Martin J, Goldstein BJ, Shi Y. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003;108:2460–2466. doi: 10.1161/01.CIR.0000099542.57313.C5. [DOI] [PubMed] [Google Scholar]
- 13.Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhoj P, Pedersen BK. A high plasma concentration of tnf-alpha is associated with dementia in centenarians. The journals of gerontology. Series A, Biological sciences and medical sciences. 1999;54:M357–364. doi: 10.1093/gerona/54.7.m357. [DOI] [PubMed] [Google Scholar]
- 14.Paolisso G, Rizzo MR, Mazziotti G, Tagliamonte MR, Gambardella A, Rotondi M, Carella C, Giugliano D, Varricchio M, D’Onofrio F. Advancing age and insulin resistance: Role of plasma tumor necrosis factor-alpha. Am J Physiol. 1998;275:E294–299. doi: 10.1152/ajpendo.1998.275.2.E294. [DOI] [PubMed] [Google Scholar]
- 15.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 16.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 17.Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in lepr(db) mice. Circulation. 2007;115:245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- 18.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 19.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, Belin de Chantemele E, Feher A, Romero MJ, Bagi Z. Peroxynitrite disrupts endothelial caveolae leading to enos uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes. 2014;63:1381–1393. doi: 10.2337/db13-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ridker PM, Rifai N, Pfeffer M, Sacks F, Lepage S, Braunwald E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation. 2000;101:2149–2153. doi: 10.1161/01.cir.101.18.2149. [DOI] [PubMed] [Google Scholar]
- 21.Gormez S, Demirkan A, Atalar F, Caynak B, Erdim R, Sozer V, Gunay D, Akpinar B, Ozbek U, Buyukdevrim AS. Adipose tissue gene expression of adiponectin, tumor necrosis factor-alpha and leptin in metabolic syndrome patients with coronary artery disease. Intern Med. 50:805–810. doi: 10.2169/internalmedicine.50.4753. [DOI] [PubMed] [Google Scholar]
- 22.Konishi M, Sugiyama S, Sugamura K, et al. Association of pericardial fat accumulation rather than abdominal obesity with coronary atherosclerotic plaque formation in patients with suspected coronary artery disease. Atherosclerosis. 2010;209:573–578. doi: 10.1016/j.atherosclerosis.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Huang A, Yang YM, Feher A, Bagi Z, Kaley G, Sun D. Exacerbation of endothelial dysfunction during the progression of diabetes: Role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2012;302:R674–681. doi: 10.1152/ajpregu.00699.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey-Downs LC, Tucsek Z, Toth P, Sosnowska D, Gautam T, Sonntag WE, Csiszar A, Ungvari Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: A paracrine mechanism contributing to vascular redox dysregulation and inflammation. J Gerontol A Biol Sci Med Sci. 2013;68:780–792. doi: 10.1093/gerona/gls238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gooz M. Adam-17: The enzyme that does it all. Critical reviews in biochemistry and molecular biology. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voros G, Maquoi E, Collen D, Lijnen HR. Differential expression of plasminogen activator inhibitor-1, tumor necrosis factor-alpha, tnf-alpha converting enzyme and adamts family members in murine fat territories. Biochim Biophys Acta. 2003;1625:36–42. doi: 10.1016/s0167-4781(02)00589-4. [DOI] [PubMed] [Google Scholar]
- 27.Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, Yang B, Rizzo V, Eguchi S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin ii. Journal of molecular and cellular cardiology. 2011;50:545–551. doi: 10.1016/j.yjmcc.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Alessio A, Esposito B, Giampietri C, Ziparo E, Pober JS, Filippini A. Plasma membrane microdomains regulate tace-dependent tnfr1 shedding in human endothelial cells. J Cell Mol Med. 2012;16:627–636. doi: 10.1111/j.1582-4934.2011.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pietri M, Dakowski C, Hannaoui S, Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A, Mouillet-Richard S, Haik S, Bailly Y, Peyrin JM, Launay JM, Kellermann O, Schneider B. Pdk1 decreases tace-mediated alpha-secretase activity and promotes disease progression in prion and alzheimer’s diseases. Nature medicine. 2013;19:1124–1131. doi: 10.1038/nm.3302. [DOI] [PubMed] [Google Scholar]
- 30.Czikora I, Feher A, Lucas R, Fulton DJ, Bagi Z. Caveolin-1 prevents sustained angiotensin ii-induced resistance artery constriction and obesity-induced high blood pressure. Am J Physiol Heart Circ Physiol. 2015;308:H376–385. doi: 10.1152/ajpheart.00649.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, Vinson C, Eckhaus M, Reitman ML. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–278. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jebelovszki E, Kiraly C, Erdei N, Feher A, Pasztor ET, Rutkai I, Forster T, Edes I, Koller A, Bagi Z. High-fat diet-induced obesity leads to increased no sensitivity of rat coronary arterioles: Role of soluble guanylate cyclase activation. Am J Physiol Heart Circ Physiol. 2008;294:H2558–2564. doi: 10.1152/ajpheart.01198.2007. [DOI] [PubMed] [Google Scholar]
- 33.Feher A, Rutkai I, Beleznai T, Ungvari Z, Csiszar A, Edes I, Bagi Z. Caveolin-1 limits the contribution of bk(ca) channel to edhf-mediated arteriolar dilation: Implications in diet-induced obesity. Cardiovasc Res. 2010;87:732–739. doi: 10.1093/cvr/cvq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fulop T, Jebelovszki E, Erdei N, Szerafin T, Forster T, Edes I, Koller A, Bagi Z. Adaptation of vasomotor function of human coronary arterioles to the simultaneous presence of obesity and hypertension. Arterioscler Thromb Vasc Biol. 2007;27:2348–2354. doi: 10.1161/ATVBAHA.107.147991. [DOI] [PubMed] [Google Scholar]
- 35.Cassuto J, Feher A, Lan L, Patel VS, Kamath V, Anthony DC, Bagi Z. Obesity and statins are both independent predictors of enhanced coronary arteriolar dilation in patients undergoing heart surgery. Journal of cardiothoracic surgery. 2013;8:117. doi: 10.1186/1749-8090-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagi Z. Mechanisms of coronary microvascular adaptation to obesity. Am J Physiol Regul Integr Comp Physiol. 2009;297:R556–567. doi: 10.1152/ajpregu.90817.2008. [DOI] [PubMed] [Google Scholar]
- 37.Bagi Z, Feher A, Cassuto J. Microvascular responsiveness in obesity: Implications for therapeutic intervention. Br J Pharmacol. 2012;165:544–560. doi: 10.1111/j.1476-5381.2011.01606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in zucker obese and zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2006;291:H1780–1787. doi: 10.1152/ajpheart.01297.2005. [DOI] [PubMed] [Google Scholar]
- 39.Payne GA, Kohr MC, Tune JD. Epicardial perivascular adipose tissue as a therapeutic target in obesity-related coronary artery disease. Br J Pharmacol. 2012;165:659–669. doi: 10.1111/j.1476-5381.2011.01370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imaizumi T, Itaya H, Fujita K, Kudoh D, Kudoh S, Mori K, Fujimoto K, Matsumiya T, Yoshida H, Satoh K. Expression of tumor necrosis factor-alpha in cultured human endothelial cells stimulated with lipopolysaccharide or interleukin-1alpha. Arterioscler Thromb Vasc Biol. 2000;20:410–415. doi: 10.1161/01.atv.20.2.410. [DOI] [PubMed] [Google Scholar]
- 42.Chan EL, Haudek SB, Giroir BP, Murphy JT. Human coronary endothelial cell activation by endotoxin is characterized by nf-kappa b activation and tnf-alpha synthesis. Shock. 2001;16:349–354. doi: 10.1097/00024382-200116050-00005. [DOI] [PubMed] [Google Scholar]
- 43.Freyer D, Manz R, Ziegenhorn A, Weih M, Angstwurm K, Docke WD, Meisel A, Schumann RR, Schonfelder G, Dirnagl U, Weber JR. Cerebral endothelial cells release tnf-alpha after stimulation with cell walls of streptococcus pneumoniae and regulate inducible nitric oxide synthase and icam-1 expression via autocrine loops. J Immunol. 1999;163:4308–4314. [PubMed] [Google Scholar]
- 44.Winkler G, Kiss S, Keszthelyi L, et al. Expression of tumor necrosis factor (tnf)-alpha protein in the subcutaneous and visceral adipose tissue in correlation with adipocyte cell volume, serum tnf-alpha, soluble serum tnf-receptor-2 concentrations and c-peptide level. Eur J Endocrinol. 2003;149:129–135. doi: 10.1530/eje.0.1490129. [DOI] [PubMed] [Google Scholar]
- 45.Takayanagi T, Crawford KJ, Kobayashi T, Obama T, Tsuji T, Elliott KJ, Hashimoto T, Rizzo V, Eguchi S. Caveolin 1 is critical for abdominal aortic aneurysm formation induced by angiotensin ii and inhibition of lysyl oxidase. Clin Sci (Lond) 2014;126:785–794. doi: 10.1042/CS20130660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (nos) and caveolin. Functional significance of the nos caveolin binding domain in vivo. The Journal of biological chemistry. 1997;272:25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- 48.Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by ca2+-calmodulin and caveolin. The Journal of biological chemistry. 1997;272:15583–15586. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- 49.Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. The Journal of biological chemistry. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- 50.Guo Q, Shen N, Yuan K, Li J, Wu H, Zeng Y, Fox J, 3rd, Bansal AK, Singh BB, Gao H, Wu M. Caveolin-1 plays a critical role in host immunity against klebsiella pneumoniae by regulating stat5 and akt activity. Eur J Immunol. 2012;42:1500–1511. doi: 10.1002/eji.201142051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M. Elevated inflammatory response in caveolin-1-deficient mice with pseudomonas aeruginosa infection is mediated by stat3 protein and nuclear factor kappab (nf-kappab) J Biol Chem. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 53.Zou H, Stoppani E, Volonte D, Galbiati F. Caveolin-1, cellular senescence and age-related diseases. Mech Ageing Dev. 2011;132:533–542. doi: 10.1016/j.mad.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lucitti JL, Mackey JK, Morrison JC, Haigh JJ, Adams RH, Faber JE. Formation of the collateral circulation is regulated by vascular endothelial growth factor-a and a disintegrin and metalloprotease family members 10 and 17. Circ Res. 2012;111:1539–1550. doi: 10.1161/CIRCRESAHA.112.279109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asterholm IW, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rutkowski JM, Davis KE, Scherer PE. Mechanisms of obesity and related pathologies: The macro- and microcirculation of adipose tissue. FEBS J. 2009;276:5738–5746. doi: 10.1111/j.1742-4658.2009.07303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.