

Abstract
TAK1 (transforming growth factor-β-activated kinase 1) is an essential intracellular mediator of cytokine and growth factor signaling and a potential therapeutic target for the treatment of immune diseases and cancer. Herein we report development of a series of 2,4-disubstituted pyrimidine covalent TAK1 inhibitors that target Cys174 , a residue immediately adjacent to the ‘DFG-motif’ of the kinase activation loop. Co-crystal structures of TAK1 with candidate compounds enabled iterative rounds of structure-based design and biological testing to arrive at optimized compounds. Lead compounds such as 2 and 10 showed greater than 10-fold biochemical selectivity for TAK1 over the closely related kinases MEK1 and ERK1 which possess an equivalently positioned cysteine residue. These compounds are smaller, more easily synthesized, and exhibit a different spectrum of kinase selectivity relative to previously reported macrocyclic natural product TAK1 inhibitors such as 5Z-7-oxozeanol.
Graphical Abstract
1. Introduction
TAK1 (transforming growth factor-β-activated kinase 1) is a serine/threonine kinase belonging to the MAPK kinase kinase (MAP3K) family initially identified because of its responsiveness to TGF-β and bone morphogenetic protein (BMP) in preosteoblast cells [1]. Knockout of TAK1 in mice is embryonically lethal, causing severe neural tube deformities early in gestation [2, 3]. TAK1 mediates responsiveness to environmental stress to control transcription and apoptosis. TAK1 activity also appears to be involved in multiple inflammatory conditions and cancers motivating interest in the development of TAK1 inhibitors for therapeutic purposes.
TAK1 mediates activation of immune processes stimulated by pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα), toll-like receptor (TLR) ligands and interleukin-1 (IL-1) [4–7]. In B cells, conditional TAK1 knockout shows that TAK1 is essential for mitogenic responses to receptor-mediated stimuli including TLR, anti-CD40 and anti-IgM antibodies [8]. In T cells, conditional TAK1 knockout reduces the development of Treg cells expressing Foxp3 [9–11]. In macrophages, TAK1 has been shown to function in innate immune responses, whereby pattern recognition receptors utilize TAK1 to activate NFκB through MyD88 [12]. TAK1 is associated with inflammatory disorders such as kidney fibrosis [13] and Crohn’s disease [14] and depletion of TAK1 decreases levels of inflammatory infiltrates and damps cytokine responses. TAK1 has also been studied in ischemic stroke models, where short-term inhibition of TAK1 blocked activation of p38 and JNK following oxygen and glucose deprivation [15].
Additionally, TAK1 is associated with multiple cancers including lymphoma [16], ovarian cancer [17], colon cancer [18], neuroblastoma [19] and pancreatic cancer [20], possibly related to modulation of inflammation in the cellular microenvironment [21]. Work by Singh and colleagues has shown that TAK1 is required for survival of some KRAS-dependent colon cancer cell lines and demonstrated that TAK1 inhibition induces apoptosis via modulation of WNT signaling [18]. Recent work by Ansell and colleagues revealed that TAK1 is an essential mediator of activated MyD88 signaling, a protein commonly mutated and constitutively active in a subtype of non-Hodgkin lymphomas called Waldenstrom’s Macroglobulinemia (WM) [22]. In addition, TAK1 activity has been associated with tumor aggressiveness in ovarian cancer [17].
A number of small molecule inhibitors of TAK1 kinase activity have been reported. 5Z-7- oxozeaenol (5Z7), a natural resorcylic lactone isolated from fungi, was identified as a TAK1 inhibitor in a screen searching for inhibitors of TAK1 catalytic activity. Subsequent studies showed 5Z7 prevents IL-1 induced activation of TAK1, JNK, MAPK and NFκB in cell culture by irreversible covalent binding to Cys174, located in the ATP-binding pocket of TAK1 [23]. Anti-TAK1 activity by 5Z7 has been demonstrated in multiple experimental systems [24, 25]. However, resorcylic acids lactones are known to inhibit multiple kinases [26], and broad-based kinase profiling has demonstrated that 5Z7 is a potent inhibitor of MEK1/2, FLT3, KIT, PDGFR, TGFRB and other kinases [27]. Improving the selectivity of 5Z7 and related molecules through chemical modification is synthetically challenging, although reversible resorcylic acid lactones were recently reported to have improved selectivity and pharmacokinetic properties [28]. AZ-TAK1 is a thiophenecarboxamide reported to inhibit TAK1 signaling in mantle cell lymphoma cancer cells and promote cell death [16]. ABC-FP, an aminofuropyridine, was reported as a biochemically potent TAK1 inhibitor with good activity in a mouse ovarian tumor model [29]. Finally, LYTAK1, an orally available pyrrolopyrimidine, was reported to inhibit NF-κB activity and potentiate the cytotoxicity of chemotherapeutic agents in pancreatic cancer [20]. Herein, we report a new series of covalent TAK1 inhibitors based on a 2,4-disubstituted pyrimidine scaffold that is well suited to further chemical modification.
2. Results and Discussion
2.1. Rationale
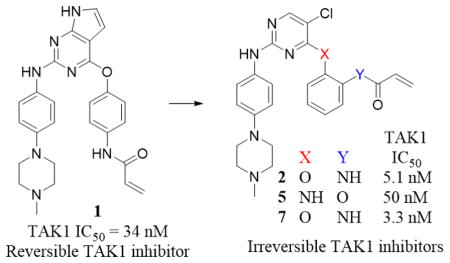
Previously we reported a series of reversible type-II kinase inhibitors including NG25, which potently inhibit TAK1 [30]. These studies were motivated by the hypothesis that alternate chemotypes might improve upon the selectivity and potency of existing TAK1 inhibitors such as 5Z7. In addition to NG25, kinome profiling of our kinase inhibitor library identified compound 1 as a potent TAK1 inhibitor with an enzymatic IC50 of 34 nM in a fixed time-point LanthaScreen binding assay (Life Technology, SelectScreen) [31]. 1 is similar to the 2,4-disubstituted pyrimidine scaffold that we used to make WZ4002, a previously reported covalent inhibitor of EGFR (Scheme 1A) [32].
Scheme 1.

Structures of WZ4002, 1 and 2 (A) and synthesis of 2 (B). Reagents and conditions: i) K2CO3, DMSO, RT; ii) TFA, 2-BuOH, 100 °C; iii) Raney nickel, H2, MeOH; iv) acryloyl chloride, sat. NaHCO3, THF, 0 °C ~RT.
To understand the mechanism of action of 1 as a TAK1 inhibitor and further evolve the compound we solved a crystal structure of the TAK1-TAB1 protein in complex with 1 (PDB 5J9L). The data collection and refinement statistics are given in Table S1. 1 exhibited a binding mode to TAK1 similar to that of WZ4002 bound to EGFR (Fig. S1). The pyrrolopyrimidine base of 1 is seen at the ATP binding site of TAK1 forming hydrogen bonds with hinge region residues Glu105 and Ala107, and hydrophobic contacts with Val50, Val90 and Phe176. The aniline ring with piperazinyl tail of 1 is bent 120º from this base and stabilized via hydrophobic contacts with the α-carbon of Gly110 on one side and nonpolar aliphatic side chain of Val42 on the other. The reactive acrylamide arm with its connecting phenyl ring is turned 114º extending towards solvent channel with hydrophobic contacts to Pro160 and Leu163. There is no electron density connecting Cys174 to the acrylamide moiety of 1 presumably due to an inappropriate trajectory of the two groups (Fig. S2A). Nevertheless, we noted that the 2-position of the 4-acrylamidophenyl group of 1 is located only 4.5 Å away from the thiol side chain of Cys174 of TAK1 (Fig. 1A) suggesting that introducing an acrylamide at this position might provide a better trajectory for approaching the cysteine residue. Based on this rationale we designed compound 2 with a structure similar to WZ4002 but changed the acrylamide from the meta-position as found in WZ4002 to the ortho-position. With this change in substitution pattern we anticipated 2 would react with Cys174, which is located immediately before the DFG-motif (DFG-1 position), whereas WZ4002 was designed to target the gatekeeper-plus-7 (GK+7) cysteine [33]. The 5-chlorine atom of the pyrimidine core in 2 was introduced to interact with the methionine GK of TAK1 in a similar manner to WZ4002 interacting with EGFR T790M GK mutants [32]. We chose not to include a 2-methoxy in the aniline tail, as in WZ4002, to avoid a potentially disadvantageous collision with the bulky side chain of Tyr106 of TAK1 (Scheme 1A).
Fig. 1.

X-ray co-crystal structure of TAK1-1 (yellow stick-cyan ribbon) (PDB 5J9L) overlaid with TAK1-2 (purple stick-green ribbon) (PDB 5J8I). (A) TAK1-2 covalently binds to Cys174 and (B) hydrogen bonds (blue dashes) extend between the anilinopyrimidine moiety of 1 or 2 and the hinge region of TAK1.
Synthesis of compound 2 was readily achieved with a high overall yield in four steps starting with 2,4,5-trichloropyrimidine. The 4-chloride group was substituted with a 2-nitrophenol under basic conditions, followed by substitution of the 2-chloride with a 4-(4-methylpiperazin-1-yl) aniline under acidic conditions to give 4. The nitro group of 4 was reduced using hydrogenation and the resulting aniline was acrylated to create 2 (Scheme 1B). Compared to 1, 2 showed potent TAK1 inhibition in a fixed time-point assay with an apparent IC50 of 5.1 nM. Mass spectrometry analysis of purified TAK1 incubated with 2 showed exclusive labeling of Cys174 (Fig S3). To investigate the structural basis for covalent binding we solved the co-crystal structure of the TAK1-TAB1 kinase domain in complex with 2 at a resolution of 2.4 Å (Fig.1B). In this structure (PDB 5J8I), the anilinopyrimidine moiety of 2 makes the expected bidentate hinge hydrogen bonds with Ala107, and continuous electron density is observed between the acrylamide warhead and Cys174, indicative of covalent bond formation (Fig. S2, 1B). The orientation and interaction of the anilinopyrimidine portion of the compound closely resemble the interactions observed in the TAK1-1 co-crystal structure, however, 1 with its pyrrole hydrogen donor forms an additional hydrogen bond with Glu105. The chlorine of 2 interacts with the gatekeeper Met104, and also with the reacted covalent warhead. The phenyl linker of 2 is orientated more co-planar to the pyrimidine core and closer to Cys174 compared with 1. The reacted ortho-acrylamide interacts with the oxygen linkage and the chlorine on the pyrimidine core intramolecularly, whereas the unreacted para-acrylamide of 1 protrudes toward the solvent.
2.2. Structure-activity relationship (SAR)
Given that MEK, ERK and TAK1 kinases possess analogous cysteine residues at the DFG-1 position [33], and 5Z7 is also capable of inhibiting MAP kinases [23, 27], we also tested the biochemical activity of 2 against MEK1 and ERK2. 5Z7 was used for comparison given that it is widely used in the literature to investigate TAK1-mediated signaling and evaluate the therapeutic potential of TAK1 inhibition as explained above. 5Z7 is also relevant to the currently reported compounds because it also operates by a covalent mechanism. Relative to TAK1, 2 exhibited 15-fold lower potency against MEK1 (LanthaScreen, IC50 = 78 nM) whereas 5Z7 showed slightly greater potency against MEK1 (IC50 = 2.9 nM) than TAK1 (IC50 = 5.6 nM). Against ERK2, 2 exhibited weak activity with an IC50 of 7.0 μM (Life Technologies, SelectScreen) [34] while 5Z7 was about 10-fold more active (IC50 = 738 nM). To systematically elucidate the structural requirements for potent inhibition of TAK1, 2 was divided into three moieties: the covalent linker (R1), the pyrimidine core (R2), and the aniline tail (R3). Each of these moieties was varied sequentially and over 20 analogs synthesized.
The first series of analogs focused on modifying the covalent linker (R1) (Table 1). Replacing the linkage between the phenyl group and the pyrimidine core from ether to amine decreased the potency against TAK1 and reduced the selectivity window between TAK1 and MEK1 (5). During our SAR studies, AstraZeneca reported a series of selective and potent ERK1/2 inhibitors, which are also based on the 2,4-disubstituted pyrimidine scaffold, and possess the same covalent linker as 5 [35]. Interestingly, these ERK1/2 inhibitors were also reported to inhibit MEK1, but not TAK1, whereas 5 exhibits only weak activity against ERK2. The thioether linkage decreased the potency even more, but exhibited better selectivity for TAK1 relative to MEK1 (6). 7, utilizing an acrylate instead of an acrylamide as the covalent warhead, exhibited good potency but less selectivity over MEK1 or ERK2 as compared to 2. Replacing the acrylamide with a propionamide (8, 9) resulted in inactivity against either TAK1 or MEK1, as expected. With a more reactive α-chloroacetamide warhead, 10 showed slightly decreased potency but much better selectivity over MEK1 (100-fold). Addition of para-acetamide in the phenyl ring results in a dramatic loss of potency (11).
Table 1.
SAR of R1
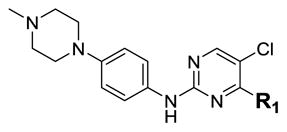
| ||||
|---|---|---|---|---|
| ID | R1 | Enzymatic IC50s (nM)a | ||
| TAK1 | MEK1 | ERK2 | ||
| 5Z7 | 5.6 | 2.9 | 738 | |
| 2 |

|
5.1 | 78 | 7890 |
| 5 |
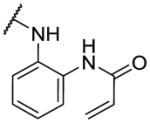
|
50 | 142 | 1090 |
| 6 |
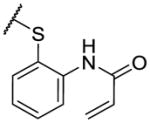
|
83 | 2480 | |
| 7 |

|
3.3 | 16 | 277 |
| 8 |
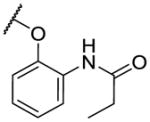
|
>10000 | >10000 | |
| 9 |
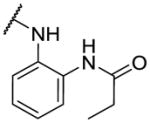
|
1630 | ||
| 10 |
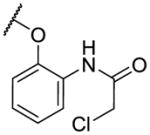
|
25 | 2500 | >10000 |
| 11 |
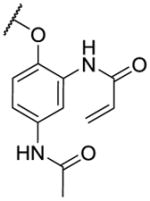
|
1640 | 4810 | |
IC50s against TAK1 and MEK1 were obtained with LanthaScreen binding assays, IC50s against ERK2 were obtained with Z’-Lyte activity assays; all assays were performed with a compound incubation time of 60 min.
In addition to TAK1-2, we also determined the co-crystal structures of TAK1-5 (PDB 5J7S), TAK1-7 (PDB 5JH6) and TAK1-10 (PDB 5E7R). Comparing these structures we observed that positions of the carbonyl groups varied due to differences in the linkers. In 2 and 10, the carbonyl of the acrylamide interacts with the side chain of Asn-161 along with an interaction with its oxygen linkage (Fig. 2A). In both 7 and 5, the carbonyl group can be seen protruding away from Asn161 making interactions with the nitrogen linkage (Fig 2B). Despite the conformational differences between these compounds, the covalent bonds with Cys174 of TAK1 are still formed due to the flexibility of the warheads and the DFG-motif. Interestingly, due to a shorter covalent warhead, 10 induces a shift in the position of the DFG-motif, which causes a shift in the conformation of the TAK1 kinase upon covalent binding. The carbonyl of the warhead in 10 specifically interacts with the side chain of Asn161 (Fig. 3). Cumulatively, the biochemical and structural information suggest that the covalent bond is indispensable for affinity, and that different linkages for the electrophiles result in conformational changes in the inhibitors, consequently affecting their potency and selectivity.
Fig. 2.

(A) X-ray co-crystal structures of TAK1-2 (green, PDB 5J8I) overlaid with TAK1-10 (orange, PDB 5E7R) showing interactions between the oxygen linkage for warhead and Asn161 (blue dashes). (B) X-ray co-crystal structures of TAK1-5 (cyan, PDB 5J7S) and TAK1-7 (gray, PDB 5JH6). Nitrogen linkage results in loss of interactions with Asn161.
Fig. 3.
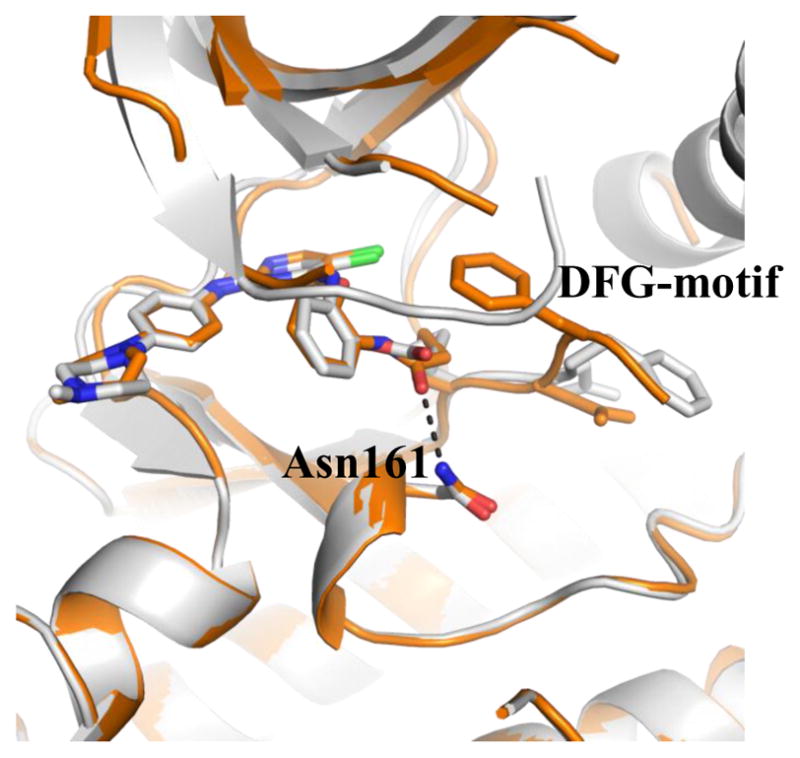
Changes in the overall conformation of DFG-motif between TAK1-7 (gray, PDB 5JH6) and TAK1-10 (orange, PDB 5E7R). The carbonyl warhead group interaction is shown (black lines).
After optimization of the covalent linker moiety, SAR of the pyrimidine core (R2) was investigated (Table 2). We found that removing the 5-chlorine resulted in a dramatic decrease in potency (12), and substituents at 5-position such as methyl, methoxy or cyclopropyl all led to a greater than 10-fold decrease in potency (13–15). Addition of a 6-amino group, which provides an additional hydrogen bond donor, did not help the hinge binding as in 1 but instead abrogated anti-TAK1 activity (16). Cyclization of the substitutions at 4- and 5-positions with a fused ring is a common strategy to optimize 2,4-disubstituted pyrimidine scaffolds [36]. However, the pyrrolopyrimidine core led to reduced activity in this case (17). In combination with the SAR of R1, these results suggest that interactions between the 5-chlorine and the acrylamide warhead or the gatekeeper residue facilitate the warhead’s reaching Cys174, whereas the bicyclic core probably disfavors the warhead from adapting the preferred conformation for a covalent reaction.
Table 2.
SAR of R2
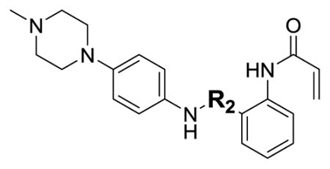
| ||||
|---|---|---|---|---|
| ID | R2 | Enzymatic IC50s (nM)a | ||
| TAK1 | MEK1 | ERK2 | ||
| 2 |
|
5.1 | 78 | 7890 |
| 12 |
|
950 | 4470 | |
| 13 |
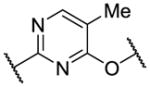
|
92 | 1630 | 9230 |
| 14 |
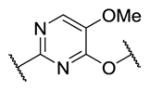
|
75 | 1300 | 1670 |
| 15 |
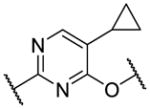
|
57 | 763 | 5600 |
| 16 |
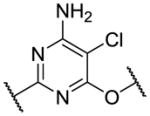
|
2750 | >10000 | |
| 17 |
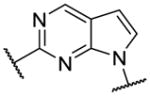
|
3010 | 6810 | >10000 |
IC50s against TAK1 and MEK1 were obtained with LanthaScreen binding assays, IC50s against ERK2 were obtained with Z’-Lyte activity assays; all assays were performed with a compound incubation time of 60 min.
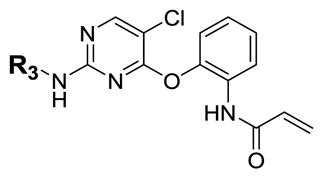
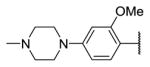
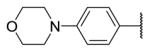
In addition, we investigated the SAR of the aniline tail (R3) (Table 3). Addition of a methoxy group to the 2-position of the aniline (18) led to loss of potency as expected, likely due to steric clashes with Tyr106. Substitution with a 4-morpholinylaniline tail (19) maintained potency against TAK1 but showed less selectivity over MEK1. Switching the substitutions from 4- to 3-position slightly increased potency (20, 21). Next we exploited 1-substituted-pyrazol-3-amine tails, which often maintain the potency for covalent inhibitors while reducing interaction with non-covalent off-targets compared to the aniline tails [36]. 1-methyl-1-H-pyrazol-3-amine (20) showed similar potency but decreased selectivity compared to 19. Compounds with further substitutions at the 1-methyl of the amine tails maintained potency (23–25) with slightly improved selectivity over MEK1 (23, 25). As the reversible counterpart of 25, 26 showed dramatically decreased potency, which is consistent with the aniline series (8, 9). The co-crystal structure of TAK1-22 was determined (PDB 5JK3) and compared to the structures of 2, 5, 7 or 10 bound to TAK1. The major difference between binding modes of these analogs involved the 4-methylpiperazin-1-yl group, which adopts the two lowest energy conformations (boat and chair), demonstrating flexibility, and is approximately 4.5Å towards the side chain of Asn114, whereas the methylpyrazole ring of 22 is planar, making hydrophobic contacts with Gly110 and Val42 from either side. Interactions with Asn114 can be exploited for in future studies as a means to optimize selectivity since the analogous residues differ in MEK1 (Gln153) or ERK2 (Lys114) (Fig. 4).
Table 3.
SAR of R3
| ||||
|---|---|---|---|---|
| ID | R3 | Enzymatic IC50s (nM)a | ||
| TAK1 | MEK1 | ERK2 | ||
| 2 |
|
5.1 | 78 | 7890 |
| 18b |
|
342 | ||
| 19b |
|
4.4 | 8.9 | 361 |
| 20b |
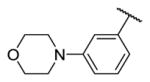
|
3.2 | 11 | 736 |
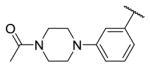
| 21 |
|
1.7 | 9.5 | 225 |
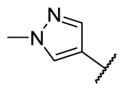
| 22 |
|
7.3 | 9.8 | 3140 |
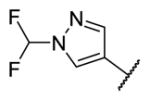
| 23 |
|
7.0 | 26 | 242 |
| 24 |
|
4.6 | 8.2 | 4620 |
| 25 |

|
2.4 | 8.5 | 90.3 |
IC50s against TAK1 and MEK1 were obtained with LanthaScreen binding assays, all assays were performed with a compound incubation time of 60 min;
R1 of 18 is same as R1 of 7.
Fig. 4.
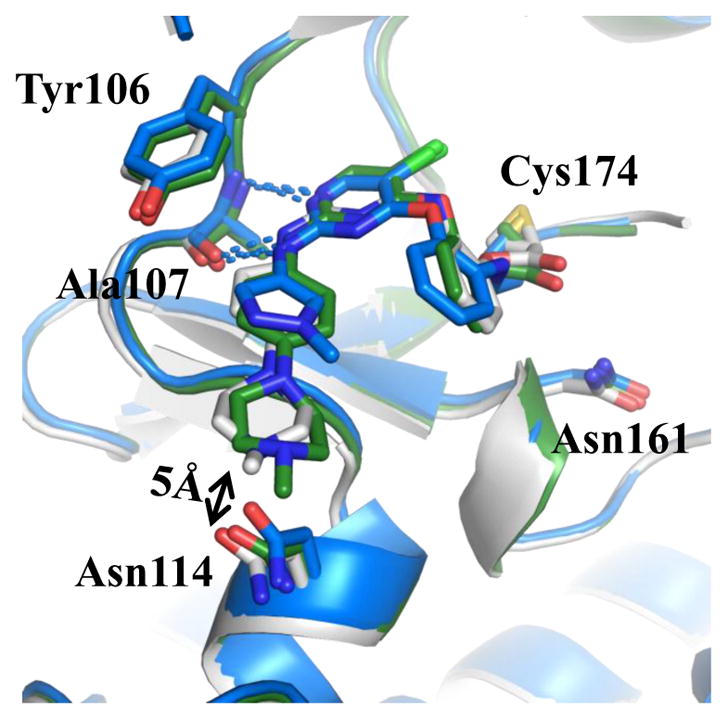
X-ray co-crystal structure of TAK1-22 (blue, PDB 5JK3), overlaid with TAK1-2 (green, PDB 5J8I) and TAK1-7 (gray, PDB 5JH6) showing key interactions (blue lines). The flexibility of the piperazinyl moiety adopting different conformations extending towards Asn114 is also seen.
Although IC50 values are time-dependent for covalent compounds, for practical reasons IC50 values were used to rank order compounds for SAR. We considered these values to be a reasonable first estimate of potency and reactivity because reaction times were strictly maintained at 60 min across assays. However, to further characterize key compounds and as an additional demonstration of their covalent nature we performed additional time-dependent experiments. We tested 2, 5 and 25 at a shorter incubation time of 10 min and observed decreased potency with short incubation time, with IC50s of 360, 125 and 7.6 nM, respectively, consistent with covalent binding. For one of our most promising lead compounds, 5, we determined a Kinact/Ki value of 6.87 x 10−6 nM−1 s−1 (Fig. S4) which is in the range of other clinically used covalent inhibitors such as afatinib or neratinib [37].
2.3. Kinome selectivity
Approximately 50 human kinases possess a cysteine immediately preceding the DFG-motif [33]. We therefore surveyed the selectivity of inhibitor 5 across a panel of 468 kinases and mutants using an in vitro ATP-site competition binding assay (KINOMEscan [38]) at a concentration of 1 μM (Fig S5, Table S2). This analysis revealed that 5 binds to TAK1 and ten other kinases with a cysteine at the DFG-1 position (Fig S5), and possesses a selectivity score, defined as the percentage of kinases with scores less than 1 (S(1)) [39], of 0.10. In contrast, its non-covalent counterpart, 9, possesses a same S(1) of 0.10 as 5 but hits no kinases with a cysteine at the DFG-1 position. Instead 9 shows strong inhibition against new targets such as MAP3K2/3, WEE1/2 and PYK2. We also profiled 7, 23 and 25 for their kinome selectivity at 1 μM. 7 was a more promiscuous inhibitor with an S(1) of 0.19. However, 23 and 25 showed significantly improved selectivity with S(5) of 0.1 and 0.03 respectively, although, TAK1 was not ranked among the strongest hits (Fig S5, Table S4–6).
In these data we also noted that 5Z7 did not display strong binding to TAK1 at a concentration of 1 μM, and only exhibited a moderate inhibition score against TAK1 at 10 μM (Table S7). These effects are likely related to the nature of the various binding assays. KINOMEscan utilizes a common “tracer” with variable binding affinity across kinases which can influence relative inhibition scores [38]. On the other hand the SelectScreen assays is optimized for each kinase. In this case SelectScreen assays were performed in a dose-response format which provides more reliable measurements than single-point KINOMEscan results. Taken together, the fact that 5 and 9 show distinct inhibitory profiles despite having nearly identical chemical structures, except in the electrophile portion, strongly supports that covalent binding is a determining factor for the kinome selectivity of these inhibitors. Along these lines 5, 23, 25 and 5Z7 preferentially bind to kinases that possess a cysteine at the DFG-1 position as compared to 7 suggesting the 2,4-substituted pyrimidine template may provide a general scaffold for further optimization of kinase selectivity (Table S2–7).
2.4. Mouse liver microsomal stability
The in vitro metabolic stability of some analogs was measured using mouse liver microsomes (MLM) to calculate half-lives (T1/2) [40]. Based on the data in Table 4, for the current 2,4-disubstituted pyrimidine scaffold it can be concluded that amine linkage is more stable than the ether linkage; the α-chloroacetamide and the acrylate warheads are more labile than the acrylamide; electron-donor groups at the 5-position of the pyrimidine core make the inhibitors more stable; and modifications of the aniline tail moiety dramatically decrease stability with half-lives ranging from less than 1 min to 23 min.
Table 4.
Mouse liver microsomal stability
| ID | MLM T1/2 (min) | ID | MLM T1/2 (min) | ID | MLM T1/2 (min) |
|---|---|---|---|---|---|
| 2 | 6.1 | 14 | 15.3 | 21 | <1 |
| 5 | 10.8 | 15 | 12.8 | 22 | 8.1 |
| 7 | 5.7 | 17 | 10.5 | 23 | 4.2 |
| 10 | 3 | 19 | 3.8 | 24 | 4 |
| 13 | 8.8 | 20 | 23 | 25 | 6.9 |
3. Conclusion
In summary, we report a structure-guided approach for development of TAK1 inhibitors with single or double-digit nanomolar enzymatic IC50 values based on a common 2,4-disubstituted pyrimidine scaffold as our previously described EGFRT790M covalent inhibitors [32]. Several of our inhibitors exhibited greater than 10-fold selectivity for TAK1 over MEK1 with compound 10 exhibiting around 100-fold selectivity. Most of the inhibitors we described were weakly active or inactive against ERK2, despite the fact that potent and selective ERK1/2 inhibitors have been developed and reported recently based on a very similar scaffold [35]. The five newly determined co-crystal structures of TAK1 bound to newly synthesized compounds guided design choices for covalent targeting of the DFG-1 cysteine in TAK1 and for improving potency and selectivity. The inhibitors described here provide a synthetically straight-forward pharmacophore and a solid structural basis for future optimization of selective covalent TAK1 inhibitors. In the accompanying communication we discuss the application of these inhibitors to TAK1-centered polypharmacology.
4. Experimental
4.1. Chemistry
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on 600 or 500 MHz (Varian AS600 or Bruker A500), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 x 50 mm, 5μM) using a gradient of 15–95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
2,5-dichloro-4-(2-nitrophenoxy)pyrimidine (3)
2-Nitrophenol (840 mg, 6.0 mmol) and potassium carbonate (800 mg, 6.0 mmol) were combined in dimethyl sulfoxide (DMSO) (10 mL) and stirred for 15 min, then 2,4,5-trichloropyrimidine (560 μL, 5.0 mmol) was added and the mixture was stirred overnight. The mixture was then diluted with ethyl acetate and washed with water and brine, dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (hexane:ethyl acetate = 3:1) to yield 1.2 g (70%) of 3 as a white solid. MS (ESI) m/z 286 (M+H)+.
5-chloro-N-(4-(4-methylpiperazin-1-yl)phenyl)-4-(2-nitrophenoxy)pyrimidin-2-amine (4)
To 3 (570 mg, 2.0 mmol) and 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (390 mg, 2.0 mmol) in sec-butanol (4 mL) was added trifluoroacetic acid (154 μL, 2.0 mmol) and the mixture was stirred overnight at 75 °C. The mixture was then concentrated, neutralized with ammonia in methanol and purified by column chromatography (dichloromethane:methanol = 10:1) to yield 265 mg (60%) of 4 as a pale-yellow solid. MS (ESI) m/z 441 (M+H)+.
N-(2-((5-chloro-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)acrylamide (2)
To 4 (88 mg, 0.2 mmol) in methanol (10 mL) was added 1 mL Raney nickel suspension in methanol. The reaction mixture was stirred for 3 h under 1 atm of hydrogen. The mixture was then filtered with celite, and the filtrate was concentrated and dried under vacuum to give a crude product as a white solid. To the obtained white solid in tetrahydrofuran (3 mL) was added saturated NaHCO3 solution (3 mL), the stirred mixture was then cooled to 0 °C, and acryloyl chloride was added (25 μL, 0.3 mmol) dropwise. The reaction mixture was stirred at 0 °C for 10 min, another batch of acryloyl chloride was added (8 μL, 0.1 mmol) dropwise. After another 5 min the mixture was allowed to recover to room temperature (RT), and diluted with ethyl acetate and washed with water and brine, dried over Na2SO4, filtered and concentrated. The crude product was then purified by reverse phase HPLC to give 58 mg (63% for 2 steps) of 2 as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 9.68 (br, 1H), 9.58 (s, 1H), 9.52 (br, 1H), 8.40 (s, 1H), 8.05 (m, 1H), 7.32 (dd, J = 8.4, 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 6.72 (m, 2H), 6.55 (dd, J = 16.8, 10.8 Hz, 1H), 6.20 (d, J = 16.8 Hz, 1H), 5.71 (d, J = 10.8 Hz, 1H), 3.66 (m, 2H), 3.50 (m, 2H), 3.14 (m, 2H), 2.85 (s, 3H), 2.83 (m, 2H). MS (ESI) m/z 465 (M+H)+.
2,5-dichloro-N-(2-nitrophenyl)pyrimidin-4-amine (28)
To 2,4,5-trichloropyrimidine (560 μL mg, 5.0 mmol) and 2-nitroaniline (700 mg, 5.0 mmol) in sec-butanol (25 mL) was added trifluoroacetic acid (383 μL, 5.0 mmol) and the mixture was stirred overnight at 60 °C. The mixture was then concentrated, neutralized with ammonia in methanol and purified by column chromatography (hexane:ethyl acetate = 2:1) to yield 640 mg (45%) of 28 as a yellow solid. MS (ESI) m/z 285 (M+H)+.
5-chloro-N2-(4-(4-methylpiperazin-1-yl)phenyl)-N4-(2-nitrophenyl)pyrimidine-2,4-diamine (29)
To 28 (570 mg, 2.0 mmol) and 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (390 mg, 2.0 mmol) in sec-butanol (4 mL) was added trifluoroacetic acid (154 μL, 2.0 mmol) and the mixture was stirred overnight at 85 °C. The mixture was then concentrated, neutralized with ammonia in methanol and purified by column chromatography (dichloromethane:methanol = 10:1) to yield 680 mg (76%) of 29 as a pale-yellow solid. MS (ESI) m/z 440 (M+H)+.
N-(2-((5-chloro-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)acrylamide (5)
To 29 (88 mg, 0.2 mmol) in methanol (10 mL) was added 1 mL Raney nickel suspension in methanol. The reaction mixture was stirred for 3 h under 1 atm of hydrogen. The mixture was then filtered with celite, and the filtrate was concentrated and dried under vacuum to give a crude product as a white solid. To the obtained white solid in tetrahydrofuran (3 mL) was added saturated NaHCO3 solution (3 mL), the stirred mixture was then cooled to 0 °C, and acryloyl chloride was added (25 μL, 0.3 mmol) dropwise. The reaction mixture was stirred at 0 °C for 10 min, another batch of acryloyl chloride was added (8 μL, 0.1 mmol) dropwise. After another 5 min the mixture was allowed to recover to room temperature (RT), and diluted with ethyl acetate and washed with water and brine, dried over Na2SO4, filtered and concentrated. The crude product was then purified by reverse phase HPLC to give 55 mg (60% for 2 steps) of 5 as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.67 (br, 1H), 9.20 (br, 1H), 8.56 (s, 1H), 8.09 (s, 1H), 7.74 (d, J = 7.2 Hz, 1H), 7.46 (d, J = 6.6 Hz, 1H), 7.40 (d, J = 9.0 Hz, 2H), 7.32 (dd, J = 7.2, 7.2 Hz, 1H), 7.24 (dd, J = 7.8, 7.2 Hz, 1H), 6.78 (d, J = 9.6 Hz, 1H), 6.50 (dd, J = 16.8, 10.8 Hz, 1H), 6.30 (d, J = 17.4 Hz, 1H), 5.79 (d, J = 10.8 Hz, 1H), 3.66 (m, 2H), 3.49 (m, 2H), 3.14 (m, 2H), 2.84 (s, 3H), 2.83 (m, 2H). MS (ESI) m/z 464 (M+H)+.
2-((2,5-dichloropyrimidin-4-yl)amino)phenol (30)
To 2-aminophenol (550 mg, 5.0 mmol) in dioxane (10 mL) was added 2,4,5-trichloropyrimidine (560 μL mg, 5.0 mmol), the mixture was stirred overnight at RT. The mixture was then diluted with ethyl acetate and washed with water and brine, dried over Na2SO4, filtered and concentrated to give 1.02 g (80%) of the crude product 30 as a white solid, which was directly used in the next step. MS (ESI) m/z 256 (M+H)+.
2-((5-chloro-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)amino)phenol (31)
To 30 (510 mg, 2.0 mmol) and 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (575 mg, 3.0 mmol) in sec-butanol (5 mL) was added trifluoroacetic acid (230 μL, 3.0 mmol) and the mixture was stirred overnight at 100 °C. The mixture was then concentrated, neutralized with ammonia in methanol and purified by column chromatography (dichloromethane:methanol = 10:1) to yield 680 mg (76%) of 31 as a white solid. MS (ESI) m/z 411 (M+H)+.
2-((5-chloro-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl acrylate (7)
To 31 (82 mg, 0.2 mmol) in dimethylformamide (2 mL) was added diisopropylethylamine (53 μL, 0.3 mmol), the stirred mixture was then cooled to −60 °C, and acryloyl chloride (17.8 μL, 0.22 mmol) was added dropwise. The reaction mixture was stirred at −60 °C for 10 min, allowed to recover to RT (room temperature) gradually in 30 min, and purified by reverse phase HPLC to give 64 mg (TFA salt, 56% for 2 steps) of 7 as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 8.96 (br, 1H), 8.36 (s, 1H), 8.04 (s, 1H), 7.72 (br, 1H), 7.34 (d, J = 9.0 Hz, 2H), 7.30 (m, 3H), 6.70 (d, J = 9.0 Hz, 2H), 6.37 (d, J = 16.8, 1H), 6.26 (dd, J = 17.4, 10.2 Hz, 1H), 5.79 (d, J = 10.8 Hz, 1H), 3.02 (m, 4H), 2.54 (m, 4H), 2.28 (br, 3H), 2.84 (s, 3H). MS (ESI) m/z 465 (M+H)+.
4.2. X-Ray crystallography procedures
TAK1–TAB1 fusion protein (TAK1 kinase domain residues 31–303 fused with C-terminal TAB1 domain residues 468–497) was expressed in insect cells, purified and crystallized as reported previously [30]. Crystals grew within 2 days by vapor diffusion in hanging drop at 20ºC by mixing protein with equal volumes of reservoir solution consisting of 0.65–0.75 M sodium citrate, 0.2 M NaCl, 0.1 M Tris, pH 7.0, and 5 mM adenosine. Crystals containing adenosine were then back-soaked with excess of inhibitors at 250 μM - 500 μM dissolved in reservoir solution for 6–8h. The crystals were then flash frozen with 20% ethyleneglycol as cryoprotectant for data collection. Diffraction data were collected at Argonne Advanced Photon Source (beamline 19-D) and processed with HKL2000 and HKL3000 [41]. The structure was determined by molecular replacement using Phaser [42] with reported TAK1 structure (PDB code: 2YIY or 4O91) as search model. Coot was used for model building [43] and refinement was carried out using both Phenix, version 1.10–2155 [44] and Refmac, version 5.8.0049. [45–47], PyMol (The PyMOL Molecular Graphics System, version 1.6.0.0) and Meastro (version 1.5.014) from Schrödinger, LLC. For demonstration of key interactions, LIGPLOT software was used [48].
4.3. Enzymatic assays
Enzymatic inhibition of MEK2 and ERK1 was tested in Z’-Lyte assays with ATP concentrations near the Km for each kinases. Compound activity against TAK1 was tested in LanthaScreen binding assays. All protocols are available from Life Technologies [49]. The MLM assays were previously reported and are commercially available from Scripps Florida [40].
Supplementary Material
Acknowledgments
NSG and PKS were supported by LINCS Grant U54-HL127365. KDW was supported by CPRIT R1207 and CPRIT RP140233. LT and NSG were supported by NIH CA 154303-05 and the Pediatric Low Grade Astrocytoma (PLGA) foundation. Results shown in this article are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by U Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DEAC02-06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science (New York, NY) 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 2.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nature immunology. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 3.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes & Development. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shirakabe K, Yamaguchi K, Shibuya H, Irie K, Matsuda S, Moriguchi T, Gotoh Y, Matsumoto K, Nishida E. TAK1 Mediates the Ceramide Signaling to Stress-activated Protein Kinase/c-Jun N-terminal Kinase. Journal of Biological Chemistry. 1997;272:8141–8144. doi: 10.1074/jbc.272.13.8141. [DOI] [PubMed] [Google Scholar]
- 5.Sakurai H, Shigemori N, Hasegawa K, Sugita T. TGF-beta-activated kinase 1 stimulates NF-kappa B activation by an NF-kappa B-inducing kinase-independent mechanism. Biochemical and biophysical research communications. 1998;243:545–549. doi: 10.1006/bbrc.1998.8124. [DOI] [PubMed] [Google Scholar]
- 6.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 7.Sakurai H, Miyoshi H, Toriumi W, Sugita T. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. The Journal of biological chemistry. 1999;274:10641–10648. doi: 10.1074/jbc.274.15.10641. [DOI] [PubMed] [Google Scholar]
- 8.Schuman J, Chen Y, Podd A, Yu M, Liu HH, Wen R, Chen ZJ, Wang D. A critical role of TAK1 in B-cell receptor–mediated nuclear factor κB activation. Blood. 2009;113:4566–4574. doi: 10.1182/blood-2008-08-176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nature immunology. 2006;7:851–858. doi: 10.1038/ni1355. [DOI] [PubMed] [Google Scholar]
- 10.Liu HH, Xie M, Schneider MD, Chen ZJ. Essential role of TAK1 in thymocyte development and activation. Proceedings of the National Academy of Sciences. 2006;103:11677–11682. doi: 10.1073/pnas.0603089103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Takeuchi O, Akira S. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. International immunology. 2006;18:1405–1411. doi: 10.1093/intimm/dxl082. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Ma FY, Tesch GH, Ozols E, Xie M, Schneider MD, Nikolic-Paterson DJ. TGF-beta1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. American journal of physiology Renal physiology. 2011;300:F1410–1421. doi: 10.1152/ajprenal.00018.2011. [DOI] [PubMed] [Google Scholar]
- 14.Grillo AR, Scarpa M, D'Inca R, Brun P, Porzionato A, De Caro R, Martines D, Buda A, Angriman I, Palu G, Sturniolo GC, Castagliuolo I. TAK1 is a key modulator of the profibrogenic phenotype of human ileal myofibroblasts in Crohn's disease. American journal of physiology Gastrointestinal and liver physiology. 2015;309:G443–454. doi: 10.1152/ajpgi.00400.2014. [DOI] [PubMed] [Google Scholar]
- 15.Neubert M, Ridder DA, Bargiotas P, Akira S, Schwaninger M. Acute inhibition of TAK1 protects against neuronal death in cerebral ischemia. Cell death and differentiation. 2011;18:1521–1530. doi: 10.1038/cdd.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buglio D, Palakurthi S, Byth K, Vega F, Toader D, Saeh J, Neelapu SS, Younes A. Essential role of TAK1 in regulating mantle cell lymphoma survival. Blood. 2012;120:347–355. doi: 10.1182/blood-2011-07-369397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai PC, Shi L, Liu VW, Tang HW, Liu IJ, Leung TH, Chan KK, Yam JW, Yao KM, Ngan HY, Chan DW. Elevated TAK1 augments tumor growth and metastatic capacities of ovarian cancer cells through activation of NF-kappaB signaling. Oncotarget. 2014;5:7549–7562. doi: 10.18632/oncotarget.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–650. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan Y, Cheng J, Vasudevan SA, Patel RH, Liang L, Xu X, Zhao Y, Jia W, Lu F, Zhang H, Nuchtern JG, Kim ES, Yang J. TAK1 inhibitor 5Z-7-oxozeaenol sensitizes neuroblastoma to chemotherapy. Apoptosis. 2013;18:1224–1234. doi: 10.1007/s10495-013-0864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melisi D, Xia Q, Paradiso G, Ling J, Moccia T, Carbone C, Budillon A, Abbruzzese JL, Chiao PJ. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. Journal of the National Cancer Institute. 2011;103:1190–1204. doi: 10.1093/jnci/djr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends in pharmacological sciences. 2012;33:522–530. doi: 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Ansell SM, Hodge LS, Secreto FJ, Manske M, Braggio E, Price-Troska T, Ziesmer S, Li Y, Johnson SH, Hart SN, Kocher JP, Vasmatzis G, Chanan-Kahn A, Gertz M, Fonseca R, Dogan A, Cerhan JR, Novak AJ. Activation of TAK1 by MYD88 L265P drives malignant B-cell Growth in non-Hodgkin lymphoma. Blood cancer journal. 2014;4:e183. doi: 10.1038/bcj.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. The Journal of biological chemistry. 2003;278:18485–18490. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 24.Omori E, Matsumoto K, Zhu S, Smart RC, Ninomiya-Tsuji J. Ablation of TAK1 upregulates reactive oxygen species and selectively kills tumor cells. Cancer research. 2010;70:8417–8425. doi: 10.1158/0008-5472.CAN-10-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan Y, Cheng J, Vasudevan SA, Patel RH, Liang L, Xu X, Zhao Y, Jia W, Lu F, Zhang H. TAK1 inhibitor 5Z-7-oxozeaenol sensitizes neuroblastoma to chemotherapy. Apoptosis. 2013;18:1224–1234. doi: 10.1007/s10495-013-0864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schirmer A, Kennedy J, Murli S, Reid R, Santi DV. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4234–4239. doi: 10.1073/pnas.0600445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Powell F, Larsen NA, Lai Z, Byth KF, Read J, Gu RF, Roth M, Toader D, Saeh JC, Chen H. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS chemical biology. 2013;8:643–650. doi: 10.1021/cb3005897. [DOI] [PubMed] [Google Scholar]
- 28.Sogabe Y, Matsumoto T, Hashimoto T, Kirii Y, Sawa M, Kinoshita T. 5Z-7-Oxozeaenol covalently binds to MAP2K7 at Cys218 in an unprecedented manner. Bioorganic & medicinal chemistry letters. 2015;25:593–596. doi: 10.1016/j.bmcl.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 29.Hornberger KR, Chen X, Crew AP, Kleinberg A, Ma L, Mulvihill MJ, Wang J, Wilde VL, Albertella M, Bittner M, Cooke A, Kadhim S, Kahler J, Maresca P, May E, Meyn P, Romashko D, Tokar B, Turton R. Discovery of 7-aminofuro[2, 3-c]pyridine inhibitors of TAK1: optimization of kinase selectivity and pharmacokinetics. Bioorganic & medicinal chemistry letters. 2013;23:4511–4516. doi: 10.1016/j.bmcl.2013.06.054. [DOI] [PubMed] [Google Scholar]
- 30.Tan L, Nomanbhoy T, Gurbani D, Patricelli M, Hunter J, Geng J, Herhaus L, Zhang J, Pauls E, Ham Y, Choi HG, Xie T, Deng X, Buhrlage SJ, Sim T, Cohen P, Sapkota G, Westover KD, Gray NS. Discovery of type II inhibitors of TGFbeta-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2) Journal of medicinal chemistry. 2015;58:183–196. doi: 10.1021/jm500480k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lebakken CS, Riddle SM, Singh U, Frazee WJ, Eliason HC, Gao Y, Reichling LJ, Marks BD, Vogel KW. Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. Journal of biomolecular screening. 2009;14:924–935. doi: 10.1177/1087057109339207. [DOI] [PubMed] [Google Scholar]
- 32.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.For ERK2 Z'-Lyte activity assay. http://www.thermofisher.com/content/dam/LifeTech/migration/files/drug-discovery/pdfs.par.60256.file.dat/20130430%20ssbk%20customer%20protocol%20and%20assay%20conditions.pdf.
- 35.Ward RA, Colclough N, Challinor M, Debreczeni JE, Eckersley K, Fairley G, Feron L, Flemington V, Graham MA, Greenwood R, Hopcroft P, Howard TD, James M, Jones CD, Jones CR, Renshaw J, Roberts K, Snow L, Tonge M, Yeung K. Structure-Guided Design of Highly Selective and Potent Covalent Inhibitors of ERK1/2. Journal of medicinal chemistry. 2015;58:4790–4801. doi: 10.1021/acs.jmedchem.5b00466. [DOI] [PubMed] [Google Scholar]
- 36.Tan L, Akahane K, McNally R, Reyskens KM, Ficarro SB, Liu S, Herter-Sprie GS, Koyama S, Pattison MJ, Labella K, Johannessen L, Akbay EA, Wong KK, Frank DA, Marto JA, Look TA, Arthur JS, Eck MJ, Gray NS. Development of Selective Covalent Janus Kinase 3 Inhibitors. Journal of medicinal chemistry. 2015;58:6589–6606. doi: 10.1021/acs.jmedchem.5b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proceedings of the National Academy of Sciences. 2014;111:173–178. doi: 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nature reviews Drug discovery. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- 39.Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, Chung E, Yang Q, Schwaller J, Knapp S, King RW, Lee JD, Herrgard S, Zarrinkar P, Gray NS. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem Biol. 2011;18:868–879. doi: 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:1242–1250. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta crystallographica. Section D, Biological crystallography. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 42.Bunkoczi G, Echols N, McCoy AJ, Oeffner RD, Adams PD, Read RJ. Phaser.MRage: automated molecular replacement. Acta crystallographica Section D, Biological crystallography. 2013;69:2276–2286. doi: 10.1107/S0907444913022750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholls RA, Long F, Murshudov GN. Low-resolution refinement tools in REFMAC5. Acta crystallographica Section D, Biological crystallography. 2012;68:404–417. doi: 10.1107/S090744491105606X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta crystallographica Section D, Biological crystallography. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta crystallographica Section D, Biological crystallography. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 48.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 49.Z'-Lyte assays. http://www.lifetechnologies.com/content/dam/LifeTech/migration/files/drug-discovery/pdfs.par.60256.file.dat/20130430%20ssbk%20customer%20protocol%20and%20assay%20conditions.pdfTTK assay: http://tools.lifetechnologies.com/content/sfs/manuals/TTK_LanthaScreen_Binding.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.