

Abstract
Krabbe Disease (KD) is a severe neuro-degenerative disorder affecting white matter in the brain and peripheral nerves. Transplantation of hemato-poetic stem cells (HSCT), although not curative, has been shown to extend survival and alleviate neurodevelopmental symptoms, if treatment precedes the onset of symptoms. Existing evidence, although not tested statistically, seems to clearly show that post-symptomatic transplantation does not improve neurodevelopmental outcomes. The impact of post-symptomatic HSCT treatment on survival, however, is an open question. We used a KD registry to examine the effect of HSCT upon survival of symptomatic KD patients. 16 transplanted patients were matched by age of onset to 68 non-transplanted patients. The potential confounding effect of age of onset was, therefore, avoided. To quantify the effect of HSCT over time, we used Cox regression analysis and observed a sustained and nearly 2.2-fold risk of death from KD in patients who were not transplanted relative to those who were (one-tailed p=0.0365; 95% lower bound=1.07). The improvement of survival due to HSCT did not appear to depend on the age of symptom onset. Thus, these results establish a long-term, quantitative benefit of HSCT even in patients who are already experiencing symptoms. They also provide a benchmark for improved survival that can be used for potential new treatments for KD.
Keywords: Newborn screening, leukodystrophies, neuro-degenerative diseases, early diagnosis
Graphical Abstract
Krabbe Disease is a neurodegenerative disorder that can be treated only with transplantation of umbilical cord blood. Earlier studies have not shown statistically significant improvement of survival of patients treated after they develop symptoms. By using Cox regression analysis we establish a 2.2 fold improvement of survival in symptomatic patients.
INTRODUCTION
Krabbe Disease (KD), also known as globoid cell leukodystrophy (OM IM#245200), is a rare but devastating illness that causes degeneration of white matter in the brain and also in peripheral nerves (Hagberg et al., 1963; Wenger and Luzi, 2014.). It is caused in autosomal recessive fashion by deficiency of the enzyme galacto-cerebrosidiase (GALC) (Suzuki and Suzuki, 1970; Suzuki, 1984). KD is characterized pathologically by widespread demyelination, dys-myelination and inflammation (Suzuki and Suzuki, 1970; Wenger and Luzi, 2014). In the most frequent early-onset variant of the disease, infants are afflicted before 6 months of life with irritability, fisting, as well as progressive and often marked spasticity. These infants eventually develop seizures, visual loss and significant medical problems that result in death by around 2 years of age (Wenger and Luzi, 2014; Hagberg et al., 1963; Duffner et al., 2011).
However, KD is clinically pleomorphic. In addition to this early infantile form (EIKD), some infants develop progressive symptoms between 6 and 12 months of age and are therefore considered to have late infantile Krabbe disease–LIKD (Lyon et al., 1991; Duffner et al., 2012). Later onset (LOKD) is comprised of a heterogeneous group of clinical syndromes that can manifest in later childhood, adolescence or even in adulthood. The symptoms of LOKD include spastic paraparesis, optic atrophy, ataxia, and cognitive decline (Lyon et al., 1991; Lyon et al., 1991; Bajaj et al., 2002). While the progression of LOKD certainly is more indolent than EIKD or LIKD, Krabbe Disease is in every instance considered to be a progressive degenerative disorder variably affecting the white matter of the brain, spinal cord, and peripheral nerves (Suzuki and Suzuki, 1970; Suzuki,1984; Wenger and Luzi, 2014.). Several specific mutations of the GALC gene have been reported in KD, but with the exception of a single homozygous deletion that is associated with EIKD (Wenger et al., 1997), genotype/phenotype correlations are not well-established in the several clinical forms of this disorder (Rafi et al., 1995; Wenger et al., 2014).
Transplantation of hematopoietic stem cells (HSCT) has been used for several leukodystrophies. HSCT is presumably effective because cells from the graft enter the CNS and produce deficient enzymes (Krivit et. al, 1998; Martin et. al, 2006; Shapiro et. al, 2000) Allogenic transplantation of cells from banked cord blood has been established as a therapy for those conditions that respond to HSCT (Escolar et al., 2005;Martin et. al, 2006) Treatment of infants who are predicted to develop EIKD or who have later onset symptoms has been achieved by use of HSCT(Krivit et al, 1998;Escolar et al., 2005). Availability of a partially effective treatment resulted in the inception of newborn screening (NBS) for KD in New York State in 2006 (Duffner et al., 2009a,b; Orsini et al., 2009). However, in the ensuing 9 years, screening has become established in only one additional state, Missouri. The US Department of Health and Human Services (USDHHS) Secretary’s Advisory Committee on Heritable Disorders indeed declined to recommend KD for NBS, citing concerns about accuracy of the diagnostic algorithm, phenotypic definition of the condition, and the benefits and potential harms of treatment (Kemper et al., 2010). The current diagnostic algorithm in fact is not specific and, thus, has many false positives (Duffner et al., 2009b; Orsini et al., 2009; Jalal et al., 2012; Turgeon et al., 2015). It therefore cannot be used for clinical decision-making and many of the infants who eventually manifest as Krabbe patients are not transplanted prior to symptom onset. Although not statistically confirmed, there is seemingly compelling evidence that HSCT treatment of pre-symptomatic patients does not improve neurodevelopmental outcomes. Evidence for or against a survival benefit, however, is inconclusive (Escolar et al., 2005). Thus, while additional treatments that at least improve the quality of life are needed, they also must exceed a survival benchmark that is determined by survival outcome for HSCT. Further study of survival outcome in symptomatic patients treated with HSCT is needed. HSCT has significant morbidity and mortality, and while it improved survival and development in pre-symptomatic children, improvement was not statistically significant in children transplanted after onset of symptoms compared to patients who were not transplanted (Escolar et al., 2005; Musolino et al., 2014). The latter result, however, was based on a small sample size with presumably low power and without controlling for age at onset of symptoms. Thus, new information about the effect of post-symptomatic transplant therapy may result from further study using a larger sample size or a different statistical approach that allows one to control for covariates. The purpose of the current investigation is to re-assess the efficacy of HSCT in symptomatic Krabbe patients with increased power compared to previous studies. Because KD is rare, we have established a database, the World Wide Registry for Krabbe Disease (WWR), to facilitate research about this condition (Duffner et al., 2009a; Duffner et al., 2011; Duffner et al., 2012). We present here results of a population-based analysis of the long-term benefits of HSCT. We focus on children from our data registry who were transplanted after the onset of symptoms.
MATERIALS AND METHODS
Identification of Research Subjects
The WWR contains detailed phenotypic information regarding 180 infants affected by KD. (Duffner et al., 2009a; Duffner et al., 2011.). From this database, we were able to identify patients who received therapeutic transplantation after the onset of symptoms. These symptoms consisted mainly of irritability, or of slight motor signs, such as fisting of the hands (Duffner et al., 2011). We also identified controls who were matched to the transplanted subjects. Controls were matched to subjects by age of symptom onset, with multiple matches allowed. The opportunity to control precisely for age of onset of symptoms eliminates its potential confounding effect; which exists because survival time varies with age at onset (Duffner et al., 2009a; Jalal et al., 2012), as may the likelihood of a KD patient being transplanted.
The total sample consisted specifically of 16 transplanted cases, of whom 7 were either EIKD or LIKD, and 9 were LOKD cases having adolescent or adult onset. The 68 controls who were not transplanted were comprised of 55 EIKD/LIKD cases and 13 LOKD cases. Thus, the total population studied consisted of 84 symptomatic KD patients.
As part of our broad effort to follow symptomatic KD patients, a parent or caretaker of each subject of the current investigation was reached for a telephone interview that was conducted after the subject was identified as a participant in this study. These interviews therefore provided updated information distinct from that reported previously(Escolar et. al, 2005). IRB approval is in place for this protocol.
Statistical Analyses
The current investigation focused exclusively upon duration of survival after transplant. The preliminary analysis of a survival indicator variable (Alive/Dead) defined at the time of final follow up was performed by chi-squared analysis. Then, in order to better discern the effect of transplant on survival time, while accounting for censoring and controlling for age at onset of symptoms a Cox proportional hazards regression analysis was performed using the PHREG procedure of the SAS software package. The “age at transplant” for each control patient was defined to be the age at which their match was transplanted. A one-tailed test of the effect of treatment was performed, because the alternatives that treatment has no effect or a negative effect would both lead to the same recommendation to not treat. A post hoc illustration of the effect of treatment was obtained by employing Breslow’s estimators of survival functions and direct adjustment for age at onset of symptoms (SAS/STAT® 9.2 User’s Guide: The PHREG Procedure, 2008; Harrell et al., 1984). The majority of patients in our registry have the early infantile phenotype (Duffner et al., 2011). Thus, the results of the primary analysis of our total sample are dominated by EIKD patients. The relatively small number of late infantile cases or later onset cases in the study population, however, allow for an exploratory look at the question “Is the effect of transplant dependent on age at onset phenotype?”. A second Cox regression analysis with an age at onset by transplant interaction term added to the primary Cox model was performed to address this question.
RESULTS
We first questioned whether a beneficial effect of HSCT on survival (yes/no) at the time of last follow up could be discerned in this population from our registry. The earlier research that established the efficacy of transplantation indicated that the benefits of this treatment, while clearly apparent in asymptomatic patients, were, with regard to survival, not significant in symptomatic patients (Escolar et al., 2005). In our study population from the WWR that focused specifically on survival odds we found that 59 or 70% of the control group were no longer living at the time of last follow up. In contrast, only 5 of the 16 (31%) transplanted patients were deceased. This difference was statistically significant (See Table 1, p=0.004). Consequently, 25 of the controls and 11 of the transplanted patients’ survival times were censored. Date of the final follow-up interview became the censoring date for subsequent, more refined, proportional hazards regression analyses.
TABLE I.
Preliminary Chi-Squared Analysis
| TRANSPLANT | |||
|---|---|---|---|
|
| |||
| YES | NO | TOTAL | |
| Died | 5 | 54 | 59 |
| Censored | 11 | 14 | 25 |
| Total | 16 | 68 | 84 |
| P=0.0004 | |||
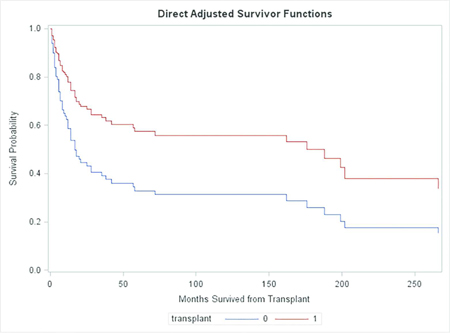
We next performed a formal analysis of hazard ratios, using the Cox proportional hazards regression model. For this analysis, the null hypothesis to be tested was that transplantation had no effect or a detrimental effect on survival. The alternative hypothesis, then, is that transplantation improved survival. Table 2 and Figure 1 show results of the Cox regression analysis. The null hypothesis was rejected (p=0.0365). Transplanted patients had a nearly 2.2 fold increase in their chance of surviving beyond each time since transplant (Table 2), with a 95% lower confidence bound of 1.07. The difference in survival curves is illustrated in Figure 1. (Note that Figure 1 shows the effect of treatment in terms of direct adjusted Breslow survival curve estimates. The plots should not be expected to have step-down drops at event times as do Kaplan-Meier estimates, and readers should not be concerned by the fact that they do not.)
TABLE II.
Cox Proportional Hazard Analysis
| Analysis of Maximum Likelihood Estimates
| |||||
|---|---|---|---|---|---|
| Name of Effecta | Estimated Effect | Standard Error of Estimate | Chi-Square Test of Significance | Pr > ChiSq | Hazard Ratio |
| Transplant | 0.77891 | 0.43443 | 3.2146 | 0.0365 | 2.179 |
| Months Symptoms Appearb | −0.02860 | 0.01056 | 7.3315 | 0.0068 | 0.972 |
Effect of transplantation measures the effect on survival in transplant versus matched non-transplant patients. It is the estimate of a parameter in the Cox proportional hazard model from which the hazard ratio relative risk is calculated.
Effect of “months symptoms appear” measures the effect of a one month difference in age at onset of symptoms between two patients who are similar in other regards. Its estimate in turn estimates the parameter in the Cox model that is related to the effect of age at onset on survival. The Hazard ratio for the effect of age at onset is calculated from this estimate.
Figure 1.

This figure shows the survival curves for transplanted patients and matched controls who were not transplanted. The difference between the levels of these curves at any given time measures the effect of transplantation at that time.
While the majority of patients in our registry have the early infantile phenotype (Duffner et al., 2011), the registry also contains information about a relatively small number of late infantile and later onset cases (Duffner et al., 2012). Hence, we turned next to the question of whether the therapeutic effect of transplantation is influenced by age at onset of KD symptoms. In this analysis, the null hypothesis is that the benefit, if any, of transplantation does not depend upon the age that symptoms first appear.
The results of our test of the interaction between the age of onset and the transplant variable on survival after transplantation showed insufficient evidence to reject the null hypothesis (p=0.71). Therefore, there is not sufficient evidence to conclude that the enhancement of survival probability due to transplantation depends on the age that symptoms first become manifest. Thus, this question needs to be addressed in a future study with a larger sample.
DISCUSSION
KD is an extremely rare disease, with an estimated incidence in the United States of 1:250,000 (Barczykowski et al., 2012). Consequently, utilization of a data registry such as the WWR enables research that would be difficult to conduct based on the experience of any single center or single care-giver (Duffner et al., 2009a; Duffner et al., 2011; Duffner et al., 2012; Jalal et al., 2012; Duffner et al., 2009b; Barczykowski et al., 2012). The current study took advantage of the capacity to identify specific patients who had undergone HSCT and to compare each of them to matched control patients who were not transplanted, but whose symptoms emerged at the same age.
Our approach therefore differs from and improves upon the earlier investigation of outcomes of symptomatic KD patients (Escolar et al., 2005). The current study used a statistical modeling approach (i.e., Cox proportional hazards modeling) that allows us to control for potential confounding effects. Age at onset of symptoms, in particular, is likely to have a confounding/biasing influence on the assessment of treatment effects if not controlled. Survival time is known to vary with age at onset of symptoms (Jalal et al., 2012; Duffner et al., 2009a) and age at onset may also have been associated with the decision to transplant or not..
Furthermore, controlling for significant covariates increases the precision with which we measure treatment effects and, therefore, increases the power of our significance test. The previous study (Escolar et al., 2005) used Kaplan-Meier log-rank analysis to determine survival probability and, thus, did not control for the potential biasing influence of age at onset of symptoms. They found no statistically significant benefit of HSCT treatment of post symptomatic patients. in contrast, when using Cox regression analysis a statistically significant effect was found (p=0.0365), with a long-term and nearly 2.2 fold enhancement of survival probability in patients who were transplanted after symptoms compared to matched controls. This result, enabled by an improved statistical approach, represents the primary finding of the current investigation.
Hence, these data provide not just the first statistically significant evidence of a benefit of transplantation on survival of symptomatic cases; but also yield an impressive, quantitative and previously undescribed measure of the magnitude of this benefit.(i.e., a 2.2 fold risk of death at any time for untransplanted compared to transplanted patients). This quantified measure of benefit of the only existing treatment for KD provides a benchmark for future treatments to achieve.
Our results also support the conclusion that patients afflicted by the later onset variants of KD benefit from transplantation even if symptomatic. But in this case the small numbers of patients having these later phenotypes do not allow a definite conclusion. It must be acknowledged that great caution is necessary in a statistical analysis applied to a small number of research subjects that fails to reject the null hypothesis. It is then best to consider the results regarding later onset variants of KD as a suggestion of the data that must await recruitment of more later onset patients for confirmation.
In contrast, the hazard ratio demonstrated by the Cox regression analysis enabled a rejection of the null hypothesis, and therefore a firm conclusion. In addition, it should be noted that establishing this enhancement of survival probability in patients who were symptomatic at the time of treatment eliminates the potential bias that would result from treatment of children who would not have developed KD symptoms.
Concern about the potential biasing effect of treating children who would otherwise remain free of symptoms is not only relevant to statistical analysis, but also to the ethics of treating asymptomatic children after newborn screening. The experience of the New York State Krabbe Consortium has indeed confirmed that there are children who fulfill the current criteria for transplantation for KD, but do not subsequently suffer from the disease. This fact was discovered when some parents of children who met the criteria for treatment declined treatment, and their children remained free of symptoms at subsequent follow-up (Wasserstein et al, 2016). These children would have been subjected unnecessarily to the potential morbidity and mortality of HSCT had they been transplanted.
It must, in addition, be emphasized that there is compelling evidence that afflicted children improve substantially if treated pre-symptomatically, while the evidence for a benefit from post-symptomatic treatment compared to no treatment is inconclusive (Escolar et al., 2005; Musolino et al., 2014). The latter question is still open. WWR data presented here provides conclusive statistical evidence for a survival benefit in symptomatic children. This analysis in no way refutes and indeed is not pertinent to the current standard of care–identification and treatment of pre-symptomatic children. We strongly affirm this standard of care In addition, since our analysis is solely statistical, any change of clinical practice will require additional prospective study of a greater number of cases.
Furthermore, we do not claim that our result should dictate the controversial question of whether a post-symptomatic patient should be transplanted or not. It does, however, provide important additional information to be considered by those who must make this difficult decision. It is important that the scientific literature provide as much relevant information as possible for consideration.
In addition, the finding of a hazard ratio establishing a decreased risk of death of 45% (100/2.2) after transplantation provides a benchmark for measuring the impact of new Krabbe therapies that may be emerging. In particular, small molecule chaperones that affect folding or localization of residual GalC protein are under study for treating KD-afflicted patients who have missense mutations of the GalC gene, representing the majority of cases (Helman et al., 2014;Wegner and Luzi, 2014). Hence, any new therapies would presumably need to improve upon either the survival ratio established by the current results, or maintain it while improving quality of life. The use of hazard ratios determined by Cox regression analysis to provide benchmarks for efficacy of treatment in improving survival is in fact well established, with use in treatment of illnesses as diverse as coronary artery disease (Harrell et al., 1984), infections (Spruance et al., 2004) and cancer (Warwick et al., 2004).
We did not here examine quality of life outcomes. Earlier studies established that there is no improvement in morbidity in patients transplanted after the emergence of symptoms. (Escolar et al., 2005; Musolino et al., 2014). Yet the very survival of these children, despite their significant burden of disability, provided an opportunity for a new quantitative analysis of the benefit of transplantation upon survival duration. The current results for the first time demonstrated a statistically significant improvement of survival in these afflicted KD patients. Further development of new treatments or improvements to the existing treatment will be needed to significantly enhance quality of life of Krabbe patients. Such studies will presumably develop therapies (Helman et al., 2014; Wegner and Luzi, 2014) to be compared to the benchmarks for survival established in the current paper and for neurodevelopmental outcomes that were established by Escolar, et al. (2005).
SIGNIFICANCE STATEMENT.
Krabbe Disease is a severe, invariably fatal disorder. Transplanted stem cells from umbilical cord blood can improve this condition, but is a risky procedure with10–20% of those treated dying from complications of treatment. The long term effects of transplantation on survival of patients with symptoms are not well understood. We showed that transplantation more than doubles the chance that these patients will survive. New and better treatments for this devastating disease will need to improve quality of life while equaling or improving this survival result. That is, the current study sets a benchmark for new treatments for post-symptomatic patients.
Acknowledgments
Contract grant sponsor: TJ Langan is the local principal investigator for the Lysosomal Disease Network (U54NS065768) a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through a collaboration between NCATS, NINDS, and NIDDK
This study was also supported by R-21 HD087818-01 from the NICHD to TJL and RLC.
We are extremely grateful for the assistance Kathleen Scott of the Hunter’s Hope Foundation for invaluable assistance with patient follow-up. The members of the New York State Krabbe Consortium must be acknowledged for freely and frankly discussing outcomes. Maureen Milligan and Barbara Craft of HJKRI provided valued assistance with the manuscript and figures.
Footnotes
CONFLICT OF INTEREST
All participants in this work declare no affiliation or financial involvement with any organization or entity with direct financial or any other interest in the subject or materials discussed here.
AUTHOR ROLES
T.J Langan and R.L. Carter designed the study. T.J. Langan, R.L. Carter, and A. Barczykowski wrote the manuscript. The results were collected by T.J. Langan, A. Barczykowski, L. Muscarella, and E.C. Pannullo. The data were analyzed by R.L Carter and J. Dare.
LITERATURE CITED
- Bajaj NP, Waldman A, Orrell R, Wood NM, Bhatia KP. Familial adult onset of Krabbe’s disease resembling hereditary spastic paraplegia with normal neuroimaging. J Neurol Neurosurg Psychiatr. 2002;72:635–638. doi: 10.1136/jnnp.72.5.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczykowski AL, Foss AH, Duffner PK, Yan L, Carter RL. Death rates in the U.S. due to Krabbe disease and related leukodystrophy and lysosomal storage diseases. Am J Med Genet A. 2012;158A:2835–2842. doi: 10.1002/ajmg.a.35624. [DOI] [PubMed] [Google Scholar]
- Carter R, Wrabetz L, Jalal K, Barczykowski A, Langan TJ. Can psychosine and galactocerebrosidase predict early infantile Krabbe disease pre-symptomatically? doi: 10.1002/jnr.23793. (See our companion paper in this issue) [DOI] [PubMed] [Google Scholar]
- Duffner PK, Jalal K, Carter RL. The Hunter’s Hope Krabbe family database. Pediatr Neurol. 2009a;40:13–18. doi: 10.1016/j.pediatrneurol.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Caggana M, Orsini JJ, Wenger DA, Patterson MC, Crosley CJ, Kurtzberg J, Arnold GL, Escolar ML, Adams DJ, Andriola MR, Aron AM, Ciafaloni E, Djukic A, Erbe RW, Galvin-Parton P, Helton LE, Kolodny EH, Kosofsky BE, Kronn DF, Kwon JM, Levy PA, Miller-Horn J. Newborn screening for Krabbe disease: the New York State model. Pediatr Neurol. 2009b;40:245–252. doi: 10.1016/j.pediatrneurol.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Jalal K, Yan L, Kay DM, Carter RL. Early infantile Krabbe disease: results of the world-wide Krabbe registry. Pediatr Neurol. 2011;45:141–148. doi: 10.1016/j.pediatrneurol.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Kay DM, Jalal K, Yan L, Abdelhalim A, Gill S, Gill AL, Carter RL. Later onset phenotypes of Krabbe disease: results of the world-wide registry. Pediatr Neurol. 2012;46:298–306. doi: 10.1016/j.pediatrneurol.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Vanderver A. Disease specific therapies in leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2014;114:527–536. doi: 10.1016/j.ymgme.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg B, Sourander P, Svennerholm L. Diagnosis of Krabbe’s infantile leucodystrophy. J Neurol Neurosurg Psychiatr. 1963;26:195–198. doi: 10.1136/jnnp.26.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell FE, Lee KL, Califf RM, Pryor DB, Rosati RA. Regression modelling strategies for improved prognostic predication. Stat Med. 1984;3:142–152. doi: 10.1002/sim.4780030207. [DOI] [PubMed] [Google Scholar]
- Helman G, Van Haren K, Bonkowsky JL, Bernard G, Pizzino A, Braverman N, Suhr D, Patterson MC, Fatemi SA, Leonard J, van der Knaap MS, Back SA, Damiani S, Goldman SA, Takanohashi A, Petryniak M, Rowitch D, Messing A, Wrabetz L, Schiffmann R, Eichler F, Escolar ML, Vanderver A. Disease specific therapies in leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2014;114:527–536. doi: 10.1016/j.ymgme.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal K, Carter R, Yan L, Barczykowski A, Duffner PK. Does galactocerebrosidase activity predict Krabbe phenotype? Pediatr Neurol. 2012;47:324–329. doi: 10.1016/j.pediatrneurol.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Kemper AR, Knapp AA, Green NS, Comeau AM, Metterville DR, Perrin JM. Weighing the evidence for newborn screening for early-infantile Krabbe disease. Genet Med. 2010;12:539–543. doi: 10.1097/GIM.0b013e3181e85721. [DOI] [PubMed] [Google Scholar]
- Krivit W, Shapiro EG, Peters C, Wagner JE, Cornu G, Kurtzberg J, Wenger DA, Kolodny EH, Vanier MT, Loes DJ, Dusenbery K, Lockman LA. Hematopoietic Stem-Cell Transplantation in Globoid-Cell Leukodystrophy. N Engl J Med. 1998;338:1119–1127. doi: 10.1056/NEJM199804163381605. [DOI] [PubMed] [Google Scholar]
- Lyon G, Hagberg B, Evrard P, Allaire C, Pavone L, Vanier M. Symptomatology of late onset Krabbe’s leukodystrophy: the European experience. Dev Neurosci. 1991;13:240–244. doi: 10.1159/000112167. [DOI] [PubMed] [Google Scholar]
- Martin PL, Carter SL, Kernan NA, Sahdev I, Wall D, Pietryga D, Wagner JE, Kurtzberg J. Results of the cord blood transplantation study (COBLT): outcomes of unrelated donor umbilical cord. Biol Blood Marrow Transplant. 2006;2:184–94. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Musolino PL, Lund TC, Pan J, Escolar ML, Paker AM, Duncan CN, Eichler FS. Hematopoietic stem cell transplantation in the leukodystrophies: a systematic review of the literature. Neuropediatrics. 2014;45:169–174. doi: 10.1055/s-0033-1364179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini JJ, Morrissey MA, Slavin LN, Wojcik M, Biski C, Martin M, Keutzer J, Zhang XK, Chuang WL, Elbin C, Caggana M. Implementation of newborn screening for Krabbe disease: population study and cutoff determination. Clin Biochem. 2009;42:877–884. doi: 10.1016/j.clinbiochem.2009.01.022. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Chen YQ, Wenger DA. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum Mol Genet. 1995;4:1285–1289. doi: 10.1093/hmg/4.8.1285. [DOI] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS/STAT® 9.2 User’s Guide. Cary, NC: SAS Institute Inc; 2008. [Google Scholar]
- Shapiro E, Krivit W, Lockman L, Jambaqué I, Peters C, Cowan M, Harris R, Blanche S, Bordigoni P, Loes D, Ziegler R, Crittenden M, Ris D, Berg B, Cox C, Moser H, Fischer A, Aubourg P. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356(9231):713–8. doi: 10.1016/S0140-6736(00)02629-5. [DOI] [PubMed] [Google Scholar]
- Spruance SL, Reid JE, Grace M, Samore M. Hazard ratio in clinical trials. Antimicrob Agents Ch. 2004;48:2787–2792. doi: 10.1128/AAC.48.8.2787-2792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Suzuki Y. Globoid cell leucodystrophy (Krabbe’s disease): deficiency of galactocerebrosidase beta-galactosidase. Proc Natl Acad Sci USA. 1970;66:302–309. doi: 10.1073/pnas.66.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K. Biochemical pathogenesis of genetic leukodystrophies: comparison of metachromatic leukodystrophy and globoid cell leukodystrophy (Krabbe’s disease) Neuropediatrics. 1984;15:32–36. doi: 10.1055/s-2008-1052380. [DOI] [PubMed] [Google Scholar]
- Turgeon CT, Orsini JJ, Sanders KA, Magera MJ, Langan TJ, Escolar ML, Duffner P, Oglesbee D, Gavrilov D, Tortorelli S, Rinaldo P, Raymond K, Matern D. Measurement of psychosine in dried blood spots - a possible improvement to newborn screening programs for Krabbe disease. J Inherit Metab Dis. 2015;38:923–929. doi: 10.1007/s10545-015-9822-z. [DOI] [PubMed] [Google Scholar]
- Warwick J, Tabàr L, Vitak B, Duffy SW. Time-dependent effects on survival in breast carcinoma. Cancer. 2004;100:1331–1336. doi: 10.1002/cncr.20140. [DOI] [PubMed] [Google Scholar]
- Wasserstein M, Andriola M, Arnold G, Aron A, Duffner P, Erbe RW, Escolar ML, Estrella L, Galvin-Parton P, Iglesias A, Kay DM, Kronn DF, Kurtzberg J, Kwon JM, Langan TJ, Levy PA, Naidich TP, Orsini JJ, Pellegrino JE, Provenzale JM, Wenger DA, Caggana M New York State Krabbe consortium. Clinical outcomes of children with abnormal newborn screens for Krabbe disease in New York State. doi: 10.1038/gim.2016.35. Submitted for publication in NEJM. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat. 1997;10:268–279. doi: 10.1002/(SICI)1098-1004(1997)10:4<268::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Luzi P, Rafi MA. Krabbe disease: are certain mutations disease-causing only when specific polymorphisms are present or when inherited in trans with specific second mutations? Mol Genet Metab. 2014;111:307–308. doi: 10.1016/j.ymgme.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Luzi P. Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease. 5. Waltham, MA: Elsevier; 2014. Krabbe Disease :Globoid Cell Leukodystrophy; pp. 337–345. [DOI] [Google Scholar]