

Abstract
BACKGROUND
Growing animal evidence demonstrates that prolonged exposure to propofol during brain development induces widespread neuronal cell death, but there is little information on the role of astrocytes. Astrocytes can release neurotropic growth factors such as brain-derived neurotropic factor (BDNF), which can exert the protective effect on neurons in paracrine fashion. We hypothesize that during propofol anesthesia, BDNF released from developing astrocytes may not be sufficient to prevent propofol-induced neurotoxicity.
METHODS
Hippocampal astrocytes and neurons isolated from neonatal Sprague Dawley rats were exposed to propofol at a clinically relevant dose of 30 μM or dimethyl sulfoxide as control for 6 h. Propofol-induced cell death was determined by propidium iodide (PI) staining in astrocyte-alone cultures, neuron-alone cultures, or co-cultures containing either low or high density of astrocytes [1:9 or 1:1 ratio of astrocytes to neurons (ANR), respectively)]. The astrocyte-conditioned medium was collected 12 h following propofol exposure and measured by protein array assay. BDNF concentration in astrocyte-conditioned medium was quantified using ELISA. Neuron-alone cultures were treated with BDNF, tyrosine receptor kinase B (TrkB) inhibitor Cyclotraxin-B, glycogen synthase kinase 3β (GSK3β) inhibitor CHIR99021, or mitochondrial fission inhibitor Mdivi-1 prior to propofol exposure. Western blot was performed for quantification of the level of Akt and GSK3β. Mitochondrial shape was visualized through TOM20 staining.
RESULTS
Propofol increased cell death in neurons by 1.8 fold [% of PI-positive cells (PI%) =18.6, 95% CI=15.2–21.9, P<0.05], but did not influence astrocyte viability. The neuronal death was attenuated by a high ANR (1:1 co-cultures) [fold change (FC) =1.17, 95% CI=0.96–1.38, P<0.05), but not with a low ANR (1:9 co-cultures)] (FC=1.87, 95% CI=1.48–2.26, P>0.05). Astrocytes secreted BDNF in a cell density-dependent way and propofol decreased BDNF secretion from astrocytes. Administration of BDNF, CHIR99021, or Mdivi-1 significantly attenuated the propofol-induced neuronal death and aberrant mitochondria in neuron-alone cultures (FC=0.8, 95% CI=0.62–0.98; FC=1.22, 95% CI=1.11–1.32; FC=1.35, 95% CI=1.16–1.54 respectively, P<0.05) and the co-cultures with a low ANR (1:9) (FC=0.85, 95% CI=0.74–0.97; FC=1.08, 95% CI=0.84–1.32; FC=1.25, 95% CI=1.1–1.39 respectively, P<0.05) Blocking BDNF receptor or Akt activity abolished astrocyte-induced neuroprotection in the co-cultures with a high ANR (1:1).
CONCLUSIONS
Astrocytes attenuate propofol-induced neurotoxicity through BDNF-mediated cell survival pathway suggesting multiple neuroprotective strategies such as administration of BDNF, astrocyte-conditioned medium, decreasing mitochondrial fission, or inhibition of GSK3β.
INTRODUCTION
Propofol (2,6-diisopropylphenol) is a short-acting intravenous anesthetic agent that has been widely used in pediatric anesthesia. However, several epidemiological studies demonstrated that there was a link between an early anesthetic exposure and a potential risk for long-term learning and memory disabilities,1,2 raising serious concern about the safety of general anesthesia. Growing evidence in rodents and non-human primates also showed that prolonged exposure of developing animals to propofol potentiated γ-aminobutyric acid types A receptor (GABAAR) activation3,4 and led to widespread neuronal cell death during the brain growth spurt through intrinsic or extrinsic pathways.5–7 However, the detailed mechanisms that underlie the propofol-induced neurotoxic effects and the rational strategies for neuroprotection are lacking.
A recent study in fetal or neonatal non-human primates showed that propofol alone induced significant apoptosis of neurons and oligodendrocytes, but not astrocytes.8 Astrocytes, the largest population of glial cells, play a pivotal role in early neurodevelopment in the central nervous system (CNS) partially through synthesizing and releasing growth factors.9,10 Brain-derived neurotrophic factor (BDNF) is a neurotrophin family of growth factor that can be largely secreted by astrocytes11,12 and critical for neurogenesis, learning, and memory.13–15 Additionally, exposure of neonatal rat brains to propofol induced neurotrophin disturbance, such as downregulation of BDNF.16 However, the cellular mechanisms linking BDNF to its potential signaling pathways in propofol-induced neurotoxic effects are largely unknown.
It has been shown that the postnatal ratio of neuronal and non-neuronal cells in rat CNS changes over time.17 Under the physiological condition, the major type of cells in cortex and hippocampus are neurons (~ 90% neurons vs. 10% non-neurons) at the first postnatal week. Then, neurons are gradually eliminated and replaced by non-neuronal cells.17,18 Compared with developing brains, the physiological ratio of astrocytes to neurons is higher (around 1:3 to 1:1) in adult animals.19,20 Thus, in this study we isolated hippocampal astrocytes and neurons from neonatal rats. We used the low-density (1:9) and high-density (1:1) of astrocytes to neurons co-culture models to mimic neonatal and adult rat brain physiological conditions in vitro, respectively. We hypothesized that insufficient levels of astrocyte-derived BDNF would contribute to propofol-induced neuron death through Akt/glycogen synthase kinase 3β (GSK3β)/mitochondrial fission pathway. An understanding of the underlying mechanisms may help to develop effective novel protective strategies for avoiding the anesthetic-induced neurotoxicity.
MATERIALS AND METHODS
Here is the outline of experimental design:
Analyzing the effect of propofol on the viability of cultured astrocytes and neurons isolated from neonatal rat hippocampus. Neurons or astrocytes alone were exposed to either propofol at clinically relevant concentrations (3, 10, or 30 μM) or DMSO as a vehicle control.
Investigating the role of astrocytes in propofol-induced neuron death. Neurons were co-cultured with low or high density of astrocytes (the ratios of astrocytes/neurons were1:9 and 1:1, respectively) prior to treatment with propofol (30 μM). The neuron viability was then analyzed.
Examining whether astrocyte-secreted growth factors contribute to the protection against propofol-induced neuron death. Neurons were exposed to propofol in the presence or absence of astrocyte-conditioned medium collected from higher density of astrocytes. Meanwhile, BDNF was added to neurons alone culture or co-cultures containing low density of astrocytes prior to propofol exposure. The protective effect of increasing density of astrocytes or exogenous astrocyte-derived BDNF was determined by analysis of cell viability.
-
Analyzing the role of increased mitochondrial fission in propofol-induced neuron death. The mitochondrial morphology was assessed using TOM20 staining. Mitochondrial fission inhibitor Mdivi-1 was used to determine the functional role of mitochondrial fission in propofol-induced neuron death.
The detailed individual approach used in this study is described below.
Co-culture of Hippocampal Neurons with Astrocytes
All rat experiments described were approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin. Neurons were isolated for primary culture21 with minor modifications. Briefly, hippocampi were isolated from 1-day-old Sprague-Dawley rats in cold Hanks’ balanced salt solution (HBSS)-HEPES buffer. The isolated hippocampi were then digested with 0.05% Trypsin-EDTA (wt/vol) (Invitrogen Corporations, Carlsbad, CA, USA) for 15 min at 37°C with gentle shaking. The digestion was terminated by washing cells three times with neuron growth medium containing Neurobasal medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (vol/vol) (Invitrogen) and 0.5 mM glutamine (Invitrogen). The cells were then gently triturated in neuron growth medium and immediately plated on the dishes pre-coated with Matrigel [2 μg/mL in Dulbecco’s Modified Eagle Medium (DMEM)/F12 (Gibco, Grand Island, NY, USA)] at the density of 1×105 cells/cm2. Following 2 h of plating, the culture media was replaced with serum-free neuronal growth medium containing Neurobasal medium supplemented with 2% B27 (Invitrogen), 0.5 mM Glutamine and [Penicillin (100 U/ml) and Streptomycin (100 μg/ml)] (Invitrogen). Twenty-four hours later, C-Arabinoside at a concentration of 5 μM (Sigma-Aldrich, St. Louis, MO, USA) was added to the medium for inhibition of cell proliferation. Half the media was changed every two days. It has been shown that hippocampal neurons are more sensitive to propofol on day 4 as compared to that on day 8 in vitro.4 Thus, 4-day-old neurons (3 days after isolation) were used for both neuron and co-culture experiments.
Astrocytes were isolated from 1-day-old Sprague-Dawley rats following a previous protocol 22 with minor modifications. The hippocampi were incubated in digestion buffer containing Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen), 20 unit/mL papain (Worthington Biochemical Corp., Freehold, NJ) and 1.5×10−4 g/ml L-cystine (Sigma-Aldrich) for 45 min at 37°C with gentle shaking. Digestion was stopped by washing cells three times with astrocyte growth medium containing DMEM supplemented with 10% FBS (vol/vol). The cells were then plated on dishes pre-coated with 0.1% poly-L-lysine (wt/vol) solution (Sigma-Aldrich) at the density of 1×105 cells/cm2. After 24 h, the medium was replaced with astrocyte growth media containing DMEM supplemented with 10% FBS and [Penicillin (100 unit/ml) and Streptomycin (100 μg/ml)]. The media were changed every two days. Prior to propofol exposure, the astrocyte culture dishes were shaken for 4 h at 300 rpm at 37°C to remove contaminated cells. Four to seven-day-old astrocytes were used for the following neuron and co-culture experiments.
For co-culture experiments, 3 days after cell isolation, neurons plated onto the bottom of the culture plates were co-cultured with astrocytes plated onto the transwell inserts (Corning, Tewksbury, MA, USA). The transwell co-culture system allows study of paracrine effects of one type of cells on another type of cells by physically separating two different types of cells. Astrocytes were co-cultured with neurons at two different ratios (the astrocyte to neuron ratio was either 1:9 or 1:1) to mimic neonatal and adult rat brain physiological conditions in vitro, respectively. The cell viability of co-cultured neurons was analyzed 12 h following 6 h of propofol exposure.
Characterization of Astrocytes and Neurons by Immunofluorescence Staining and Confocal Microscopy
Neurons and astrocytes were seeded on Matrigel pre-coated glass cover slips at the density of 5×104 cells/cover slip.23,24 After 4 days in culture, the cover slips were fixed with 1% paraformaldehyde for 30 min at room temperature and treated with 0.5% Triton X-100 (Sigma-Aldrich) for 15 min followed by incubation with 10% donkey serum for 20 min to block unspecific proteins. After blocking, neurons were incubated with the primary antibody against neuron-specific marker mouse microtubule-associated protein (MAP)2 (Abcam, Cambridge, MA, USA) while astrocytes were incubated with the primary antibody against astrocyte-specific marker rabbit glial fibrillary acidic protein (GFAP) (Abcam) for 1 h at 37°C in a humidified incubator. The cells were then incubated with the secondary antibody Alexa Fluor 594 anti-mouse or 488 anti-rabbit immunoglobulin G (Invitrogen) for 1 h. Finally, the nuclei of neurons and astrocytes were labeled with Hoechst 33342 dye for 15 min. The cover slips were mounted onto the glass slides and imaged using the laser-scanning confocal microscope (Nikon Eclipse TE 2000-U, Nikon Inc., Melville, NY, USA).
Propofol Exposure and Experimental Protocol
In children, plasma concentration of propofol to maintain and induce pediatric anesthesia is much lower (5.6–56 μM~1–10 μg/ml).25–27 Propofol is a highly lipophilic agent and therefore the concentration of propofol is relatively high (22–112 μM/4–20 μg/ml) in lipid-rich tissues such as the brain.28–32 Different doses of propofol have been reported to induce neurotoxicity. For instance, Pearn et al. observed that 3 μM of propofol induced the significant increase in cleaved caspase-3 expression in neurons isolated from BALBc mice.5 Sall et al. showed that exposure of neonatal rat hippocampal neurons to as low as 5 μM of propofol resulted in increased neuron death.33 However, only a high dose of propofol (112 μM) could lead to cell death of 14-day rat cortical neurons.34 The various doses of propofol that were able to induce neuron death in different studies may be due to the complexity factors among animal species, such as differences in ages, brain subregions, exposure duration, cell culture conditions and endpoint measurements.
In our pilot studies we tested the toxic effect of several doses of propofol (3–100 μM) on the cultured neonatal rat hippocampal neurons and astrocytes. The data showed that 6 h of propofol exposure at the concentrations of 30–100 μM induced neuron death, but did not influence astrocyte viability. Thus, 30 μM propofol (6 h) was used in the mechanism study. Propofol (Sigma-Aldrich) was prepared as stock solutions at the concentrations of 3, 10, and 30 mM in dimethyl sulfoxide (DMSO) (Sigma-Aldrich), which were then diluted to 1000 times in neuronal growth medium. An equal volume of DMSO was used as the vehicle control. Following 6h propofol exposure, the medium was washed and replaced with fresh neuronal growth medium in both DMSO-treated control and propofol-treated cultures. The cell death in neurons and astrocytes was assessed 12 h after propofol exposure. LY290004 (10 μM, PI3K/Akt inhibitor), Cyclotraxin-B (25 μM, BDNF receptor TrkB inhibitor), CHIR 99021 (10 μM, GSK3β inhibitor) and BDNF (100 ng/mL) were administered in the presence of propofol. Mdivi-1 (25 μM, mitochondrial fission inhibitor) was given 30 min prior to propofol exposure. The concentrations of the inhibitors described above were determined based on others’ reports35,36 and our pilot studies (data not shown).
Measurement of Cell Death by Propidium Iodide Staining
Cell death was determined by propidium iodide (PI) and Hoechst 33342 (Sigma-Aldrich) double staining.4 PI is a DNA-specific red fluorescence dye that is only permeant to dead cells while Hoechst 33342 is a blue fluorescence DNA-specific dye used for staining nuclei of both living and dead cells. Briefly, 12 h after the propofol exposure, neurons and astrocytes were washed with phosphate-buffered saline (PBS) and incubated with Hoechst 33342 (40 μM) diluted in neuron basal medium for 15 min at 37°C. The medium was then replaced with PI (50 μM)-contained neuron basal medium at 37°C for 10 min. Fluorescent signals in the culture were imaged under the Inverted Fluorescence Microscope (Olympus, Japan) immediately after staining. Five samples and at least twenty images in each group were randomly chosen for assessment. The numbers of PI and Hoechst 33342 were counted using ImageJ software (National Institutes of Health, Bethesda, Maryland). Cell death was expressed as the percentage of PI-positive dead cells out of Hoechst 33342-positive total cells. In all experiments, cells were imaged and analyzed by an observer blinded to the experimental conditions.
Detection of Soluble Factors Released from Astrocytes using Antibody Arrays
The astrocyte-derived soluble factors in the conditioned medium were assessed using L-Series Rat Antibody Array 90 (RayBiotech, Norcross, GA, USA) following manufacture’s protocol. This array screens 90 paracrine proteins including growth factors, cytokines, chemokines, angiogenic factors, and proteases soluble receptors released in astrocyte-conditioned medium. The layout of different proteins in the membrane is available at the website: http://www.raybiotech.com/files/manual/Antibody-Array. Briefly, astrocytes (1×105 cells/cm2) were exposed to 30 μM propofol or equal volume of DMSO for 6 h followed by 12 h incubation in neuron basal medium. The medium was collected and concentrated five times and then loaded in the dialysis column. After the overnight dialysis, the protein concentration of dialyzed sample was determined using Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA, USA). The protein (5 mg/sample) was labeled with biotin for 30 min and went through the spin column for purification. The biotin-labeled samples were loaded on the array membrane which was preprinted with capture antibodies, and incubated for 2 h at room temperature. The membrane was then washed and incubated with horseradish peroxidase (HRP)-conjugated streptavidin for 2 h with gentle shaking. The intensity of dot immunoblot signals were detected using a chemiluminescence imaging system and quantified by densitometry using ImageJ software. The data were normalized by loading control (p-3b) and expressed as % control.
ELISA Analysis of BDNF Concentration in Astrocyte-Conditioned Medium
The concentration of BDNF in astrocyte-conditioned medium was measured using rat BDNF sandwich enzyme-linked immunosorbent assay (ELISA) Assay Kit (ChemiKine Millipore, Billerica, MA, USA) following manufacture’s protocol. Briefly, 1×105 cells/cm2 and 1.1×104 cells/cm2 astrocytes were cultured in 30-mm dishes and considered as the high- and low-density of astrocytes, respectively). The medium was collected 12 h after incubation with neuron basal medium, and was subsequently concentrated ten times. Fifty μl samples were assayed in the 96-well plates coated with mouse monoclonal antibody against BDNF. BDNF- specific biotin-conjugated second antibodies were added followed by the streptavidin-enzyme and substrate incubation. The absorbance of each sample was measured at 450 nm using Epoch spectrophotometer (BioTek, Winooski, VT, USA). Protein concentrations in cell lysates were determined with Bio-Rad Protein Assay Kit. BDNF concentration in the astrocyte-conditioned medium was normalized by protein concentration and expressed as fold change of low-density astrocytes.
Measurement of Mitochondrial Fission by Confocal Microscopy
Following 6 h of propofol exposure, neurons on the cover slips from neuron-alone culture and 1:9 neuron-astrocyte co-culture were washed with PBS and fixed with 1% paraformaldehyde (wt/vol) for 30 min at room temperature. The shape of mitochondria was visualized by immunofluorescence staining (as described above) with the primary antibody against rabbit mitochondrial preprotein translocases of the outer membrane 20 (TOM20) (Santa Cruz, Dallas, TX, USA). The cover slips were then incubated with secondary antibodies Alexa Fluor 488 anti-rabbit immunoglobulin G (Invitrogen). The nuclei of neurons were labeled with Hoechst 33342 dye. The mitochondria were imaged using the laser-scanning confocal microscope (Nikon Eclipse TE 2000-U, Nikon Inc., Melville, NY, USA). Every single mitochondrial shape within neuronal processes (15 cells in each group) was evaluated by calculation of two factors: aspect ratio and form factor using ImageJ 1.41o software as described previously.24,37 Briefly, aspect ratio is a measure of the length of the mitochondria and is calculated as the ratio of the major to minor axes of the ellipse while the form factor is a measure of the degree of mitochondrial branching and is calculated using the equation: Form factor = perimeter2/4π × area. Both parameters have a minimal value of 1 representing a perfect circle. Healthy, elongated and interconnected mitochondria have a high form factor and aspect ratio while fragmented, discontinuous mitochondria have low form factor and aspect ratio values. In all experiments, neuronal cells were imaged and analyzed by an observer blinded to the experimental conditions.
Western Blot
Following 6 h of propofol exposure, neurons were collected and lysed in N-PER Neuronal Protein Extraction Reagent (Pierce, Thermo Scientific) containing protease inhibitor and phosphatase inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). Lysates were centrifuged at 10,000 g for 15 min at 4°C. The supernatant was collected and the concentration of each sample was determined using Bio-Rad Protein Assay Kit. The samples were denatured by boiling for 5 min at 97°C. A protein quantity of 15 μg/lane was loaded and separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins in the gel were then transferred to polyvinylidene fluoride (PVDF) membrane. The membrane was pre-blocked with Starting Block Buffer (Thermo Fisher Scientific, Waltham, MA, USA) and then incubated with primary antibodies against rabbit phosphorylated Akt (p-Akt) (Ser-473), rabbit anti-Akt, rabbit anti-phosphorylated GSK3β (p-GSK3β) (Ser9), rabbit anti-GSK3β (Cell Signaling, Danvers, MA, USA), and β-actin (Abcam, Cambridge, MA, USA). After washing with Tris-buffered saline with Tween-20 (TBS-T), the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (cell signaling) for 1 h at room temperature. The densities of immunoblots were quantified using ImageJ software and the data were expressed as % control.
Statistical Analysis
All data were expressed as individual values, mean values ± standard deviation (SD) and 95% confidence interval (CI). Statistical analysis was performed using GraphPad Prism® version 6.0 (GraphPad Software Inc., La Jolla, CA). The statistically significant differences of data between two groups were tested by Student t-test. The one-way analysis of variance (ANOVA) and Tukey’s post-hoc test were used for testing multiple groups. A level of P<0.05 was considered to be statistically significant. It has been reported that propofol at the concentrations ranging from 3 μM to 112 μM induced developmental neurotoxicity in animal cell model.5,33,34 Thus, we first performed a pilot study and treated the cultured astrocytes or neurons with various concentrations of propofol in order to find the best representative concentration of propofol used for the neurons and astrocytes co-culture studies. We found in our pilot study (n=4/group) that the neuron death in control and protocol (30 μM) groups was 10.2 ± 1.31 and 18.6 ± 2.08, respectively. Accordingly, a study with sample size >=4/group would be sufficient to detect the difference between the control and propofol groups at significant level of 0.05 with power of 0.8. In the current study we selected the sample size of 5 to ensure the power of study.
RESULTS
Propofol Dose-Dependently Induced Cell Death in Neurons, but not in Astrocytes
To determine whether propofol has toxic effect on developing astrocytes and neurons, cell viability was measured using PI staining. Propofol exposure did not significantly alter the numbers of PI-positive dead astrocytes (PI%=2.02, 95% CI=1.75–2.3 in DMSO control; PI%=1.93, 95% CI=1.11–2.76 in 3 μM propofol; PI%=1.92, 95% CI=1.21–2.62 in 10 μM propofol; PI%=2.06, 95% CI=1.67–2.46 in 30 μM propofol, P<0.05, Figure 1A). In contrast, the numbers of PI-positive dead neurons were significantly increased by 1.5 and 1.8 fold after 10 and 30 μM propofol exposures compared with control (PI%=15.5, 95% CI=10.7–20.3 in 10 μM propofol; PI%=18.6, 95% CI=15.3–21.9 in 30 μM propofol, P<0.05, Figure 1B), respectively. In addition, we observed that the baseline dead astrocytes were lower than the dead neurons (2% in control astrocytes vs. 10% in control neurons) (Figure 1A–b & B–b).
Figure 1.
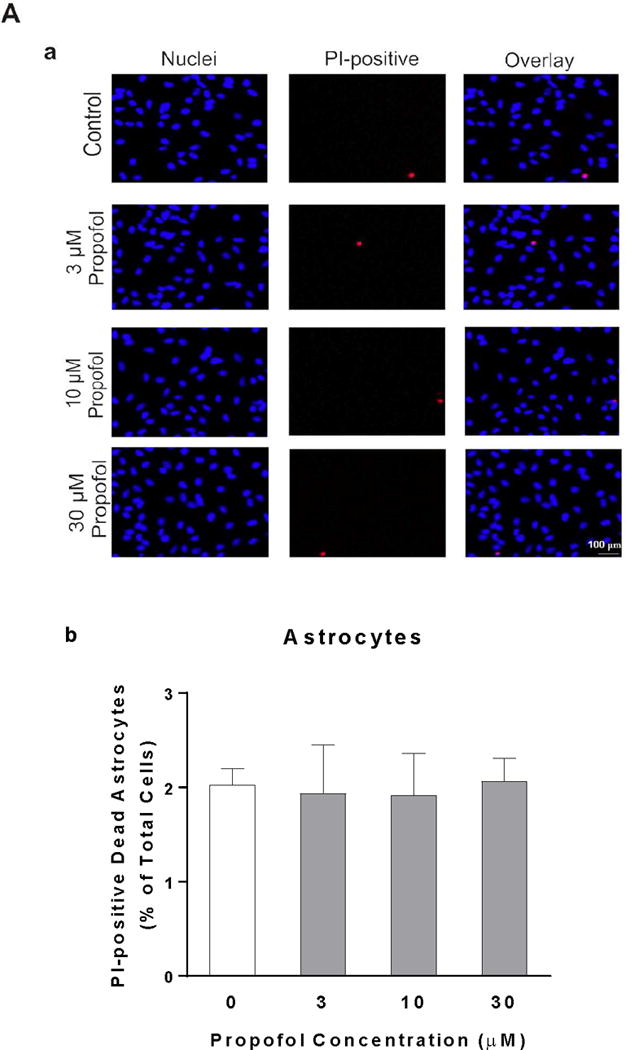
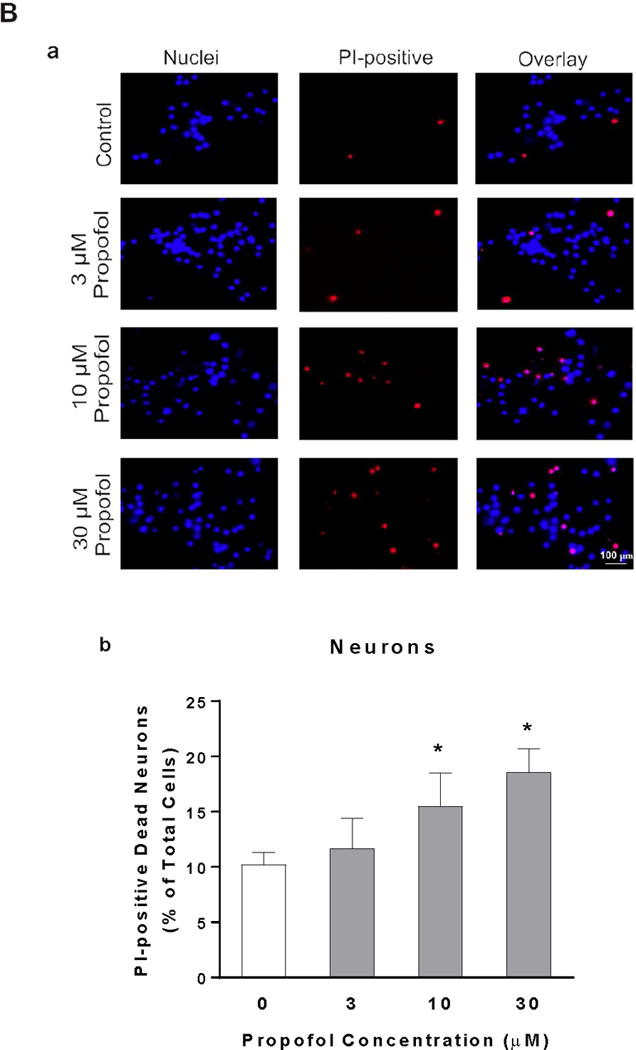
Neurons are more vulnerable than astrocytes in response to propofol. Astrocytes and neurons were exposed to propofol or dimethyl sulfoxide (DMSO) as a vehicle control for 6 h. Cell death was measured 12 h following propofol exposure and expressed as a percentage of propidium iodide (PI)-positive dead cells out of total cells. (A) The effects of different doses of propofol or DMSO on cell viability of astrocytes. (A-a) The fluorescence images of astrocytes co-stained with PI and Hoechst 33342. The PI-positive red signals indicate dead cells and Hoechst 33342-postive blue signals represent total cells. (A-b) Propofol at any doses did not significantly affect cell viability of astrocytes compared with control. (B) Propofol dose-dependently increased the cell death in neurons. (B-a) The fluorescence images of PI- and Hoechst 33342-stained neurons. (B-b) Propofol at the concentrations of 10 and 30 μM induced significant neuron death compared with control. n=4. *P<0.05 vs. Control.
Conditioned Medium from High-Density of Astrocytes Attenuates Propofol-Induced Neuron Death
To determine the contribution of astrocytes to propofol-induced neurotoxicity, neurons were co-cultured with two different densities of astrocytes (the ratios of astrocytes/neurons were either 1:1 or 1:9) prior to 30 μM propofol exposure in transwell co-culture system. Astrocytes cocultured with a higher astrocyte to neuron ratio (1:1) significantly reduced the propofol-induced neuron death [fold change (FC) =1.17, 95% CI=0.96–1.38, P<0.05, Figure 2A] while astrocytes cocultured with a lower astrocyte to neuron ratio (1:9) failed to protect the neurons against the propofol stress (FC=1.87, 95% CI=1.48–2.26, P>0.05, Figure 2A). To confirm whether the protective effect of astrocytes on neurons resulted from the secretion in the astrocyte-conditioned medium, neurons were exposed to 30 μM propofol in the presence or absence of astrocyte-conditioned medium collected from higher density of astrocytes. The conditioned medium from cultured astrocytes decreased propofol-induced neuron death by 38% (FC=1.10, 95% CI=0.77–1.43, P<0.05, Figure 2B).
Figure 2.
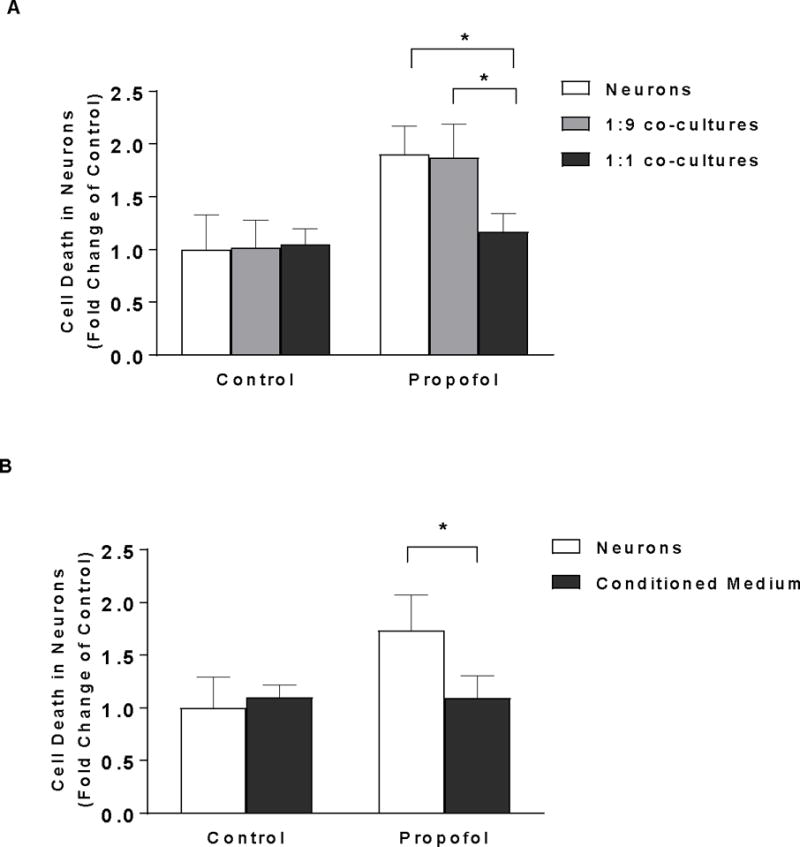
Astrocytes protect propofol-induced neuron death through conditioned medium. (A) Astrocytes cell number-dependently rescued the propofol-induced cell death in neurons when co-cultured with neurons. Neurons were co-cultured with two different densities of astrocytes (the ratios of astrocytes/neurons were either 1:1 or 1:9) in transwell plates that physically separated astrocytes from neurons. The co-culture was then exposed to 6 h of 30 μM propofol or DMSO as a control. Cell death was measured 12 h following propofol exposure and expressed as fold change of control. Co-cultured astrocytes with the higher ratio against neuron cell counts (1:1) but not with the lower ratio (1:9) attenuated the propofol-induced neuron death. n=5. *P<0.05. (B) Conditioned medium collected from the higher density of astrocytes attenuated the propofol-induced neuron death. Neurons were exposed to 6 h of 30 μM propofol in the presence or absence of astrocyte conditioned medium. Cell death was measured 12 h following propofol exposure and expressed as fold change of control. n=4. *P<0.05.
Astrocytes Secrete Many Soluble Factors into the Medium and Astrocyte-Secreted Soluble Factors Including BDNF in Conditioned Medium are Downregulated by Propofol
To dissect the mechanisms of attenuation of propofol-mediated neuron death conferred by high-density astrocyte-conditioned medium shown in Figure 3, 90 protein expressions in the conditioned medium collected from DMSO- or propofol-treated astrocytes were analyzed using dot blot array. Among 90 proteins analyzed, 34 soluble proteins were detected in both DMSO- and propofol-treated astrocyte-conditioned medium (Figure 3A-a). These 34 proteins include growth factors (BDNF, VEGF-C, PDGF-AA, and TGF-β1), cytokines [interleukin (IL)-1β, IL-6, and IL-10], chemokines (MIP-1 alpha), neurotransmitters (Orexin A), transmembrane receptors (CXCR4, NGFR, and TLR4), matricellular proteins (MMP-2), and etc. (Figure 3A-b). Of the 34 paracrine factors, propofol significantly reduced the level of nine astrocyte-released growth factors and cytokines. Propofol decreased the release of growth factors BDNF, VEGF-C, PDGF-AA, and TGF-β1 by 47.4%, 40.9%, 76.6%, and 74.3%, respectively, compared with DMSO control. The secreted cytokines such as IL-2, IL-3, IL-4, IL-5, and IL-6 were downregulated by propofol by 67.1%, 78.2%, 65.1%, 75.6%, and 71.9% (n=4/group). These nine downregulated paracrine factors have been shown to play important roles in the neuron survival, learning, and memory 38–44. Specifically, BDNF is directly involved in neuron survival.44
Figure 3.

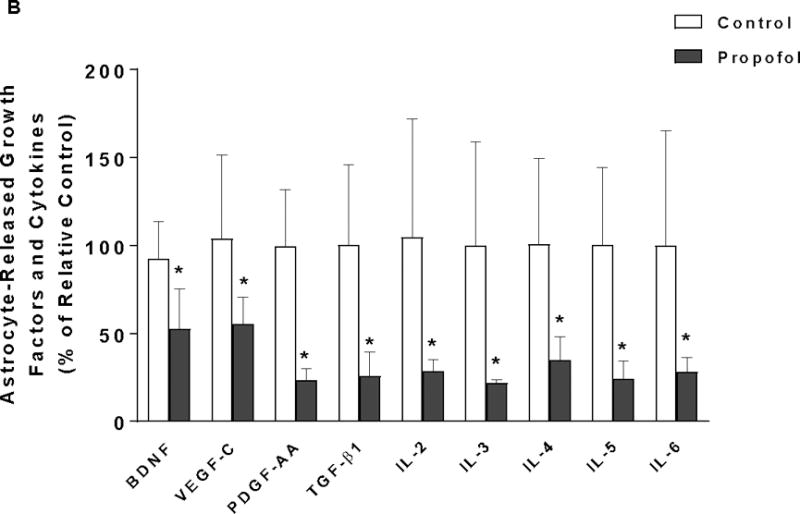
Astrocyte-derived growth factors and cytokines in conditioned medium are downregulated by propofol. (A) Analysis of paracrine factors in the conditioned medium from DMSO- or propofol-treated astrocytes using dot blot array. (A-a) The representative images of dot blot analysis of 90 soluble protein profiles in astrocyte-conditioned medium following propofol and DMSO exposure. The each double black dot indicates the individual protein that could be detected. There were 34 soluble factors detected. (A-b) The table lists the name of the 34 soluble factors [shown as black dots in (a)] that were released from both propofol- and DMSO-treated astrocytes. The protein array map refers to RayBiotech web http://www.raybiotech.com/files/manual/Antibody-Array. (B) Among 34 detected soluble factors, propofol downregulated the secretion of nine key growth factors and cytokines that are important for neuron survival [e.g., brain-derived neurotrophic factor (BDNF) was indicated in the protein array maps] from astrocytes as compared to DMSO control. Conditioned medium was collected 12 h following 6 h of 30 μM propofol exposure. The protein expression in conditioned medium was normalized as % of p-3b internal control using dot analysis. n=4. *P<0.05 vs. relative control.
High-Density Astrocytes Attenuates Propofol-Induced Cell Death through BDNF/TrKB Signaling
To assess whether astrocyte-derived BDNF is cell density-dependent, we compared the production of soluble BDNF in conditioned medium from the high-density of astrocytes with that from the low-density of astrocytes using ELISA assay. We found that firstly, BDNF secretion in the conditioned medium released from astrocytes at the higher density increased two fold compared with the lower density (FC=1.97, 95% CI=1.08–2.86 in high density, P<0.05, Figure 4A). Secondly, administration of recombinant BDNF attenuated propofol-induced cell death in both neurons alone and neurons co-cultured with the low-density of astrocytes (1:9 co-cultures) (FC=0.8, 95% CI=0.62–0.98 in neuron alone; FC=0.85, 95% CI=0.74–0.97 in 1:9 co-cultured neurons, P<0.05, Figure 4B). Thirdly, cyclotraxin-B, BDNF receptor TrkB inhibitor, increased neuron death in 1:1 co-cultured neurons. However, blockade of TrkB receptor in 1:1 co-cultured neurons did not completely abolish the high-density astrocyte-induced reduction in neuron death (FC=2.17, 95% CI=1.74–2.6 in neurons; FC=1.48, 95% CI=1.30–1.67 in 1:1 co-cultured neurons, P<0.05, Figure 4C).
Figure 4.
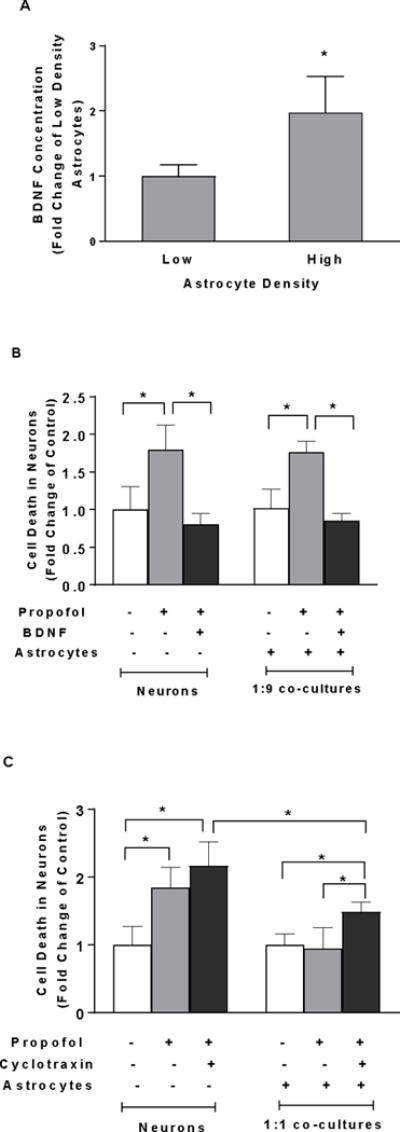
High density of astrocytes compared with low density of astrocytes in the co-culture attenuates propofol-induced neuron death through BDNF/TrkB pathway. (A) Astrocytes cell density- dependently released BDNF. BDNF secretion in conditioned medium released from the high-density astrocytes was higher than that from the low-density astrocytes. BDNF concentration was measured in astrocyte-conditioned medium by enzyme-linked immunosorbant assay (ELISA). n=4. *P<0.05 vs. low-density astrocytes. (B) Recombinant BDNF (100 ng/ml) rescued the propofol-induced neuron death both in neuron-alone culture and co-cultured neurons with low-density of astrocytes (ratio of astrocytes/neurons is 1:9). (C) Inhibition of BDNF binding receptor tropomyosin receptor kinase B (TrKB) activation in neurons co-cultured with high-density of astrocytes (ratio of astrocytes/neurons is 1:1) blocked the astrocyte-induced neuroprotection. n=5. *P<0.05.
High-Density Astrocyte-Induced Neuroprotection against Propofol is partially through Akt-Mediated GSK3β Signaling
To investigate whether high-density astrocyte-induced neuroprotection involves cell survival signaling, Akt inhibitor LY290004 was administered in 1:1 co-cultured neurons in the presence of propofol. Western blot results showed that propofol significantly decreased the activation of Akt (p-Akt) in neurons, whereas the co-cultured astrocytes attenuated the propofol-induced reduction in p-Akt (FC=0.20, 95% CI=0.07–0.33 in neurons; FC=0.11, 95% CI=0.007–0.22 in 1:1 co-cultured neurons, P<0.05, Figure 5A-a & b). Consequently, the astrocyte-induced reduction in cell death in 1:1 co-cultured neurons was abolished by Akt inhibitor LY290004 (FC=1.78, 95% CI=1.50–2.05 in 1:1 co-cultured neurons, P<0.05, Figure 5C). However, LY290004 alone did not cause the increase in neuron death (FC=1.16, 95% CI=0.63–1.69, P>0.05, Figure 5D).
Figure 5.
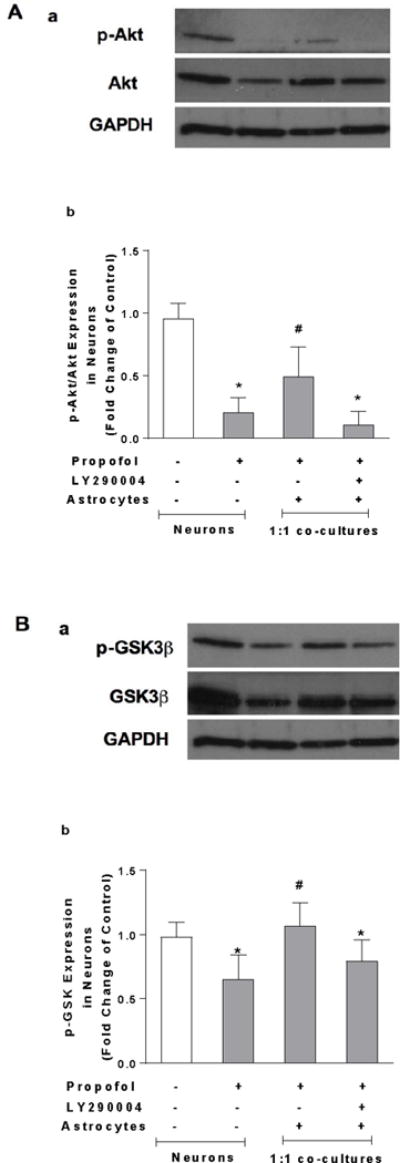
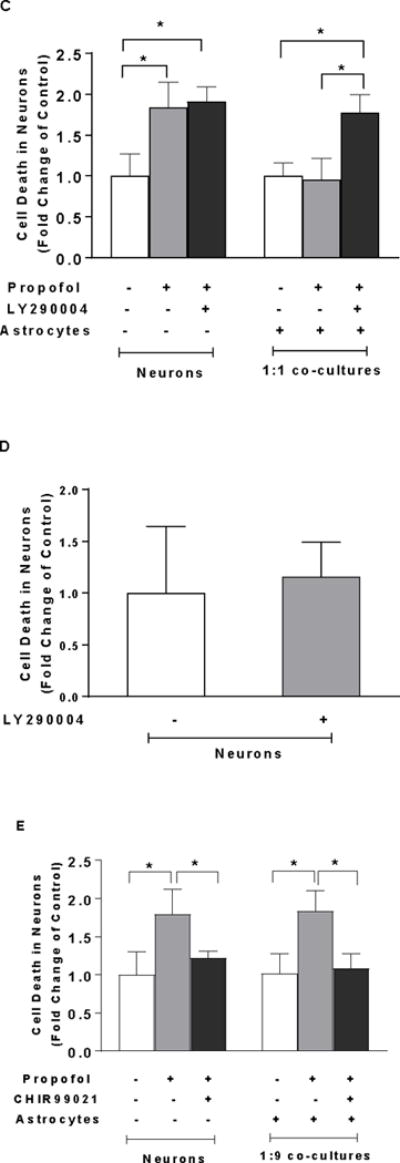
High density of astrocytes compared with low density of astrocytes in the co-culture attenuates propofol-induced neuron death through Akt/GSK3β pathway. (A) The effect of propofol, co-cultured high-density of astrocytes, and Akt inhibitor LY290004 on Akt expression in the neurons analyzed using Western blot. (A-a) Representative Western blot bands of Akt, p-Akt, and β-actin. (A-b) Astrocytes attenuated propofol-induced decrease of activation of Akt (p-Akt). This astrocytes-mediated effect was abolished by LY290004. P-Akt level was expressed as fold change of control without propofol treatment. n=6. *P<0.05 vs. Control; #P<0.05 vs Propofol. (B) The effect of propofol, co-cultured high-density of astrocytes, and LY290004 on glycogen synthase kinase-3 beta (GSK3β) expression in the neurons analyzed using Western blot. (B-a) Representative Western blot bands of GSK3β, p-GSK3β, and β-actin. (B-b) Astrocytes attenuated propofol-induced decrease of inactivation of GSK3β (p-GSK3β). This astrocytes-mediated effect was abolished by LY290004. The protein expression of p-GSK3β was expressed as fold change of control. n=6. *P<0.05 vs. Control; #P<0.05 vs Propofol. (C) The astrocyte-induced reduction in neuron death in 1:1 astrocyte-neuron co-culture was abolished by Akt inhibitor LY290004. (D) LY290004 alone did not cause the increase in cell death in neurons n=4. (E) The propofol-induced increase in neuron death in 1:9 astrocyte-neuron co-culture was rescued by GSK3β inhibitor CHIR99021. n=5. *P<0.05. p-Akt = phosphorylated (ser 473) protein kinase B; p-GSK3β = phosphorylated GSK3β (ser 9).
To examine whether GSK3β is a downstream target of Akt signaling, the effect of LY290004 on GSK-3β expression in the 1:1 co-cultured neurons was analyzed using Western blot. Propofol decreased inactivation of GSK3β (p-GSK3β) while astrocytes attenuated propofol-induced decrease of p-GSK3β. This astrocytes-mediated and altered p-GSK3β was abolished by Akt inhibitor LY290004 (FC=0.65, 95% CI=0.44–0.85 in neurons; FC=0.79, 95% CI=0.61–0.97 in 1:1 co-cultured neurons, P<0.05, Figure 5B-a & b). Additionally, the propofol-induced increase in cell death in 1:9 co-cultured neurons was rescued by GSK3β inhibitor CHIR99021 (FC=1.08, 95% CI=0.84–1.32, P<0.05, Figure 5E).
Insufficient BDNF Secretion due to Low-Density of Astrocytes Contributes to Propofol-Induced Mitochondrial Fission through GSK3β-Mediated Signaling
To determine whether the increased mitochondrial fission contributes to the propofol-induced neuron death, mitochondrial morphology was assessed using TOM20 immunofluorescence staining. Mitochondrial fission inhibitor Mdivi-1 was used to block mitochondrial fission prior to propofol administration. Following propofol exposure, shorter and fragmented mitochondria were prevalent in the neuron-alone and 1:9 co-cultured neurons as compared to the interconnected and tubular mitochondria in control neurons. Mdivi-1 attenuated the propofol-induced mitochondrial fragmentation evidenced by the elongated mitochondria (Figure 6A-a). Propofol significantly decreased the values of aspect ratio and form factor of mitochondria in the neuron-alone culture and 1:9 neuron-astrocyte co-culture, indicating enhanced mitochondrial fission [aspect ratio (AR)=2.76, 95% CI=2.28–3.25 and form factor (FF)=2.33, 95% CI=2.04–2.63 in neurons; AR=2.76, 95% CI=2.2–3.31 and FF=2.33, 95% CI=1.91–2.75 in 1:9 co-cultured neurons; P<0.05, Figure 6A-b & c], whereas Mdivi-1 prevented propofol-induced mitochondrial fragmentation by increasing both aspect ratio and form factor (AR=4.13, 95% CI=3.58–4.68 and FF=4.11, 95% CI=3.67–4.56 in neurons; AR=4.45, 95% CI=3.51–5.39 and FF=4.17, 95% CI=3.53–4.81 in 1:9 co-cultured neurons, P<0.05, Figure 6A-b & c). Consequently, inhibition of mitochondrial fission by Mdivi-1 rescued propofol-induced cell death in neuron-alone culture and 1:9 neuron-astrocyte co-cultures (FC=1.35, 95% CI=1.16–1.54 in neuron alone; FC=1.25, 95% CI=1.1–1.39 in 1:9 co-cultured neurons, P<0.05, Figure 6A-d).
Figure 6.
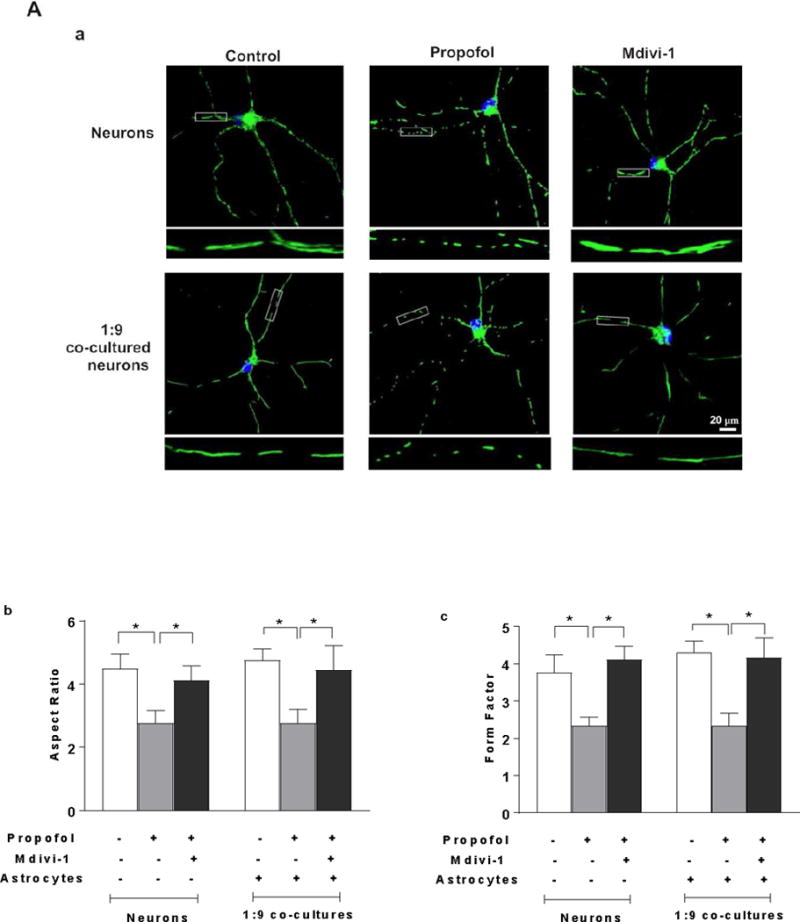
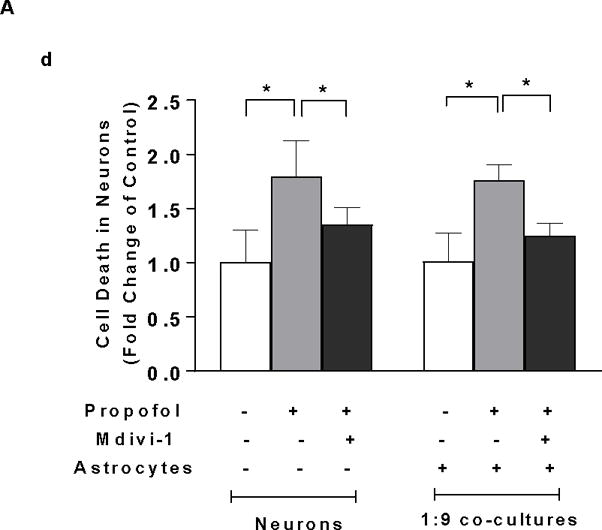
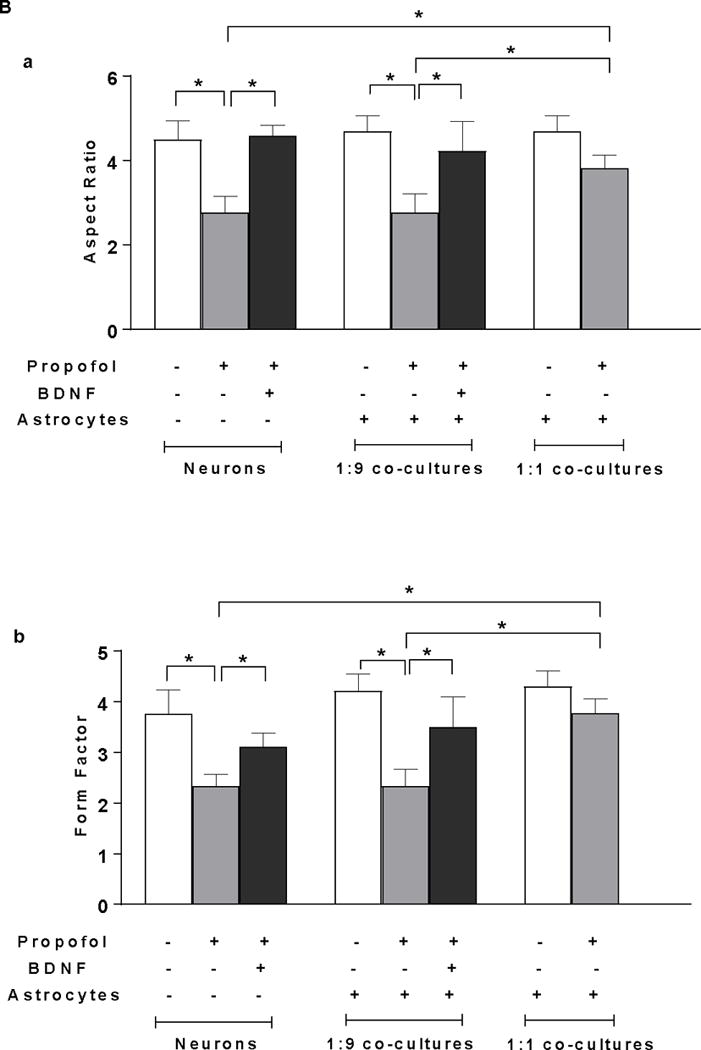
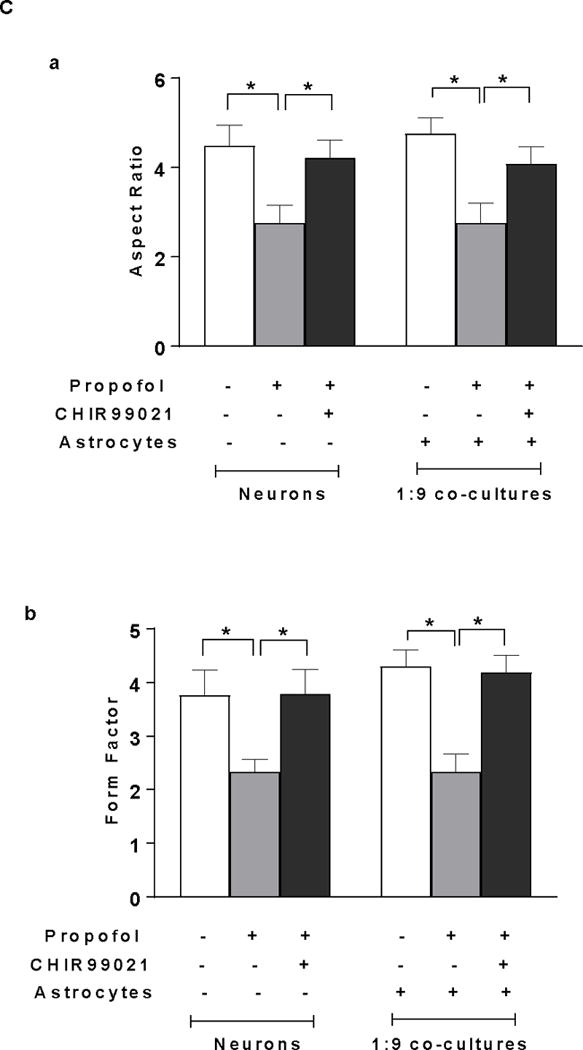
Insufficient BDNF in the low density of astrocytes in the co-culture contributes to propofol-induced neuron death through BDNF/GSK3β/mitochondrial fission pathway. (A) Propofol-induced increase of mitochondrial fission contributes to neuron death in both neuron-alone culture and astrocyte-neuron co-culture with the low-density of astrocytes (ratio of astrocytes/neurons is 1:9). (A-a) The representative confocal fluorescence images of mitochondria in neuron-alone culture and 1:9 neuron-astrocyte co-culture. Following 6 h of 30 μM propofol exposure, mitochondria were labeled with the mitochondrial dye TOM20 (green) using immunofluorescence staining and imaged by confocal microscopy. The blue are cell nuclei stained with Hoechst 33342. In the control and Mdivi-1-treated neurons, the mitochondria were interconnected and tubular. Following propofol exposure, much shorter and fragmented mitochondria exhibiting punctate signals were prevalent in the cells. (A-b & c) Quantification of mitochondrial shape in the neurons by measuring both aspect ratio and form factor represent mitochondrial length and mitochondrial branching, respectively. Propofol significantly decreased the values of aspect ratio and form factor in neuronal processes of neuron-alone culture and 1:9 astrocyte-neuron co-culture, indicating enhanced mitochondrial fission while Mdivi-1 prevented propofol-induced mitochondrial fragmentation by increasing both aspect ratio and form factor. (A-d) Inhibition of mitochondrial fission by Mdivi-1 rescued propofol-induced neuron death in neuron-alone culture and 1:9 astrocyte-neuron co-culture. (B) The effect of BDNF asministration and co-cultured high-density of astrocytes on propofol-induced mitochondrial fission in neurons. (B-a & b) BDNF administration attenuated the propofol-induced mitochondrial fission by increasing both aspect ratio and form factor in neuron-alone culture and 1:9 astrocyte-neuron co-culture. The high density of astrocytes attenuated the propofol-induced mitochondrial fragmentation in neurons (ratio of astrocytes/neurons is 1:1). (C) The effect of GSK3β inhibitor CHIR99021 on propofol-induced mitochondrial fission in neuron-alone culture and 1:9 astrocyte-neuron co-culture. (C-a & b) CHIR99021 attenuated the propofol-induced neuron mitochondrial fission by increasing both aspect ratio and form factor. n=5. *P<0.05.
To examine whether insufficient numbers of astrocytes and inadequate levels of BDNF contribute to propofol-induced mitochondrial fission, the high-density of astrocytes and recombinant BDNF were added in the neuron-alone culture and 1:9 co-cultures. Both exogenous BDNF administration and the high-density of astroyctes attenuated the propofol-induced mitochondrial fission as evidended by the increased aspect ratio and form factor in neuron-alone culture and 1:9 neuron-astrocyte co-culture (AR=4.58, 95% CI=4.27–4.89 and FF=3.11, 95% CI=2.77–3.45 in neurons treated with BDNF; AR=4.23, 95% CI=3.36–5.10 and FF=3.50, 95% CI=2.74–4.25 in 1:9 co-cultured neurons treated with BDNF; AR=3.82, 95% CI=3.44–4.20 and FF=3.78, 95% CI=3.43–4.13 in co-cultures with high-density astrocytes P<0.05, Figure 6B-a & b).
To determine whether GSK3β plays a role in propofol-induced mitochondrial fission, CHIR99021 was used to inhibit propofol-induced increase of GSK3β activation. CHIR99021 attenuated the propofol-induced decrease of aspect ratio and form factor in neuron-alone culture and 1:9 astrocyte-neuron co-culture, suggesting the decreased mitochondrial fission (AR=4.22, 95% CI=3.74–4.7 and FF=3.79, 95% CI=3.23–4.36 in neurons; AR=4.09, 95% CI=3.6–4.57 and FF=4.19, 95% CI=3.79–4.58 in 1:9 co-cultured neurons, P<0.05, Figure 6C-a & b).
DISCUSSION
In the current study, we examined the roles of astrocytes in propofol-induced cell death of the neurons isolated from neonatal rat hippocampus and the underlying mechanisms of BDNF, Akt, GSK3β, and mitochondrial fission using cultured neurons-alone, astrocytes-alone, and neurons co-cultured with low- and high-density of astrocytes models. We demonstrated for the first time that (1) Propofol at clinically relevant concentrations dose-dependently induced neuron death, but did not influence astrocyte viability; (2) Co-cultured astrocytes at the high density were able to rescue propofol-induced neuron death through astrocyte-secreted soluble factors, while astrocytes at the low density were unable to protect neurons. Specifically, astrocytes secreted BDNF in a cell number dependent fashion and subsequently prevented propofol-induced neuron death; (3) Propofol-induced neuron death occurs through Akt/GSK3β/mitochondrial pathway; and (4) Insufficient BDNF secretion due to low-density of astrocytes contributes to propofol-induced neuron death via Akt/GSK3β/mitochondrial fission pathway.
Despite many findings in animal models that anesthetics induced neurotoxicity in the developing brain,5,8,33,45–47 there is little information regarding the role of astrocytes in the neurotoxicity. A recent finding demonstrated that glial cells from neonatal rat cortex co-cultured with neurons prevented ketamine-induced neuron death48 indicating a protective role of supporting cells in ketamine-induced developmental neurotoxicity. The present study used dissociated hippocampal neurons and astrocytes in co-culture system to address this current anesthetic issue. Our data showed that neurons co-cultured with a low density of astrocytes (1:9 ratio co-culture) failed to reduce propofol-induced neuronal death. Conversely, high concentration of astrocytes (1:1 ratio co-cultures) blunted the detrimental effect of propofol (Figure 2), suggesting that astrocytes exert neuroprotective effect, but the low ratio of astrocyte to neurons may be an important contributor to the increased vulnerability of developing brain to anesthetics. As reported, the timing of great sensitivity of neurons to propofol is within postnatal two weeks in rodent brains. During this period of time, the structure of brain region and distribution of cell types differ greatly from those in adult brain. It has been reported that the glia to neuron ratio (GNR) varies widely across areas of the brain in developing rats17. In postnatal day 7 rat brain, GNR is 1:4.1 in cerebral cortex and 1:11.1 in hippocampus while in adult rat brain (postnatal day 50), GNR increases to 1.35:1 in cerebral cortex and to 1.86 in hippocampus. Since GNR varies widely across areas of the brain in developing rat brains, future study will be focused to dissect the correlations between the vulnerability of different neonatal brain regions and subregions to propofol, and the GNR. It appears that the ratio of supporting cells to neurons is extremely crucial during the critical period of brain development.
The crosstalk between astrocytes and neurons in response to propofol is largely unknown. Astrocytes exert the capability to synthesize and release paracrine soluble factors to surrounding neurons to promote neuron growth and survival. 49 This study is the first to report the protein profile of astrocyte-conditioned medium following propofol. Thirty-four cytokines and growth factors were identified out of 90 proteins. Nine neurotrophic factors secreted from astrocyte-conditioned medium that were downregulated following 6h propofol exposure (Figure 3). Among nine neurotrophic factors, BDNF, VEGF, PDGF and TGF-β are a family of secreted proteins that participate in cell growth, survival, differentiation, and synaptic plasticity during brain development.38–44 It has been shown that a single dose of propofol could change neurotrophin BDNF and NT-3 mRNA expression in mouse brain16 suggesting the propofol-induced neurotoxic effect is largely caused by neurotrophic disturbance. We for the first time reported that BDNF was secreted and released by astrocytes in a cell density-dependent manner. Given that the low-density of astrocytes released the lower level of BDNF as compared to the higher density may contribute to the sensitive vulnerability of neurons to propofol, and additional BDNF to neurons increase neuron survival (Figure 4A & B). BDNF is the most abundant neurotrophic growth factor expressed in hippocampus and is responsible for neurogenesis, learning, and memory11,12,35, we then focused on the BDNF role in propofol neurotoxicity in co-culture study. Neurotrophin has three common tyrosine kinase (Trk) receptors TrkA, TrkB, and/or TrkC. TrkA binds nerve growth factor (NGF) at a high affinity. TrkB has specific binding affinity to BDNF and TrkC binds closely to NT-3. The binding of neurotrophins to Trk receptors leads to the activation of signaling pathways.50–52 We observed that conditioned medium-induced neuron survival was partially but not completely blocked by the selective TrkB inhibitor cyclotraxin (Figure 4C). It appears that BDNF may not be the sole player in neuroprotection. We cannot exclude the possibility that other neurotropin factor such as VEGF-C, PDGF and TGF-β may contribute to astrocyte conditioned medium-induced neuron survival.
Next we studied the mechanism by which astrocyte-derived BDNF increases cell survival of developing neurons. Understanding the mechanism underlying astrocytes-induced paracrine effect can help develop potential therapeutic strategies to avoid propofol-induced neurotoxicity. PI3K/Akt and glycogen synthase kinase (GSK3β) have been shown to be involved in various cell survival pathways including developing human embryonic stem cell-derived neurons23,24,53. Blockage of Akt, a negative regulator of GSK3β or activation of GSK3β caused cells to undergo the apoptotic process54,55. Furthermore, tyrosine phosphorylation of GSK3β led to GSK3β inactivation. Ketamine was reported to induce neuroapoptosis. This toxic effect was associated with the decrease in Akt and GSK3β phosphorylation (p-GSK3β) and co-administration of lithium with ketamine attenuated the neuronal death through deactivation of GSK3β.53 Recently, evidence linking propofol-induced neurotoxicity and defective STAT3/SPOUT2/Akt pathway was reported in human neurons 23. Despite convincing evidence of involvement of Akt/ GSK3β pathway in developing neuronal death, whether astrocyte-derived BDNF regulates this pathway to increase neuronal survival in propofol-injured neurons is unknown. In developing neurons, BDNF exerts its pro-survival effects commonly via two key signaling cascades 56. In one pathway, BDNF activates Akt activation through its receptor TrkB-induced PI3K activity. In the second pathway, BDNF activates ribosomal S6 kinase (Rsk)2 or mitogen- and stress-activated protein kinase 1 (Msk1) through TrkB-induced extracellular signal-regulated kinase 1/2 (Erk) activation. In the present study, the data showed that BDNF increased cell survival through enhancing phosphorylation of Akt, and its downstream p-GSK3β (Figure 5 & 6). Although Akt inhibitor LY290004 abolished astrocytes-mediated protection, LY290004 alone did not cause the increase in cell death in neurons (Figure 5C & D). This result suggests that decreased Akt activity may not be the only consequence of propofol-induced pleiotropic effects. Indeed, Straiko et al. reported that propofol at concentrations of 50 or 100 mg/kg suppressed the phosphorylation of Erk, another important survival kinase in mouse brain57, implicating other molecular mechanisms in the dysregulation of intercellular organelles and intrinsic or extrinsic cell apoptosis process. These above findings provide new insights into the preventive strategies on the side effects of propofol in developing brain.
There are two limitations in the present study. One of the barriers lies in the relevance of the current in vitro co-culture system to the in vivo animal model. In vitro cell culture approach can provide great benefits on 1) investigating propofol toxicity under the controlled conditions and the potential mechanisms without interference of confounding physiology or pathology factors seen in the intact animal model, and 2) dissecting the contribution of individual types of neuronal cells from different brain regions to the anesthetic developing neurotoxicity. However, the in vitro system is always artificial. The real interaction among different cell types in intact animals may be missing in in vitro culture system. Under physiological condition, the interaction between neurons and astrocytes is more complex due to the participation of other types of glial cells such as oligodendrocytes. One recent study showed that propofol induced toxic effects in oligodendrocytes in non-human primate brains.8 Thus, it is important to confirm our in vitro findings using intact animal model. As these experiments rely on an entirely artificial in vitro system, without additional experimentation in vivo, it is difficult to interpret the biological significance of the results presented in the current manuscript. Another limitation underlies that in addition to Akt/GSK3β/mitochondrial fission pathway additional signaling might also be involved as the mechanisms of propofol-induced neuron death. For example, microRNA-1 has been shown to negatively regulate BDNF secretion in central nervous system,58 suggesting a potential role of microRNA-1 in the regulation of anesthetic-induced neurotoxicity. In addition, the current study only used apoptotic cell death as an endpoint. Propofol might exert other toxic effects such as abnormalities in dendritic development, neuron stem cell proliferation, and synaptogenesis, which remain for future investigation.
In conclusion, we used neurons-alone, astrocytes-alone, neurons co-cultured with low- and high-ratio of astrocyte to neuron models to dissect the mechanisms involved in propofol-induced neuron death. We demonstrated for the first time that propofol- induced neuron death was partially through Akt/GSK3β/mitochondrial fission pathway. Similarly, the insufficient levels of BDNF due to the low ratio of astrocyte to neuron contributed to the neuron death via Akt/GSK3β/mitochondrial fission pathway. The current study laid a foundation for the development of novel therapeutic strategies for insuring the safety of pediatric anesthesia by interfering in the BDNF/Akt/GSK3β/mitochondrial fission pathways.
Acknowledgments
The authors thank Terri Misorski (Program Coordinator, Department of Anesthesiology) for editorial and proofreading assistance.
Funding: This work was supported by grants P01GM066730 and R01HL034708 from the National Institutes of Health (NIH), Bethesda, MD, FP00003109 from Advancing Healthier Wisconsin Research and Education Initiative Fund, Milwaukee, WI (to Dr. Zeljko J. Bosnjak) and R01GM112696 from the NIH (to Dr. Xiaowen Bai).
Footnotes
Yanan Liu
Role: This author designed the study, conducted the study, analyzed the data, and wrote the manuscript
Conflicts: Yanan Liu reported no conflicts of interest
Attestation: Yanan Liu has seen the original study data, reviewed the analysis of the data, approved the final manuscript, and is the author responsible for archiving the study files
Yasheng Yan
Role: This author conducted the study
Conflicts: Yasheng Yan reported no conflicts of interest
Attestation: Yasheng Yan has seen the original study data, reviewed the analysis of the data, and approved the final manuscript
Yasuyoshi Inagaki
Role: This author conducted the study and analyzed the data
Conflicts: Yasuyoshi Inagaki reported no conflicts of interest
Attestation: Yasuyoshi Inagaki has seen the original study data, reviewed the analysis of the data, and approved the final manuscript
Sarah Logan
Role: This author conducted the study
Conflicts: Sarah Logan reported no conflicts of interest
Attestation: Sarah Logan approved the final manuscript
Zeljko J Bosnjak
Role: This author designed the study and wrote the manuscript
Conflicts: Zeljko J Bosnjak reported no conflicts of interest
Attestation: Zeljko J Bosnjak has seen the original study data, reviewed the analysis of the data, and approved the final manuscript
Xiaowen Bai
Role: This author designed the study and wrote the manuscript
Conflicts: Xiaowen Bai reported no conflicts of interest
Attestation: Xiaowen Bai has seen the original study data, reviewed the analysis of the data, and approved the final manuscript
For Editorial Office: This report was previously presented, in part, at the Presented in part at the International Anesthesia Research Society 2015 Annual Meeting, Honolulu, HI, USA, March 22, 2015.
Contributor Information
Yanan Liu, Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin.
Yasheng Yan, Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin.
Yasuyoshi Inagaki, Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin.
Sarah Logan, Departments of Physiology and Anesthesiology, Medical College of Wisconsin, Milwaukee, WI.
Zeljko J Bosnjak, Departments of Anesthesiology and Physiology, Medical College of Wisconsin, Milwaukee, Wisconsin.
Xiaowen Bai, Departments of Anesthesiology and Physiology, Medical College of Wisconsin, Milwaukee, Wisconsin.
References
- 1.Flick RP, Katusic SK, Colligan RC, Wilder RT, Voigt RG, Olson MD, Sprung J, Weaver AL, Schroeder DR, Warner DO. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–61. doi: 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai X, Bosnjak ZJ. Emerging model in anesthetic developmental neurotoxicity: human stem cells. International journal of clinical anesthesiology. 2013;1:1002. [PMC free article] [PubMed] [Google Scholar]
- 4.Kahraman S, Zup SL, McCarthy MM, Fiskum G. GABAergic mechanism of propofol toxicity in immature neurons. Journal of neurosurgical anesthesiology. 2008;20:233–40. doi: 10.1097/ANA.0b013e31817ec34d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearn ML, Hu Y, Niesman IR, Patel HH, Drummond JC, Roth DM, Akassoglou K, Patel PM, Head BP. Propofol Neurotoxicity Is Mediated by p75 Neurotrophin Receptor Activation. Anesthesiology. 2012;116:352–61. doi: 10.1097/ALN.0b013e318242a48c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong Y, Liang Y, Chen J, Li L, Qin Y, Guan E, He D, Wei Y, Xie Y, Xiao Q. Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons in vitro via downregulation of NF-kappaB p65 and Bcl-2 and upregulation of caspase-3. Cell biochemistry and function. 2014;32:720–9. doi: 10.1002/cbf.3077. [DOI] [PubMed] [Google Scholar]
- 7.Kargaran P, Lenglet S, Montecucco F, Mach F, Copin JC, Vutskits L. Impact of propofol anaesthesia on cytokine expression profiles in the developing rat brain: a randomised placebo-controlled experimental in-vivo study. European journal of anaesthesiology. 2015;32:336–45. doi: 10.1097/EJA.0000000000000128. [DOI] [PubMed] [Google Scholar]
- 8.Creeley C, Dikranian K, Dissen G, Martin L, Olney J, Brambrink A. Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. British journal of anaesthesia. 2013;110(Suppl 1):i29–38. doi: 10.1093/bja/aet173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development (vol 14, pg 311–321, 2013) Nat Rev Neurosci. 2013;14:443-. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cody MM, Freeman MR. Architects in neural circuit design: Glia control neuron numbers and connectivity. J Cell Biol. 2013;203:395–405. doi: 10.1083/jcb.201306099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulmer CG, VonDran MW, Stillman AA, Huang YY, Hempstead BL, Dreyfus CF. Astrocyte-Derived BDNF Supports Myelin Protein Synthesis after Cuprizone-Induced Demyelination. J Neurosci. 2014;34:8186–96. doi: 10.1523/JNEUROSCI.4267-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dougherty KD, Dreyfus CF, Black IB. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol Dis. 2000;7:574–85. doi: 10.1006/nbdi.2000.0318. [DOI] [PubMed] [Google Scholar]
- 13.Bekinschtein P, Cammarota M, Medina JH. BDNF and memory processing. Neuropharmacology. 2014;76:677–83. doi: 10.1016/j.neuropharm.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 14.Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LRM, Izquierdo I, Medina JH. Persistence of long-term memory storage requires a late protein synthesis- and BDNF-dependent phase in the hippocampus. Neuron. 2007;53:261–77. doi: 10.1016/j.neuron.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nature neuroscience. 2008;11:131–3. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- 16.Karen T, Schlager GW, Bendix I, Sifringer M, Herrmann R, Pantazis C, Enot D, Keller M, Kerner T, Felderhoff-Mueser U. Effect of propofol in the immature rat brain on short- and long-term neurodevelopmental outcome. PloS one. 2013;8:e64480. doi: 10.1371/journal.pone.0064480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bandeira F, Lent R, Herculano-Houzel S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14108–13. doi: 10.1073/pnas.0804650106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. Astrocytes and disease: a neurodevelopmental perspective. Genes & development. 2012;26:891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christensen JR, Larsen KB, Lisanby SH, Scalia J, Arango V, Dwork AJ, Pakkenberg B. Neocortical and hippocampal neuron and glial cell numbers in the rhesus monkey. Anatomical record. 2007;290:330–40. doi: 10.1002/ar.20504. [DOI] [PubMed] [Google Scholar]
- 20.Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends in neurosciences. 2003;26:523–30. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Nunez J. Primary Culture of Hippocampal Neurons from P0 Newborn Rats. Journal of visualized experiments : JoVE. 2008 doi: 10.3791/895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarkar P, Narayanan J, Harder DR. Differential effect of amyloid beta on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience. 2011;194:241–9. doi: 10.1016/j.neuroscience.2011.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Twaroski DM, Yan YS, Olson JM, Bosnjak ZJ, Bai XW. Down-regulation of MicroRNA-21 Is Involved in the Propofol-induced Neurotoxicity Observed in Human Stem Cell-derived Neurons. Anesthesiology. 2014;121:786–800. doi: 10.1097/ALN.0000000000000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai XW, Yan YS, Canfield S, Muravyeva MY, Kikuchi C, Zaja I, Corbett JA, Bosnjak ZJ. Ketamine Enhances Human Neural Stem Cell Proliferation and Induces Neuronal Apoptosis via Reactive Oxygen Species-Mediated Mitochondrial Pathway. Anesth Analg. 2013;116:869–80. doi: 10.1213/ANE.0b013e3182860fc9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viviand X, Berdugo L, De La Noe CA, Lando A, Martin C. Target concentration of propofol required to insert the laryngeal mask airway in children. Paediatr Anaesth. 2003;13:217–22. doi: 10.1046/j.1460-9592.2003.01006.x. [DOI] [PubMed] [Google Scholar]
- 26.Hume-Smith HV, Sanatani S, Lim J, Chau A, Whyte SD. The effect of propofol concentration on dispersion of myocardial repolarization in children. Anesth Analg. 2008;107:806–10. doi: 10.1213/ane.0b013e3181815ce3. [DOI] [PubMed] [Google Scholar]
- 27.Varveris DA, Morton NS. Target controlled infusion of propofol for induction and maintenance of anaesthesia using the paedfusor: an open pilot study. Paediatr Anaesth. 2002;12:589–93. doi: 10.1046/j.1460-9592.2002.00921.x. [DOI] [PubMed] [Google Scholar]
- 28.Riu PL, Riu G, Testa C, Mulas M, Caria MA, Mameli S, Mameli O. Disposition of propofol between red blood cells, plasma, brain and cerebrospinal fluid in rabbits. European journal of anaesthesiology. 2000;17:18–22. doi: 10.1046/j.1365-2346.2000.00573.x. [DOI] [PubMed] [Google Scholar]
- 29.Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Clinically relevant concentrations of propofol but not midazolam alter in vitro dendritic development of isolated gamma-aminobutyric acid-positive interneurons. Anesthesiology. 2005;102:970–6. doi: 10.1097/00000542-200505000-00016. [DOI] [PubMed] [Google Scholar]
- 30.Chung HG, Myung SA, Son HS, Kim YH, Namgung J, Cho ML, Choi H, Lim CH. In vitro effect of clinical propofol concentrations on platelet aggregation. Artificial organs. 2013;37:E51–5. doi: 10.1111/j.1525-1594.2012.01553.x. [DOI] [PubMed] [Google Scholar]
- 31.Ludbrook GL, Visco E, Lam AM. Propofol - Relation between brain concentrations, electroencephalogram, middle cerebral artery blood flow velocity, and cerebral oxygen extraction during induction of anesthesia. Anesthesiology. 2002;97:1363–70. doi: 10.1097/00000542-200212000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Costela JL, Jimenez R, Calvo R, Suarez E, Carlos R. Serum protein binding of propofol in patients with renal failure or hepatic cirrhosis. Acta anaesthesiologica Scandinavica. 1996;40:741–5. doi: 10.1111/j.1399-6576.1996.tb04521.x. [DOI] [PubMed] [Google Scholar]
- 33.Sall JW, Stratmann G, Leong J, Woodward E, Bickler PE. Propofol at clinically relevant concentrations increases neuronal differentiation but is not toxic to hippocampal neural precursor cells in vitro. Anesthesiology. 2012;117:1080–90. doi: 10.1097/ALN.0b013e31826f8d86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spahr-Schopfer I, Vutskits L, Toni N, Buchs PA, Parisi L, Muller D. Differential neurotoxic effects of propofol on dissociated cortical cells and organotypic hippocampal cultures. Anesthesiology. 2000;92:1408–17. doi: 10.1097/00000542-200005000-00032. [DOI] [PubMed] [Google Scholar]
- 35.Ryu YK, Khan S, Smith SC, Mintz CD. Isoflurane impairs the capacity of astrocytes to support neuronal development in a mouse dissociated coculture model. Journal of neurosurgical anesthesiology. 2014;26:363–8. doi: 10.1097/ANA.0000000000000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manadas B, Santos AR, Szabadfi K, Gomes JR, Garbis SD, Fountoulakis M, Duarte CB. BDNF-induced changes in the expression of the translation machinery in hippocampal neurons: protein levels and dendritic mRNA. Journal of proteome research. 2009;8:4536–52. doi: 10.1021/pr900366x. [DOI] [PubMed] [Google Scholar]
- 37.Zaja I, Bai XW, Liu YN, Kikuchi C, Dosenovic S, Yan YS, Canfield SG, Bosnjak ZJ. Cdk1, PKC delta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem Bioph Res Co. 2014;453:710–21. doi: 10.1016/j.bbrc.2014.09.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donzis EJ, Tronson NC. Modulation of learning and memory by cytokines: Signaling mechanisms and long term consequences. Neurobiol Learn Mem. 2014;115:68–77. doi: 10.1016/j.nlm.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kopec AM, Carew TJ. Growth factor signaling and memory formation: temporal and spatial integration of a molecular network. Learn Memory. 2013;20:531–9. doi: 10.1101/lm.031377.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang EJ, Reichardt LF. Neurotrophins: Roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cariboni A, Davidson K, Dozio E, Memi F, Schwarz Q, Stossi F, Parnavelas JG, Ruhrberg C. VEGF signalling controls GnRH neuron survival via NRP1 independently of KDR and blood vessels. Development. 2011;138:3723–33. doi: 10.1242/dev.063362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fruttiger M, Calver AR, Kruger WH, Mudhar HS, Michalovich D, Takakura N, Nishikawa S, Richardson WD. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron. 1996;17:1117–31. doi: 10.1016/s0896-6273(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 43.Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K. Journal of physiology. Vol. 96. Paris: 2002. TGF-beta and the regulation of neuron survival and death; pp. 25–30. [DOI] [PubMed] [Google Scholar]
- 44.Lipsky RH, Marini AM. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Annals of the New York Academy of Sciences. 2007;1122:130–43. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 45.Mintz CD, Smith SC, Barrett KM, Benson DL. Anesthetics interfere with the polarization of developing cortical neurons. Journal of neurosurgical anesthesiology. 2012;24:368–75. doi: 10.1097/ANA.0b013e31826a03a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cattano D, Young C, Straiko MM, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106:1712–4. doi: 10.1213/ane.0b013e318172ba0a. [DOI] [PubMed] [Google Scholar]
- 47.Yu D, Jiang Y, Gao J, Liu B, Chen P. Repeated exposure to propofol potentiates neuroapoptosis and long-term behavioral deficits in neonatal rats. Neuroscience letters. 2013;534:41–6. doi: 10.1016/j.neulet.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 48.Zuo D, Wang C, Li Z, Lin L, Duan Z, Qi H, Li L, Sun F, Wu Y. Existence of glia mitigated ketamine-induced neurotoxicity in neuron-glia mixed cultures of neonatal rat cortex and the glia-mediated protective effect of 2-PMPA. Neurotoxicology. 2014;44:218–30. doi: 10.1016/j.neuro.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Gomes FCA, Spohr TCLS, Martinez R, Neto VM. Cross-talk between neurons and glia: highlights on soluble factors. Braz J Med Biol Res. 2001;34:611–20. doi: 10.1590/s0100-879x2001000500008. [DOI] [PubMed] [Google Scholar]
- 50.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annual review of neuroscience. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 52.Barbacid M. The Trk family of neurotrophin receptors. Journal of neurobiology. 1994;25:1386–403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 53.Liu JR, Baek C, Han XH, Shoureshi P, Soriano SG. Role of glycogen synthase kinase-3 in ketamine-induced developmental neuroapoptosis in rats. British journal of anaesthesia. 2013;110:3–9. doi: 10.1093/bja/aet057. [DOI] [PubMed] [Google Scholar]
- 54.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. The Journal of biological chemistry. 1998;273:19929–32. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 55.Pap M, Cooper GM. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta signaling pathway. Molecular and cellular biology. 2002;22:578–86. doi: 10.1128/MCB.22.2.578-586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sossin WS, Barker PA. Something old, something new: BDNF-induced neuron survival requires TRPC channel function. Nature neuroscience. 2007;10:537–8. doi: 10.1038/nn0507-537. [DOI] [PubMed] [Google Scholar]
- 57.Straiko MM, Young C, Cattano D, Creeley CE, Wang H, Smith DJ, Johnson SA, Li ES, Olney JW. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology. 2009;110:862–8. doi: 10.1097/ALN.0b013e31819b5eab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varendi K, Kumar A, Harma MA, Andressoo JO. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell Mol Life Sci. 2014;71:4443–56. doi: 10.1007/s00018-014-1628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]