

Abstract
Hepatitis A virus (HAV) has been adapted to grow efficiently in primate and some nonprimate cell lines but not in cells of murine origin. To understand the inability of the virus to grow in mouse cells, we studied the replication of HAV in immortalized and nontransformed MMH-D3 mouse liver cells, which require growth factors and collagen to maintain their phenotype. HAV grew in MMH-D3 cells transfected with virion RNA but not in those infected with viral particles, indicating a cell entry block for HAV. However, MMH-D3 cells cultured under suboptimal conditions in the absence of growth factors acquired susceptibility to HAV infection. Serial passages of the virus in MMH-D3 cells under suboptimal growth conditions resulted in the selection of HAV variants that grew efficiently in MMH-D3 cells cultured under both optimal and suboptimal conditions. Nucleotide sequence analysis of the MMH-D3 cell-adapted HAV revealed that N1237D and D2132G substitutions were present in the capsid regions of six viral clones. These two mutations are most likely located on the surface of the virion and may play a role in the entry of HAV into the mouse liver cells. Our results demonstrate that mouse hepatocyte-like cells code for all factors required for the efficient growth of HAV in cell culture.
Hepatitis A virus (HAV), a member of the Picornaviridae family, causes acute hepatitis in humans (for a review, see reference 14). The mature viral particle consists of a coat of 60 copies of at least three viral proteins (VP1, VP2, and VP3), which encapsidates a positive-strand RNA genome of approximately 7,500 nucleotides. It is not clear whether the fourth capsid protein, VP4, which is found in all other picornaviruses, is also present in mature HAV particles. In general, wild-type HAV grows inefficiently in vitro but accumulates attenuating mutations during cell culture adaptation (for a review, see reference 14). HAV has been adapted to grow in cells of primate (1, 5-7, 10, 12, 28) and nonprimate (9) origin. Therefore, the cellular factors required for HAV growth are not restricted to primate cells.
The pathogenesis of HAV is poorly understood, and experimentation is difficult because primates are the only animal model for this virus (8, 17, 21). Development of a small-animal model to study the pathogenesis of HAV is highly desirable, especially in light of recent findings showing an inverse association between HAV infection and the development of asthma (22, 23). It is likely that this inverse association is mediated by the human HAV cellular receptor 1 (hhavcr-1) (11), an ortholog of Tim1 and Tim2, which have been described as asthma determinant genes in mice (24, 37). Moreover, it has recently been shown that infection with HAV may protect individuals from atopy if they carry a variant of hhavcr-1 (25).
HAV does not grow efficiently in mouse cell lines (9, 35), and our initial attempts to infect mice with HAV were also unsuccessful (J. Lu, D. Feigelstock, and G. Kaplan, unpublished data). Adaptation of this virus to mouse cells may be required to develop a mouse model for HAV. We have recently shown that mouse Ltk- cells transfected with the infectious cDNA of HAV can support intracellular virus replication (19). However, the HAV grown in Ltk- cells was not capable of reinfecting naïve mouse cells. Since it is possible that liver-specific factors are required for cell entry of HAV, we attempted to adapt HAV to grow in mouse liver cells. Few immortalized mouse hepatocyte cell lines have been generated to date, and most present a transformed phenotype and loss of liver cell characteristics (3, 4). Recently, Amicone et al. (2) generated nontransformed immortalized cell lines from livers of transgenic mice carrying a truncated cytoplasmic form of the human MET gene. One of these cell lines, termed MMH-D3, was derived from a liver of a 3-day-old MET transgenic mouse and has a homogeneous epithelial morphology (34). Here, we report that HAV grew in MMH-D3 cells transfected with virion RNA but not in those infected with viral particles. Interestingly, the MMH-D3 cells gained susceptibility to HAV infection after withdrawal of epidermal growth factor (EGF) from the culture medium. Serial passages of HAV in MMH-D3 cells grown under suboptimal conditions, i.e., in the absence of growth factors and in uncoated plates, resulted in the selection of HAV variants that grew more efficiently in mouse cells than the parental virus. Surprisingly, this mouse-adapted HAV also grew efficiently in MMH-D3 cells cultured under optimal conditions. Nucleotide sequence analysis of the capsid region revealed that the mouse-adapted HAV contained two mutations, N1237D and D2132G, which most likely map to the surfaces of the virions and may allow the entry of HAV into mouse liver cells.
This is the first report to demonstrate that mouse cells code for functional HAV receptors and other factors required for the efficient growth of HAV in cell culture. The experimental system described in this paper will help identify cellular and viral determinants required for the efficient growth of HAV in murine cells, which could lead to the development of a mouse model for study of the pathogenesis of HAV.
MATERIALS AND METHODS
Antisera.
Anti-HAV antiserum was produced in rabbits immunized with commercially available HAV vaccine (32). Peroxidase-labeled goat anti-rabbit antibodies (Kirkegaard and Perry Laboratories, Inc.) were used as recommended by the manufacturer.
Cells and viruses.
The GL37 continuous clone of African green monkey kidney cells (GL37 cells) (15) was grown in Eagle's minimal essential medium (EMEM) containing 10% fetal bovine serum (FBS) at 37°C in a CO2 incubator. Fetal rhesus monkey kidney (FRhK-4) cells, a kind gift of S. Emerson, National Institutes of Health (NIH), were grown in monolayer cultures in EMEM supplemented with 10% horse serum. MMH-D3 cells were obtained from Frank Chisari, The Scripps Research Institute, La Jolla, Calif. The MMH-D3 cells were cultured between passages 3 and 20 by using plates treated for 5 min with 50 μg of type 1 rat collagen (BD Biosciences)/ml. The MMH-D3 cells were grown in RPMI 1640 medium containing 10% FBS, 10 μg of insulin (Roche)/ml, 50 ng of EGF (BD Biosciences)/ml, and 30 ng of insulin-like growth factor II (IGF II) (Sigma)/ml at 37°C in a CO2 incubator. A mouse liver cell line was derived from MMH-D3 cells after 40 serial passages under suboptimal conditions in uncoated plates using a growth medium without EGF and IGF II; these were termed high-passage MMH (HP-MMH) cells. Murine Ltk- cells were grown in modified Eagle's minimal essential medium (DMEM) containing 10% FBS at 37°C in a CO2 incubator.
The cell culture-adapted HM175 strain of HAV, derived from an infectious cDNA clone (6), was termed HAV/7. This cDNA-derived virus was grown for approximately 100 passages in BS-C-1 cells and was termed HAV PI (a kind gift of S. Feinstone, U.S. Food and Drug Administration). The HAV viral stocks were produced in FRhk-4 cells.
To produce viral stocks, cell monolayers were detached with trypsin, growth medium was added, HAV was released from the cells by three freeze-thaw cycles, cell debris was pelleted, and supernatants containing the virus were stored at −80°C in sterile tubes.
Transfection of cells with HAV RNA.
HAV/7 was purified through a 40% sucrose cushion (36). Virion RNA was extracted with phenol-chloroform-1% sodium dodecyl sulfate, precipitated with ethanol in the presence of 0.3 M sodium acetate, washed with 70% ethanol, dried, and resuspended in DEP (diethyl pyrocarbonate)-treated water. A transfection mix consisting of 1 μg of HAV/7 virion RNA and 5 μl of Lipofectine transfection reagent (Invitrogen, Inc.) in 0.1 ml of OptiMEM serum-free medium (Invitrogen, Inc.) was incubated for 30 min at room temperature. Cells seeded 24 h earlier in 25-cm2 flasks were washed with OptiMEM and inoculated with the transfection mix in 2 ml of OptiMEM. At 24 h posttransfection, monolayers were washed, and growth medium containing 10% FBS was added to the cells.
Determination of HAV titers.
Infected monolayers were subjected to three freeze-thaw cycles, cellular debris was pelleted, and supernatants containing HAV were stored at −70°C. HAV titers were determined by an endpoint enzyme-linked immunosorbent assay (ELISA) in 96-well plates containing confluent monolayers of GL37 cells (32). Briefly, six replicate wells were inoculated with 100 μl of 10-fold dilutions of HAV in EMEM-10% FBS, and plates were incubated at 35°C for 2 weeks in a CO2 incubator. Cell monolayers were fixed with 90% methanol, washed, and stained with a 1:2,500 dilution of rabbit anti-HAV antibodies and a 1:25,000 dilution of peroxidase-labeled goat anti-rabbit antibodies. TMB one-component substrate (Kirkegaard and Perry Laboratories, Inc.) was added, and the reaction was stopped with 1% H2SO4. Viral titers were determined by the method of Reed and Muench (29) using the ID50 program (version 5.0) developed by John L. Spouge (National Center for Biotechnology Information, NIH).
Quantitative RT-PCR.
HAV RNA was quantitated by a Taqman assay (33) using the Platinum Quantitative RT-PCR (reverse transcription-PCR) ThermoScript one-step system as recommended by the manufacturer (Invitrogen, Inc.). In vitro-synthesized full-length HAV RNA transcribed from linearized pT7HAV by using T7 RNA polymerase (38) was used as the standard for the quantitative PCR.
Isolation of HAV clones and nucleotide sequencing analysis of the capsid region.
Viral clones were obtained by endpoint dilution in 96-well plates containing confluent monolayers of GL37 cells. Plates were inoculated with 10-fold dilutions of an HAV-MMH passage-21 stock that had 6 additional passages in MMH-D3 cells. Forty-eight wells were inoculated per virus dilution, and plates were incubated at 35°C for 12 days in a CO2 incubator. Supernatants were transferred to a new plate, and cells were fixed and stained with anti-HAV antibodies as described above for the determination of viral titers. To ensure that each clone was derived from a single infectious particle, we sonicated the virus before inoculating the cells and selected clones from the highest possible dilution, which had 8 ELISA-positive wells out of the 48 inoculated wells. Supernatants of six ELISA-positive wells were harvested, and total RNA was extracted from 100 μl of each clone by using Trizol-LS (Invitrogen, Inc.) as recommended by the manufacturer. After ethanol precipitation, RNA was resuspended in 20 μl of DEP-treated water containing 1 mM dithiothreitol and 1 U of RNasin/μl. The cDNA of the capsid region of uncloned HAV-MMH, the six clones as well as the parental HAV PI, was amplified by RT-PCR. First-strand viral cDNA was synthesized by using 10 μl of the extracted RNA, primer 5′-TTTTCCACATCTTGGATTTGCAAAATGCAA-3′, corresponding to nucleotides 4219 to 4190 of HAV, and Superscript II reverse transcriptase as recommended by the manufacturer (Invitrogen, Inc.). The cDNA fragment of the capsid region was amplified by using primers 5′-GGCATTTAGGTTTTTCCTCATTCTTA-3′ and 5′-AACCAATATCTGCATAATTCA-3′, corresponding to nucleotides 702 to 727 and 3900 to 3879 of the HAV genome, respectively, and the Expand High Fidelity PCR System (Roche) as recommended by the manufacturer. Amplified cDNA fragments were purified by electrophoresis in Tris-acetate-EDTA-1% agarose gels, bands were cut and extracted from the gel by using GenElute agarose spin columns (Sigma), and cDNA was precipitated with ethanol. PCR products were sequenced by using an ABI Prism automatic sequencer and the ABI Prism Big Dye Terminator cycle-sequencing ready reaction kit (Perkin-Elmer Cetus, Inc.) as recommended by the manufacturer. Synthetic oligonucleotides 5′-GGCATTTAGGTTTTTCCTCATTCTTA-3′, 5′-GCTGAGGTTGGATCACACCAG-3′, 5′-GAGAGGAGATCTTGTCTTTGA-3′, 5′-CACAATAAGCTTCCCAATGGCAGTGTACTCACCTTT-3′, 5′-ATTGGGAAGCTTATTGTGTATTGTTATAACAGATTG-3′, 5′-CATCATAGATTTTAAAGTTCCAGC-3′, 5′-AGCTCTGGTCACCAGGAACCATAGCACAGATCAATC-3′, and 5′-TGGAAGTACTTCACTTGACA-3′, corresponding to nucleotides 702 to 727, 900 to 929, 1802 to 1822, 2081 to 2046, 2064 to 2099, 3743 to 3720, 1206 to 1171, and 3209 to 3189 of the HAV genome, respectively, were used as sequencing primers.
RESULTS
MMH-D3 cells do not support the efficient growth of HAV PI.
To assess whether HAV could infect mouse hepatocyte-like MMH-D3 cells grown under optimal conditions (2) (using collagen-coated plates and a medium containing EGF and IGF II), subconfluent monolayers of MMH-D3 cells were infected with HAV PI at a multiplicity of infection (MOI) of 1 to 2 50% tissue culture infective doses (TCID50)/cell. HAV-susceptible GL37 and non-HAV-susceptible Ltk- cells were infected under similar conditions. After 24 h, cells were washed three times, split 1:3, and grown at 35°C for 1 week. Cells were split weekly, one-third of the cells were seeded into a new T25 flask, and one-third of the cells were used to produce a viral stock that was titrated in monkey GL37 cells by the ELISA endpoint dilution assay (Fig. 1). As expected, HAV PI grew in positive-control GL37 cells and did not grow in negative-control mouse Ltk- cells. HAV PI did not grow in mouse MMH-D3 cells, which indicated that these cells were not susceptible to HAV PI infection.
FIG. 1.
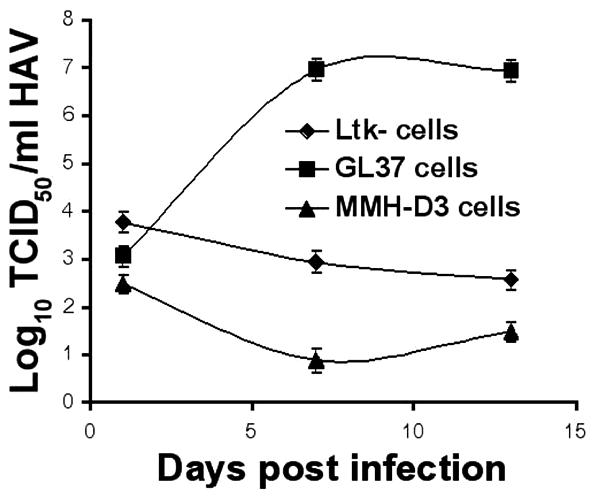
Growth of HAV in cell lines after infection. Monolayers of MMH-D3, Ltk-, and GL37 cells grown in 25-cm2 flasks were infected with HAV PI at an MOI of 1 to 2 TCID50/cell. At 24 h postinfection, cells were washed three times, split 1:3, and grown at 35°C in a CO2 incubator. Cells were split 1:3 weekly; approximately one-third of the collected cells were subjected to three freeze-thaw cycles, cell debris was pelleted, and the supernatant containing HAV was stored at −70°C. HAV was titrated by an endpoint ELISA in 96-well plates containing GL37 cell monolayers. Values are log10 HAV titers determined by the method of Reed and Müench (29). Error bars, standard deviations.
HAV grows in MMH-D3 cells transfected with virion RNA.
To analyze whether HAV failed to grow in MMH-D3 cells because of a cell entry block(s), MMH-D3 cells were transfected with HAV virion RNA. GL37 and Ltk- cells were used as positive and negative transfection controls, respectively. Cells were split 1:3 weekly, and one-third of the cells was used to prepare a viral stock. At 2 weeks posttransfection, HAV titers were determined by ELISA using an endpoint dilution assay in GL37 cells (Fig. 2). Interestingly, MMH-D3 cells transfected with virion RNA produced 103 to 104 TCID50 of HAV/ml. HAV did not grow in Ltk- cells transfected with virion RNA under the same conditions, a finding that is consistent with our previous results (9) and further confirms that Ltk- cells contain an intracellular block(s) to HAV replication. HAV grew to higher titers (106 to 107 TCID50/ml) in GL37 cells, a result that is expected after multiple rounds of infection in these susceptible cells, and was not detected in mock-transfected cells. Since the virus grew in MMH-D3 cells transfected with virion RNA but not in MMH-D3 cells infected with HAV particles (Fig. 1), we concluded that these cells were permissive for HAV PI replication but not susceptible to HAV PI infection.
FIG. 2.

Growth of HAV in cell lines transfected with virion RNA. Monolayers of MMH-D3, Ltk-, and GL37 cells grown in 25-cm2 flasks were transfected with virion RNA. At 6 h posttransfection, cells were washed three times, split 1:3, and grown at 35°C in a CO2 incubator. Cells were split 1:3 weekly; one-third of the collected cells were subjected to three freeze-thaw cycles, cell debris was pelleted, and the supernatant containing HAV was stored at −70°C. HAV was titrated as described in the legend to Fig. 1. Error bars, standard deviations.
MMH-D3 cells grown under suboptimal conditions gained susceptibility to HAV PI infection.
It was of interest to determine whether changes in cell culture conditions could affect the susceptibility of MMH-D3 cells to HAV infection. To do so, we cultured MMH-D3 cells under suboptimal conditions in the absence of EGF and IGF II for 40 passages, and we termed the resulting cell line HP-MMH cells. It should be pointed out that the morphology of this cell line was different from that of the parental MMH-D3 line, i.e., the cells looked smaller and lost contact inhibition. We compared the growth of HAV in MMH-D3 cells cultured under optimal conditions, MMH-D3 cells cultured under suboptimal conditions for 3 days, HP-MMH cells, and control GL37 cells. Cells were infected with HAV PI at an MOI of 2 TCID50/cell and were incubated at 35°C under 5% CO2. At 24 h postinfection, cells were washed and trypsinized. One-third of the cells was seeded in a T25 flask for further culture, one-third was used to produce a viral stock, and the remaining one-third was used to extract total RNA for a viral quantitative RT-PCR assay (Taqman). Surprisingly, the MMH-D3 cells cultured under suboptimal conditions for 3 days and HP-MMH cells gained susceptibility to HAV infection and produced approximately 105 TCID50/ml (Fig. 3A). As expected, HAV grew in GL37 cells to approximately 106 TCID50/ml and did not grow in MMH-D3 cells cultured under optimal conditions. Indirect immunofluorescence analysis using human anti-HAV antibodies showed that approximately 1 to 2% of the HAV PI-infected mouse cells had the characteristic cytoplasmic granular fluorescence of HAV-infected cells compared to 100% of the GL37-infected cells (data not shown). This limited growth of HAV in the mouse cells indicated that HAV infection was still partially blocked in MMH-D3 cells cultured under suboptimal conditions for 3 days and HP-MMH cells. A Taqman assay (Fig. 3B) performed at 32 days postinfection showed that approximately 105 and 3 × 104 HAV genome equivalents per μg of total cellular RNA were produced in MMH-D3 cells cultured under suboptimal conditions for 3 days and in HP-MMH cells, respectively. Viral RNA was not detected in MMH-D3 cells grown under optimal conditions or in mock-infected cells. Positive-control GL37 cells produced approximately 2 to 3 log units more RNA than MMH-D3 cells grown under suboptimal conditions. These Taqman results confirmed that the MMH-D3 cells grown under suboptimal conditions gained susceptibility to HAV infection. Further analysis showed that approximately 105 TCID50 of HAV/ml were detected in cells grown without EGF or without both EGF and IGF II (Fig. 4). A low level of HAV (103 TCID50/ml) was detected in MMH-D3 cells cultured with both growth factors (complete medium) or in the absence of IGF II, which most likely represents input virus or background levels of HAV replication. Consequently, withdrawal of EGF alone was sufficient to allow HAV PI infection of the MMH-D3 cells. Since HAV replicated in MMH-D3 cells transfected with virion RNA (Fig. 2), our results clearly showed that withdrawal of EGF eliminated the block(s) to HAV PI infection in these cells.
FIG. 3.

Susceptibility to HAV infection of MMH-D3 cells grown under suboptimal conditions. Monolayers of MMH-D3 cells grown under optimal conditions [MMH-D3 (+) IGF II/EGF] or without IGF II and EGF [MMH-D3 (−) IGF II/EGF] were infected with HAV PI at an MOI of 1 TCID50/cell. HP MMH cells, which were derived from MMH-D3 cells after 40 passages under suboptimal conditions, and positive-control GL37 cells were also infected with HAV under the same conditions. At 24 h postinfection, cells were washed three times, split 1:3, and grown at 35°C in a CO2 incubator. Cells were split 1:3 weekly and grown at 35°C in a CO2 incubator. (A) An aliquot of the remaining cells corresponding to approximately one-third of the infected monolayer was subjected to three freeze-thaw cycles. Cellular debris was pelleted, and the virus in the supernatant was stored at −70°C. HAV was titrated as described in the legend to Fig. 1. Error bars, standard deviations. (B) At 32 days postinfection, approximately one-third of the collected cells were used to prepare total RNA. The number of HAV genomes per microgram of total RNA was determined by a quantitative Taqman RT-PCR. Bars represent mean HAV RNA copies per microgram of total RNA obtained from four independent RT-PCRs. The limit of detection of the Taqman PCR is indicated by a dashed line. Error bars, standard deviations.
FIG. 4.

Growth of HAV in MMH-D3 cells cultivated without EGF. MMH-D3 cells grown under optimal conditions (with IGF II and EGF [complete medium]), without IGF II [(−) IGF II], without EGF [(−) EGF], or without both growth factors [(−) IGF II/EGF] were infected with HAV PI at an MOI of a 2 TCID50/cell. At 24 h postinfection, cells were washed three times, split 1:3, and grown at 35°C in a CO2 incubator. Cells were split 1:3 weekly; approximately one-third of the remaining cells were subjected to three freeze-thaw cycles, cellular debris was pelleted, and the HAV in the supernatant was titrated as described in the legend to Fig. 1. Error bars, standard deviations.
Adaptation of HAV to grow in MMH-D3 cells cultured under optimal conditions.
We hypothesized that passages of HAV in HP-MMH cells could result in the accumulation of host range mutations that would allow HAV infection of MMH-D3 cells cultured under optimal conditions. Therefore, we grew HAV PI in HP-MMH cells for an extended period. Cells were infected with HAV PI at an MOI of 2 TCID50/cell, incubated at 37°C under 5% CO2, and split 1:3 weekly. At 83 days postinfection, a viral stock was prepared and termed HAV-MMH. In addition, we performed 21 serial passages of HAV-MMH in naïve HP-MMH cells grown in T25 flasks. Viral stocks of each serial passage were prepared and stored at −80°C. We then analyzed the growth of HAV-MMH passage 20 (P20), HAV-MMH passage 21 (P21), and parental HAV PI in MMH-D3 cells cultured under optimal conditions (Fig. 5). Cells were infected with HAV at an MOI of 0.2 TCID50/cell and incubated at 37°C under 5% CO2. At 24 h postinfection, monolayers were washed and trypsinized. One-third of the cells was seeded into a T25 flask for further culture, and one-third of the cells was used to produce a viral stock. Titration of the viral stocks showed that HAV-MMH P20 and P21 grew 5 log units in MMH-D3 cells whereas HAV PI did not grow in these cells. Hence, extensive passage of HAV PI in HP-MMH cells resulted in the selection of HAV-MMH variants that grew in MMH-D3 cells cultured under optimal conditions. These variants overcame the cell entry blocks(s) that prevented infection of MMH-D3 cells cultured under optimal conditions with the parental HAV PI.
FIG. 5.

Growth of HAV-MMH in MMH-D3 cells cultivated under optimal conditions. Monolayers of MMH-D3 cells grown in six-well plates with a medium containing EGF and IGF II were infected with parental HAV PI, HAV-MMH P20, or HAV-MMH P21 at an MOI of 0.2 TCID50/cell. At 24 h postinfection, cells were washed three times and incubated at 35°C in a CO2 incubator. Cells were split 1:3 weekly; approximately one-third of the remaining cells were subjected to three freeze-thaw cycles, cellular debris was pelleted, and the HAV in the supernatant was titrated as described in the legend to Fig. 1. Error bars, standard deviations.
HAV variants that grow in MMH-D3 cells contain mutations in the capsid region.
To determine whether HAV-MMH P21 contained mutations in the capsid region that allowed it to grow in MMH-D3 cells cultured under optimal conditions, we analyzed the nucleotide sequences of six viral clones obtained by endpoint dilution (clones 3 to 7 and 9). To do so, cDNA fragments of the capsid region of the uncloned HAV-MMH P21, the six clones, and the parental HAV PI were amplified by RT-PCR, gel purified, and subjected to automated nucleotide sequencing. The deduced amino acid sequences of the capsid region (Table 1) revealed that the uncloned HAV-MMH and the six clones contained N1237D and D2132G substitutions in VP1 and VP2, respectively, compared to HAV PI. In addition, clone 5 contained an A3190V mutation in VP3, and clone 7 contained a A1176D mutation in VP1. Since all the clones infected MMH-D3 cells (data not shown), our results suggested that the N1237D and D2132G mutations were responsible for the adaptation of HAV to mouse hepatocyte-like MMH-D3 cells.
TABLE 1.
Amino acida substitutions in the capsid proteins VP1, VP2, and VP3 of uncloned HAV-MMH and six HAV-MMH clones compared to HAV PI
| Virus | VP1
|
VP2 residue 132 | VP3 residue 190 | |
|---|---|---|---|---|
| Residue 176 | Residue 237 | |||
| HAV PI | A | N | D | A |
| HAV-MMH | D | G | ||
| HAV-MMH clone 3 | D | G | ||
| HAV-MMH clone 4 | D | G | ||
| HAV-MMH clone 5 | D | G | V | |
| HAV-MMH clone 6 | D | G | ||
| HAV-MMH clone 7 | D | D | G | |
| HAV-MMH clone 9 | D | G | ||
One-letter amino acid code is used.
DISCUSSION
The determinants of HAV replication and pathogenesis are poorly understood. Experimentation with HAV is difficult because it grows poorly in cell culture. Since primates are the only animal models for this virus, development of a small-animal model to study the pathogenesis of HAV is highly desirable. A mouse model for HAV would be ideal, but unfortunately, HAV does not replicate efficiently in cells of murine origin.
To circumvent this problem, we attempted to adapt HAV to grow efficiently in MMH-D3 cells, a nontransformed mouse hepatocyte cell line (2). To maintain optimal proliferation and retain their hepatocyte-like characteristics, the MMH-D3 cells were cultivated in the presence of insulin, EGF, and IGF II. Our results showed that HAV can grow in MMH-D3 cells transfected with virion RNA but not in MMH-D3 cells infected with HAV particles, which implies a problem at the cell entry level. The cell entry block(s) was overcome by withdrawal of EGF from the culture medium of the MMH-D3 cells. This acquired susceptibility to HAV infection could be due to several reasons, for instance, (i) the expression of an endogenous mouse gene(s) capable of serving as a cellular receptor for HAV, (ii) the activation of transcriptional or posttranscriptional events that allowed normally expressed genes to function as HAV cellular receptors, or (iii) the repression of a mouse gene(s) that prevented cell entry of HAV. More research will be required to elucidate the molecular basis for the gain of susceptibility to HAV infection after withdrawal of EGF from the cell culture medium of the MMH-D3 cells.
Interestingly, hepatitis A (HA) is an age-dependent disease. Children 6 years old or younger usually develop subclinical forms of HA whereas older individuals develop more-severe manifestations of the disease (13, 16). Because aging reduces the response to EGF treatment and the phosphorylation of the EGF receptor (18, 27; reference 31 and references therein), it is tantalizing to postulate an inverse relationship between the severity of HA and the response to EGF. The response to EGF may also play a role in the severity of HA. Further research will be required to determine whether EGF could also influence the susceptibility of human hepatocytes to HAV infection and to determine whether EGF plays a role in the severity of HA.
It has previously been reported that HAV can grow efficiently in cells of primate origin and some cells of nonprimate origin but not in mouse cells. Takeda et al. (35) detected negative-strand HAV RNA in mouse L929 and NIH/3T3 cells after infection with HAV. However, low levels of HAV (101.8 TCID50s) were produced after transfection of viral RNA, suggesting that a very limited infection occurred in these cells. We have recently shown that mouse Ltk- cells can support HAV growth after transfection with HAV infectious cDNA (19). It should be pointed out that Ltk- cells were not susceptible to HAV infection and that the cDNA-transfected Ltk- cells that supported efficient levels of HAV growth had to be selected by several rounds of single-cell cloning. This paper is the first report of mouse cells that are susceptible to HAV infection and support the efficient growth of this virus. We showed that MMH-D3 cells grown under suboptimal conditions gained susceptibility to HAV infection and produced approximately 105 TCID50 of virus/ml. Additionally, serial passages of HAV in HP-MMH cells, which were derived from MMH-D3 cells cultured under suboptimal conditions, resulted in the selection of viral variants that acquired the capability of infecting hepatocyte-like MMH-D3 cells cultured under optimal conditions. These mouse-adapted HAV-MMH variants contained four amino acid substitutions in the capsid region. Mutations N1237D and D2132G were present in the six clones that we analyzed, which suggested that these two substitutions were responsible for the adaptation of HAV to mouse hepatocyte-like cells. We are currently constructing HAV mutants to determine whether these two changes are indeed responsible for the adaptation of HAV to mouse hepatocyte-like cells. Although the structure of HAV has not yet been resolved, a model of the HAV capsid predicted that both mutations are located at the viral surface (20). Interestingly, N1237D is adjacent to Q1232, a residue that is within the major antigenic site of HAV (26). In addition, Y1236 has been shown to be accessible to iodination in highly purified virions (30), which also suggests that this residue is exposed at the virion surface. Therefore, it is possible that amino acid residues 237 of VP1 and 132 of VP2 are indeed located at the surface of the HAV particle and that mutations N1237D and D2132G allowed the interaction of HAV-MMH with a mouse receptor that mediated cell entry. Further research and the elucidation of the structure of HAV will be required to validate this hypothesis.
A multigene family of mouse orthologs of hhavcr-1 has recently been identified (24). Two members of this multigene family, Tim1 and Tim2, share a high degree of homology with hhavcr-1. It is presently unknown whether members of the mouse Tim family have HAV receptor function. However, preliminary results (D. Feigelstock and G. Kaplan, unpublished data) indicated that withdrawal of EGF from the cell culture medium of MMH-D3 cells did not affect the levels of Tim1 and Tim2 expression. Further work will be required to determine whether members of the Tim family have HAV receptor function.
In this paper, we showed for the first time that mouse cells code for all the cellular factors required for the efficient growth of HAV. We also showed that MMH-D3 cells gained susceptibility to HAV infection upon withdrawal of EGF from the cell culture medium. Our data also suggest that mutations at the viral surface allowed the entry of HAV into mouse hepatocyte-like cells. The experimental system reported here will contribute to the identification of cellular factors required for HAV growth and possibly to the development of a mouse model for study of the pathogenesis of HAV.
Acknowledgments
We thank Susan Zullo for critical review of the manuscript, Frank Chisari for encouraging discussions on the adaptation of HAV to mouse cells and for providing the MMH-D3 cells, and Michael Klutch for automatic sequencing.
REFERENCES
- 1.Almog, R., M. Low, D. Cohen, G. Robin, S. Ashkenazi, H. Bercovier, M. Gdalevich, Y. Samuels, I. Ashkenazi, J. Shemer, A. Eldad, and M. S. Green. 1999. Prevalence of anti-hepatitis A antibodies, hepatitis B viral markers, and anti-hepatitis C antibodies among immigrants from the former USSR who arrived in Israel during 1990-1991. Infection 27:212-217. [DOI] [PubMed] [Google Scholar]
- 2.Amicone, L., F. M. Spagnoli, G. Spath, S. Giordano, C. Tommasini, S. Bernardini, V. De Luca, C. Della Rocca, M. C. Weiss, P. M. Comoglio, and M. Tripodi. 1997. Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J. 16:495-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An, G., G. Luo, and R. Wu. 1994. Expression of MUC2 gene is down-regulated by vitamin A at the transcriptional level in vitro in tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10:546-551. [DOI] [PubMed] [Google Scholar]
- 4.Antoine, B., F. Levrat, V. Vallet, T. Berbar, N. Cartier, N. Dubois, P. Briand, and A. Kahn. 1992. Gene expression in hepatocyte-like lines established by targeted carcinogenesis in transgenic mice. Exp. Cell Res. 200:175-185. [DOI] [PubMed] [Google Scholar]
- 5.Arankalle, V. A., S. A. Tsarev, M. S. Chadha, D. W. Alling, S. U. Emerson, K. Banerjee, and R. H. Purcell. 1995. Age-specific prevalence of antibodies to hepatitis A and E viruses in Pune, India, 1982 and 1992. J. Infect. Dis. 171:447-450. [DOI] [PubMed] [Google Scholar]
- 6.Cohen, J. I., B. Rosenblum, J. R. Ticehurst, R. J. Daemer, S. M. Feinstone, and R. H. Purcell. 1987. Complete nucleotide sequence of an attenuated hepatitis A virus: comparison with wild-type virus. Proc. Natl. Acad. Sci. USA 84:2497-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daemer, R. J., S. M. Feinstone, I. D. Gust, and R. H. Purcell. 1981. Propagation of human hepatitis A virus in African green monkey kidney cell culture: primary isolation and serial passage. Infect. Immun. 32:388-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dienstag, J. L., S. M. Feinstone, A. Z. Kapikian, and R. H. Purcell. 1975. Faecal shedding of hepatitis-A antigen. Lancet i:765-767. [DOI] [PubMed] [Google Scholar]
- 9.Dotzauer, A., S. M. Feinstone, and G. Kaplan. 1994. Susceptibility of nonprimate cell lines to hepatitis A virus infection. J. Virol. 68:6064-6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emerson, S. U., Y. K. Huang, and R. H. Purcell. 1993. 2B and 2C mutations are essential but mutations throughout the genome of HAV contribute to adaptation to cell culture. Virology 194:475-480. [DOI] [PubMed] [Google Scholar]
- 11.Feigelstock, D., P. Thompson, P. Mattoo, Y. Zhang, and G. G. Kaplan. 1998. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J. Virol. 72:6621-6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funkhouser, A. W., R. H. Purcell, E. D'Hondt, and S. U. Emerson. 1994. Attenuated hepatitis A virus: genetic determinants of adaptation to growth in MRC-5 cells. J. Virol. 68:148-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadler, S. C., and L. McFarland. 1986. Hepatitis in day care centers: epidemiology and prevention. Rev. Infect. Dis. 8:548-557. [DOI] [PubMed] [Google Scholar]
- 14.Hollinger, F. B., and S. U. Emerson. 2001. Hepatitis A virus, p. 799-840. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 15.Kiyohara, T., T. Satoh, H. Yamamoto, A. Totsuka, and Y. Moritsugu. 1997. The latest seroepidemiological pattern of hepatitis A in Japan. Jpn. J. Med. Sci. Biol. 50:123-131. [DOI] [PubMed] [Google Scholar]
- 16.Lednar, W. M., S. M. Lemon, J. W. Kirkpatrick, R. R. Redfield, M. L. Fields, and P. W. Kelley. 1985. Frequency of illness associated with epidemic hepatitis A virus infections in adults. Am. J. Epidemiol. 122:226-233. [DOI] [PubMed] [Google Scholar]
- 17.LeDuc, J. W., S. M. Lemon, C. M. Keenan, R. R. Graham, R. H. Marchwicki, and L. N. Binn. 1983. Experimental infection of the New World owl monkey (Aotus trivirgatus) with hepatitis A virus. Infect. Immun. 40:766-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, J., and N. J. Holbrook. 2003. Common mechanisms for declines in oxidative stress tolerance and proliferation with aging. Free Radic. Biol. Med. 35:292-299. [DOI] [PubMed] [Google Scholar]
- 19.Lu, J., G. Dveksler, and G. G. Kaplan. 2004. Rescue of hepatitis A virus from cDNA-transfected but not virion RNA-transfected mouse Ltk- cells. Arch. Virol. 149:759-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo, M., M. G. Rossmann, and A. C. Palmenberg. 1988. Prediction of three-dimensional models for foot-and-mouth disease virus and hepatitis A virus. Virology 166:503-514. [DOI] [PubMed] [Google Scholar]
- 21.Mathiesen, L. R., F. Hardt, O. Dietrichson, R. H. Purcell, D. Wong, P. Skinhoj, J. O. Nielsen, H. Zoffmann, and K. Iversen. 1980. The role of acute hepatitis type A, B, and non-A non-B in the development of chronic active liver disease. Scand. J. Gastroenterol. 15:49-54. [DOI] [PubMed] [Google Scholar]
- 22.Matricardi, P. M., F. Rosmini, L. Ferrigno, R. Nisini, M. Rapicetta, P. Chionne, T. Stroffolini, P. Pasquini, and R. D'Amelio. 1997. Cross sectional retrospective study of prevalence of atopy among Italian military students with antibodies against hepatitis A virus. BMJ 314:999-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matricardi, P. M., F. Rosmini, V. Panetta, L. Ferrigno, and S. Bonini. 2002. Hay fever and asthma in relation to markers of infection in the United States. J. Allergy Clin. Immunol. 110:381-387. [DOI] [PubMed] [Google Scholar]
- 24.McIntire, J. J., S. E. Umetsu, O. Akbari, M. Potter, V. K. Kuchroo, G. S. Barsh, G. J. Freeman, D. T. Umetsu, and R. H. DeKruyff. 2001. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat. Immunol. 2:1109-1116. [DOI] [PubMed] [Google Scholar]
- 25.McIntire, J. J., S. E. Umetsu, C. Macaubas, E. G. Hoyte, C. Cinnioglu, L. L. Cavalli-Sforza, G. S. Barsh, J. F. Hallmayer, P. A. Underhill, N. J. Risch, G. J. Freeman, R. H. DeKruyff, and D. T. Umetsu. 2003. Immunology: hepatitis A virus link to atopic disease. Nature 425:576. [DOI] [PubMed] [Google Scholar]
- 26.Nainan, O. V., M. A. Brinton, and H. S. Margolis. 1992. Identification of amino acids located in the antibody binding sites of human hepatitis A virus. Virology 191:984-987. [DOI] [PubMed] [Google Scholar]
- 27.Palmer, H. J., C. T. Tuzon, and K. E. Paulson. 1999. Age-dependent decline in mitogenic stimulation of hepatocytes. Reduced association between Shc and the epidermal growth factor receptor is coupled to decreased activation of Raf and extracellular signal-regulated kinases. J. Biol. Chem. 274:11424-11430. [DOI] [PubMed] [Google Scholar]
- 28.Provost, P. J., and M. R. Hilleman. 1979. Propagation of human hepatitis A virus in cell culture in vitro. Proc. Soc. Exp. Biol. Med. 160:213-221. [DOI] [PubMed] [Google Scholar]
- 29.Reed, L. J., and H. Muench. 1938. A simple method of estimating fifty per cent end points. Am. J. Hyg. 27:493-497. [Google Scholar]
- 30.Robertson, B. H., V. K. Brown, B. P. Holloway, B. Khanna, and E. Chan. 1989. Structure of the hepatitis A virion: identification of potential surface-exposed regions. Arch. Virol. 104:117-128. [DOI] [PubMed] [Google Scholar]
- 31.Roth, G. S. 1995. Changes in tissue responsiveness to hormones and neurotransmitters during aging. Exp. Gerontol. 30:361-368. [DOI] [PubMed] [Google Scholar]
- 32.Silberstein, E., G. Dveksler, and G. G. Kaplan. 2001. Neutralization of hepatitis A virus (HAV) by an immunoadhesin containing the cysteine-rich region of HAV cellular receptor-1. J. Virol. 75:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silberstein, E., L. Xing, W. van de Beek, J. Lu, H. Cheng, and G. G. Kaplan. 2003. Alteration of hepatitis A virus (HAV) particles by a soluble form of HAV cellular receptor 1 containing the immunoglobulin- and mucin-like regions. J. Virol. 77:8765-8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spagnoli, F. M., L. Amicone, M. Tripodi, and M. C. Weiss. 1998. Identification of a bipotential precursor cell in hepatic cell lines derived from transgenic mice expressing cyto-Met in the liver. J. Cell Biol. 143:1101-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda, Y., M. Ashida, and C. Hamada. 1996. Growth of hepatitis A virus in murine cells. Acta Virol. 40:201-208. [PubMed] [Google Scholar]
- 36.Thompson, P., J. Lu, and G. G. Kaplan. 1998. The Cys-rich region of hepatitis A virus cellular receptor 1 is required for binding of hepatitis A virus and protective monoclonal antibody 190/4. J. Virol. 72:3751-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umetsu, D. T., J. J. McIntire, O. Akbari, C. Macaubas, and R. H. DeKruyff. 2002. Asthma: an epidemic of dysregulated immunity. Nat. Immunol. 3:715-720. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, Y., and G. G. Kaplan. 1998. Characterization of replication-competent hepatitis A virus constructs containing insertions at the N terminus of the polyprotein. J. Virol. 72:349-357. [DOI] [PMC free article] [PubMed] [Google Scholar]