

A recent paper by J. R. St-Jean and colleagues reported the complete genome sequences of the human coronavirus OC43 (HCoV-OC43) laboratory strain from the American Type Culture Collection (ATCC) and an HCoV-OC43 clinical isolate, designated Paris (7). The ATCC HCoV-OC43 (VR759) strain originated in 1967 and was passaged several times in suckling mouse brain and in cell culture. The contemporary HCoV-OC43 Paris strain was isolated in 2001 and was subsequently cultured in an HRT-18 cell line by the authors. A high degree of genetic stability was stated for HCoV-OC43 since only six nucleotide variations in the whole genome could be observed between both HCoV-OC43 strains, with isolation dates 34 years apart. There are, however, some arguments to suggest that the HCoV-OC43 genome is not as stable as suggested by the authors and that the HCoV-OC43 Paris strain might not be a contemporary strain but might be a result of cross-contamination with the ATCC HCoV-OC43 strain.
A first argument is based on the reported evolutionary rates of RNA viruses in general and coronaviruses in particular. An evolutionary rate of 4.0 × 10−4 nucleotide substitutions per site per year was estimated for severe acute respiratory syndrome (SARS) coronavirus by using ORF1ab sequence data (5), and an evolutionary rate of 7.5 × 10−4 nucleotide substitutions per site per year was estimated for porcine transmissible gastroenteritis virus by using S gene sequence data (6). Most RNA viruses have been reported to have evolutionary rates in the range of 10−4 to 10−3 substitutions per site per year, although slightly slower mutation rates have also been described (1, 2). Here, we estimated the evolutionary rate for the complete genome sequence data of HCoV-OC43 (one strain; GenBank accession number NC_005147 [isolated in 1967]) and BCoV (four strains; GenBank accession numbers U00735 and AF220295 [isolated in 1972] and NC_003045 and AF391542 [isolated in 1998]) using a maximum likelihood (ML) approach. A ML phylogenetic tree for the complete genomes was reconstructed with PAUP version 4.10 (8), and the rate of nucleotide substitution was estimated by using Rhino software version 1.2 (http://evolve.zoo.ox.ac.uk/), which implements a molecular clock model accommodating serially sampled sequences (4). This approach resulted in an estimate of 1.54 × 10−4 nucleotide substitutions per site per year (95% confidence interval, 0.97 × 10−4 to 2.12 × 10−4) (4), which is 30-fold higher then the mutation rate (5.7 × 10−6 nucleotide substitutions per site per year) corresponding to the data presented by St-Jean et al. The maximum likelihood evolutionary rate was used to assess the probability that the contemporary HCoV-OC43 strain described by St-Jean and colleagues was effectively sampled in 2001. Assuming that nucleotide substitutions follow a Poisson process (3), the probability of observing only six mutations (n = 6) between two isolates sampled 34 years apart (t = 34) can be calculated by using the following model:
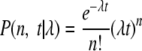 |
where λ denotes the evolutionary rate in nucleotide substitutions per year.
For the maximum likelihood estimate of the evolutionary rate, the expected number of mutations after 34 years of evolution is 163. The probability of observing only six mutations after 34 years of evolution is 4.98 × 10−61 (the probability of observing six or fewer mutations is 5.17 × 10−61). Even for the lower 95% confidence interval limit of the evolutionary rate, the probability of observing only six mutations or fewer during this time is extremely low (P = 4.92 × 10−36) and therefore we believe that it seems unlikely that the Paris HCoV-OC43 strain is truly a circulating strain from 2001. It should be noted that this approach is conservative since it ignores any shared ancestry of the two viral strains.
Furthermore, in a paper by Vabret and colleagues from the same laboratories as St-Jean et al. describing an HCoV-OC43 outbreak in Normandy, France, in 2001, M gene sequence data analysis revealed an estimated genetic distance of up to approximately 32 nucleotide changes per 1,000 nucleotides between some of the clinical isolates (9). For the whole M gene, this observation would apply to up to approximately 22 nucleotide variations between some of the 2001 isolates, whereas St-Jean and coworkers found only one nucleotide change in the M gene between two HCoV-OC43 strains isolated 34 years apart. Also, St-Jean and colleagues found only one variation in the S gene, the most variable coronavirus gene. Comparison of this observation to the data of the Normandy outbreak would imply that the S gene of two HCoV-OC43 strains isolated 34 years apart is more conserved than the M gene of several clinical HCoV-OC43 isolates, all from the same year (2001), which seems improbable.
In conclusion, we believe that the findings of St-Jean et al. regarding the genetic stability of human coronavirus OC43 are unlikely. We doubt that the HCoV-OC43 contemporary strain presented by the authors is a circulating strain from 2001, which is supported by an extremely low probability value. We propose that this might be due to contamination with the HCoV-OC43 ATCC strain of the cell line used to propagate the clinical isolate.
Acknowledgments
This work was supported by a fellowship from the Flemish Fonds voor Wetenschappelijk Onderzoek (FWO) to Leen Vijgen and by FWO grant G.0288.01.
REFERENCES
- 1.Domingo, E., and J. J. Holland. 1988. High error rates, population equilibrium and evolution of RNA replication systems, p. 3-36. In E. Domingo, J. J. Holland, and P. Ahlquist (ed.), RNA genetics, vol. 3. CRC Press, Boca Raton, Fla. [Google Scholar]
- 2.Jenkins, G. M., A. Rambaut, O. G. Pybus, and E. C. Holmes. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156-165. [DOI] [PubMed] [Google Scholar]
- 3.Kimura, M. 1983. The neutral allele theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom.
- 4.Rambaut, A. 2000. Estimating the rate of molecular evolution: incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics 16:395-399. [DOI] [PubMed] [Google Scholar]
- 5.Salemi, M., W. M. Fitch, M. Ciccozzi, M. J. Ruiz-Alvarez, G. Rezza, and M. J. Lewis. 2004. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J. Virol. 78:1602-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez, C. M., F. Gebauer, C. Sune, A. Mendez, J. Dopazo, and L. Enjuanes. 1992. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 190:92-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.St-Jean, J. R., H. Jacomy, M. Desforges, A. Vabret, F. Freymuth, and P. J. Talbot. 2004. Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J. Virol. 78:8824-8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swofford, D. L. 1998. PAUP* 4.0: phylogenetic analysis using parsimony (*and other methods). Sinauer Associates, Sunderland, Massachusetts.
- 9.Vabret, A., T. Mourez, S. Gouarin, J. Petitjean, and F. Freymuth. 2003. An outbreak of coronavirus OC43 respiratory infection in Normandy, France. Clin. Infect. Dis. 36:985-989. [DOI] [PMC free article] [PubMed] [Google Scholar]