

Abstract
The iron-sulfur (Fe-S) cluster (ISC) biogenesis pathway is indispensable for many fundamental biological processes and pathogenic variations in genes encoding several components of the Fe-S biogenesis machinery, such as NFU1, BOLA3, IBA57 and ISCA2 are already implicated in causing four types of multiple mitochondrial dysfunctions syndromes (MMDS). We report on two unrelated families, with two affected children each with early onset neurological deterioration, seizures, extensive white matter abnormalities, cortical migrational abnormalities, lactic acidosis and early demise. Exome sequencing of two affected individuals, one from each family, revealed a homozygous c.259G>A [p.(Glu87Lys)] variant in ISCA1 and Mendelian segregation was confirmed in both families. The ISCA1 variant lies in the only shared region of homozygosity between the two families suggesting the possibility of a founder effect. In silico functional analyses and structural modeling of the protein predict the identified ISCA1 variant to be detrimental to protein stability and function. Notably the phenotype observed in all affected subjects with the ISCA1 pathogenic variant is similar to that previously described in all four types of MMDS. Our findings suggest association of a pathogenic variant in ISCA1 with another MMDS.
Keywords: Multiple mitochondrial dysfunctions syndrome, ISCA1, Iron-sulfur biogenesis, Neuroregression, Leukodystrophy
INTRODUCTION
Iron-sulfur (Fe-S) clusters (ISCs) are small inorganic cofactors of metalloproteins found in bacteria and eukaryotes that are indispensable for cellular processes such as respiration, protein translation, purine metabolism, DNA repair and gene expression regulation.1–3 IscA proteins are highly conserved and fundamental to the physiology of prokaryotes and eukaryotes. Loss of function of several Fe-S cluster assembly components have been known to result in human diseases including multiple mitochondrial dysfunctions syndromes (MMDS).4–7
Currently four types, MMDS1 (MIM #605711), MMDS2 (MIM #614299), MMDS3 (MIM #615330) and MMDS4 (MIM #616370) have been mapped to NFU1 (MIM #608100), BOLA3 (MIM #613183), IBA57 (MIM #615316) and ISCA2 (MIM #615317) respectively. All MMDS share variable neurodevelopmental delay, regression, seizures, lactic acidosis and leukodystrophy resulting in early death of affected individuals.4, 8–10 Here we report another likely MMDS resulting from a biallelic mutation in ISCA1, a key component of the Fe-S biogenesis process, from two unrelated Indian families.
MATERIALS AND METHODS
Patients
Family 1 consulted us for prenatal counseling. They were in a consanguineous marriage and had two healthy daughters and lost two children (Figure 1A). Both P1 and P2 had normal birth and antenatal history followed by inconsolable cry and feeding difficulties since newborn period. Both of them did not achieve any other developmental milestone except partial head control at 3 months of age. P1 and P2 developed seizures at the age of 5 months and 4 months respectively followed by progressive neurological deterioration and spasticity. P1 died at 1 year and 7 months while P2 succumbed at 5 years of age. Stored DNA was obtained for patient P2 and blood samples were obtained for parents and healthy children. Brain imaging of both children showed pachygyria, extensive cerebral and cerebellar white matter disease and dilated cerebral ventricles. Increased lipid lactate peak was seen on magnetic resonance spectroscopy of brain in P1 (Figure 1B). Normal hematological and biochemical investigations were noted except for elevated creatinine phosphokinase in P2.
Figure 1.
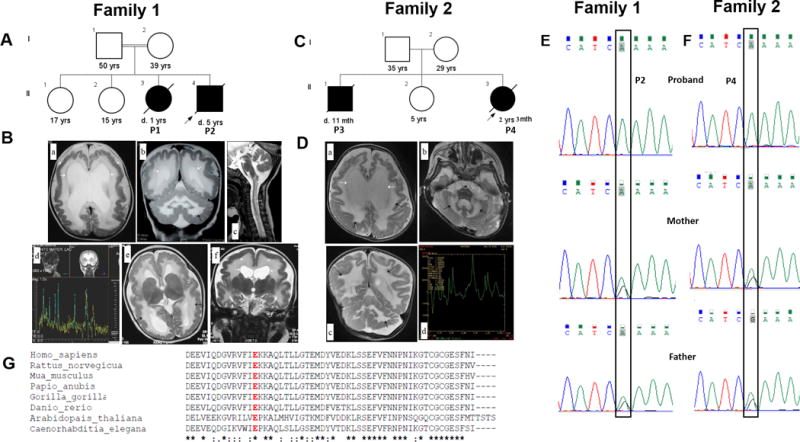
(A) Pedigree of Family 1. (B) Brain MRI images of family 1, P1 and P2. (a,b) T2 weighted MRI shows pachygyria (arrowhead), moderate dilatation of the ventricles (white arrows) and diffuse T2 hyperintensity in the cerebral and cerebellar white matter (black arrows) and posterior limb of internal capsule in P1 at 6 months. (c) MRI performed for the same child at 9 months shows myelination abnormality involving the medulla and the cervical cord (white arrows) in the white matter suggestive of delayed myelination and (d) MR spectroscopy shows increased lipid-lactate peaks. (e,f) T2 weighted MRI in P2 shows pachygyria (arrowhead), moderate dilatation of the ventricles (white arrows) and diffuse T2 hyperintensity (black arrows) at the age of 5 months. (C) Pedigree of Family 2. (D) Brain MRI images of family 2, P4. (a–c) T2 weighted MRI in P4 shows diffuse T2 hyperintensity in the cerebral and cerebellar white matter (black arrows), dilated ventricles (white arrows) with (d) raised lipid-lactate peaks on MR spectroscopy at the age 1 year 11 months, consistent with dysmyelination. (E,F) Sanger validation of the ISCA1 variant in family 1 and 2. The pathogenic variation c.259G>A of ISCA1 is found in homozygous state in (upper panel) probands of family 1 and 2 and is heterozygous in their parents (middle and lower panel). (G) Comparison of ISCA1 orthologs from H. sapiens, R. norvegicus, M. musculus, P. anubis, G. gorilla, D. rerio, A. thaliana and C. elegans reveals the high conservation of the Glu87 residue (highlighted in red).
Family 2 was ascertained from our in-house exome data for children with neurodevelopmental disorders. P3 and P4 (Figure 1C) were born to non-consanguineous couple and were asymptomatic till first two months of life. They did not achieve any developmental milestones. Seizures developed at the age of 3 months and 2 months in P3 and P4 respectively following which neurologic deterioration was noted in them. P3 had lactic acidosis and white matter disease was documented in medical records of P3 on computed tomography of brain. P4 had extensive leukodystrophy involving cerebral and cerebellar white matter with dilated ventricles. Magnetic resonance spectroscopy showed elevated lipid lactate peak in her brain (Figure 1D). They succumbed to the disease at the age of 11 months and 2 years 3 months respectively. Complete clinical details for both families are provided in Table 1.
Table 1.
Clinical findings observed in our patients.
| Clinical Findings | Family 1 | Family 2 |
|---|---|---|
| Individual | P2 | P4 |
| Origin | Indian | Indian |
| Sex | Male | Female |
| Age at assessment | 8 months | 1 year 11 months |
| Age at demise | 5 years | 2 years 3 months |
| Coding DNA change | c.259G>A | c.259G>A |
| Amino acid change | p.E87K | p.E87K |
| Zygosity | Homozygous | Homozygous |
| Exon | 4 | 4 |
| Birth weight (gm/SD) | 3600/−1 | 2700/−1 |
| Anthropometry | ||
| Weight (gm/SD) | NA | 5800/−6 |
| Length (cm/SD) | NA | 76/−5 |
| OFC (cm/SD) | 42/−2 | 43.5/−6 |
| Neurological abnormalities | ||
| Seizures | + | + |
| Onset of seizures | 4 months | 2 months |
| Developmental delay | + | + |
| Milestones achieved | Partial head control | No milestones achieved |
| Feeding difficulty | + | + |
| Neurological deterioration | + | + |
| Tone | Spasticity | Spasticity |
| Deep tendon reflexes | Exaggerated | Exaggerated |
| Strabismus | − | + |
| Other clinical findings | History of incessant cry, tremors in the hands | − |
| MRI findings | ||
| Pachygyria | + | − |
| Cerebral white matter | Leukodystrophy | Leukodystrophy |
| Cerebellar white matter | Leukodystrophy | Leukodystrophy |
| Cerebral ventriculomegaly | + | + |
| MRS findings | Elevated lipid-lactate peak | Elevated lipid-lactate peak |
| EEG findings | NA | Normal |
| Ophthalmological findings | Stippled pigmentation of fundus | NA |
| Hearing evaluation | NA | Normal |
| Blood lactate (mg/dl) | 36 | 40.5 |
| CPK (IU/L) | 568 | 42 |
| Hematology | Normal | Normal |
| Renal function tests | Normal | Normal |
| Liver function tests | Normal | Normal |
| Metabolic profile1 | Normal | NA |
Abbreviations: NA: not available; +: present; −: absent; CPK: creatine phosphokinase; EEG: electroencephalogram; MRI: Magnetic resonance imaging; MRS:Magnetic Resonance spectroscopy; OFC: occipitofrontal circumference; SD: standard deviation;
Metabolic profile: serum amino acids, urine organic acids and serum carnitine profiles
This research work has the approval of the institutional ethics committee. Specific parental informed consent was obtained for the use of photographs, clinical and research findings for publication.
Whole exome sequencing
Whole exome sequencing (WES) was carried out as described previously to achieve an average coverage depth of 100–130×, such that ~95% of the bases are covered at >20x, with a sensitivity of >90%.11 WES raw data was processed using SeqMule and the called variants were annotated with ANNOVAR.12, 13 The overall variant filtering strategy is outlined in Supplementary Table S1.
Homozygosity mapping
Using FILTUS software, homozygosity mapping was performed for both the probands with default input file settings while loading the exome VCF files.14 The minimum segment threshold and the posterior threshold parameters were set to 3Mb and 0.5 respectively for running the AutEx algorithm, that is built on the method of hidden Markov model for detecting the autozygous regions.15
Protein modeling
Structures of wild-type and mutant p.(Glu87Lys) human ISCA1 protein (UniProt accession: Q9BUE6) was predicted using the online web-server SWISS-MODEL (https://swissmodel.expasy.org/).16 Multiple sequence alignment was performed using Clustal Omega.17 Amino acid conservation was also evaluated using the ConSurf server (http://consurf.tau.ac.il/2016/).18
RESULTS
We ascertained two families with two affected children each with a neurodevelopmental disorder. The phenotype mainly consists of early onset progressive neurodegeneration, seizures, extensive white matter disease and elevated lactate levels. A non-synonymous missense pathogenic variation, c.259G>A [p.(Glu87Lys)] in ISCA1 (NM_030940.3) in the homozygous state was found in family 1 (ClinVar accession SCV000328646). The same variant was also observed in proband P4 of family 2 from the in-house exome sequencing data (Supplementary Figure S2). The variant was validated by Sanger sequencing in affected subjects from both families (upper panel in Figure 1E, 1F). Targeted testing of parents and healthy siblings in both the families using Sanger sequencing confirmed the variation to be heterozygous in them (middle and lower panel in Figure 1E, 1F). Unaffected sibs in family 1 were heterozygous carriers of the variant.
The p.(Glu87Lys) variant of ISCA1 reported here is not present in a homozygous state in 1000 Genomes project, the Exome Variant Server, CentoMD and in our in-house exome data of 139 unrelated individuals from the local population. However, it is present in the heterozygous state in 1/118662 individuals (AF = 0.000008427) in ExAC database. ISCA1 has a positive Z score (z = 1.29) for missense constraint in ExAC database, which embodies its intolerance to variations. In the recently made available gnomAD browser,19 the variant p.(Glu87Lys) is observed in heterozygous state in a single individual out of 240,238 (AF = 0.000004163). This variant appears to have an impact on the functional Fe-S biogenesis domain of ISCA1 (Supplementary Figure S1B), at a residue that is highly conserved, as indicated by Clustal Omega multiple sequence alignment (Figure 1G) and ConSurf (Figure 2A, 2B, 2C), with a PhyloP score of 7.442. This was further supported by Position Specific Evolutionary Preservation (PANTHER-PSEP)20 analysis that predicts this SNV to be probably damaging. Several functional in silico prediction tools were also used to interrogate the damaging consequences of the p.(Glu87Lys) variation. It was also predicted to be highly deleterious with a CADD score of 22.5.21 Screening for Nonacceptable Polymorphisms (SNAP2)22, 23, a web-based tool that combines a variety of information pertaining to evolutionary conservation, secondary structure and solvent accessibility of the protein predicted the p.(Glu87Lys) variant to have a severe detrimental effect with a score of 80 and accuracy of 91%.
Figure 2.
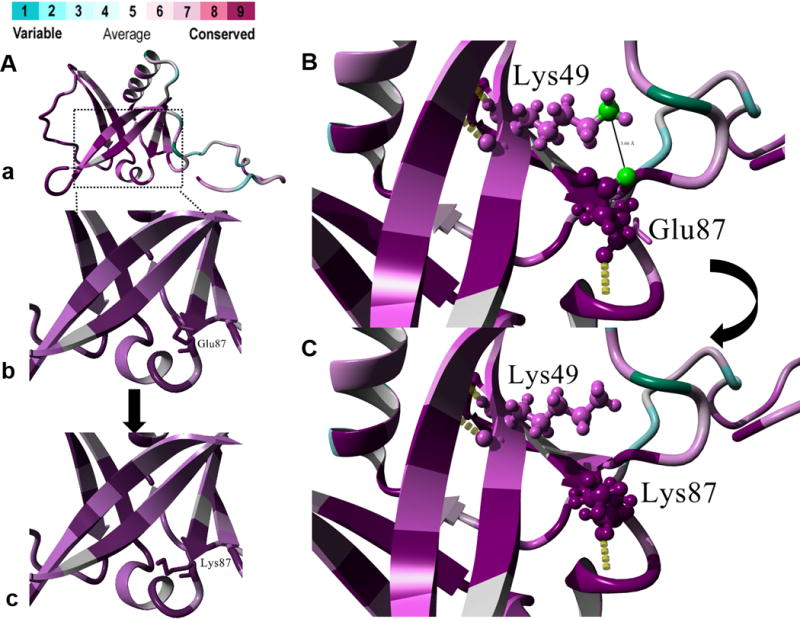
(A) Protein structure prediction shows replacement of (a,b) the native glutamic acid residue that has an acidic side-chain with (c) lysine bearing a basic side-chain. Color spectrum indicates high conservation for the Glu87 residue. (B) Predicted salt-bridge formation between Glu87 and Lys49 where the distance between the hydrogen donor atom (NZ) of Lys49 and the hydrogen acceptor atom (OE1) of Glu37 is 3.66 Å. (C) Replacement of the Glu87 with mutant Lys87 is predicted to lead to the loss of the salt-bridge between the native residue and Lys49. (D,E)
Since the two probands are of Indian origin, from the same geographic location (family 1 was consanguineous and family 2 denied any consanguinity) and the same pathogenic variant is identified in both of them, we explored the possibility of a founder effect. After examining the homozygous regions around the ISCA1 variant in both the probands, generated by the exome sequencing data, we identified only one overlapping region of homozygosity (ROH) spanning 3.3 Mb in chromosome 9 (Chr9: 85613354-88925774) flanking the variant (Supplementary Figure S3).
We used 3D protein modeling to assess the structural impact of the replacement of native acidic Glu87 by the mutant basic Lys87 residue (Figure 2A–C). The human ISCA1 gene encodes for a 129 amino acid product involved in the biogenesis of iron–sulfur (Fe-S) clusters and contains a conserved C-terminal HESB signature. The missense substitution p.(Glu87Lys) occurs at a highly conserved residue in the essential functional domain for Fe-S biogenesis in the ISCA1 protein (Supplementary Figure S1B). HOPE and ConSurf analyses indicated salt-bridge formation between Glu87 and Lys49 residues in wild-type ISCA1, which is lost in the mutant (Figure 2B and Figure 2C). VADAR analysis predicted that the side-chain Hydrogen bonding interaction of Glu87 with Ser73 and Lys88 are potentially lost in the ISCA1 mutant. Analyses of protein stability via I-Mutant (score 7) and Eris web-server (score 0.64 ΔΔG kcal/mol) are also consistent in predicting that this variant leads to destabilization of ISCA1 protein. Detailed results of protein modeling are available as a supplementary note in supplementary data.
DISCUSSION
We describe two independent families, both with two affected children each, with a severe neurodevelopmental disorder associated with a homozygous c.259G>A variant in ISCA1. The major clinical features include severe neurodevelopmental delay, seizures, spasticity and regression of milestones. Extensive leukodystrophy, lactate peaks and ventricular dilatation were observed on magnetic resonance imaging of brain. We note the phenotype observed in all affected subjects in both families is similar to that previously described for all four types of MMDS (Supplementary Table S2). Neuronal migrational abnormalities seen in both children in Family 1 has not been previously described in other subtypes of MMDS. Pigmentary retinopathy, impaired hearing and vision, elevated creatine kinase, lactic acidosis are other likely features of this condition, usually considered to suggest a mitochondrial disease.
In addition, ISCA1 was observed in the only common ROH in the two families suggesting the possibility of a founder mutation in the local population (Supplementary Table S3 and Supplementary Table S4). The genes within the shared region are given in Supplementary Table S5.
The Fe-S protein biogenesis in eukaryotes occurs in mitochondria, cytosol, or nucleus and is carried out in two steps. The first step is the formation of [2Fe-2S] cluster by proteins forming the core ISC assembly components. This cluster is transferred to monothiol glutaredoxin 5 (GRX5), which acts as a Fe/S cluster transfer protein inserting the [2Fe-2S] cluster into mitochondrial [2Fe- 2S]-requiring proteins. The second step involves the generation of [4Fe-4S] components and their integration into the required metalloproteins (Supplementary Figure S1A). Unlike the core ISC assembly components, proteins mediating the second step are not involved in formation of mitochondrial [2Fe-2S] and cytosolic [4Fe-4S] clusters.24, 25 This delivery step requires highly conserved and functionally non-redundant A-type ISC proteins, ISCA1 and ISCA2 in eukaryotes.26 Pathogenic variations in genes encoding for several components of this machinery, NFU1, BOLA3, IBA57 and ISCA2 are already implicated in causing four types of multiple mitochondrial dysfunctions syndromes MMDS.
Additionally, we note that the deleterious nature of this variation is suggested by its occurrence at residue 87 that lies within a region of 48–90 residues of ISCA1, delineated previously to be instrumental for mediating ISCA1 interaction with IOP1 (iron-only hydrogenase-like protein 1), that plays a role in cytosolic Fe-S protein assembly pathway.27 Our protein structural analyses are also consistent in predicting that the missense substitution Glu87Lys has severe consequences disrupting crucial molecular interactions, reducing protein stability and is likely debilitating for ISCA1 function.
To conclude, the existing literature on the diseases caused by pathogenic variants in various components of Fe-S biogenesis pathway, in silico functional analyses, structural modeling predictions, founder effect, the common phenotype in all affected subjects from two families, which is similar to that described in all four types of MMDS that occur due to mutations in genes encoding proteins involved in Fe-S biogenesis, strongly suggest that the presently reported ISCA1 variant likely results in another MMDS. Early demise of patients limited our access to cell lines for further functional studies.
Supplementary Material
Acknowledgments
We thank the families who cooperated with the evaluation of the subjects and consented for participation in this study.
Funding: This work was supported by National Institutes of Health funded the project titled ‘Genetic Diagnosis of Heritable Neurodevelopmental Disorders in India: Investigating the Use of Whole Exome Sequencing and Genetic Counseling to Address the High Burden of Neurodevelopmental Disorders' (1R21NS094047-01).
Footnotes
Conflict of interest
Authors declare no conflict of interest.
Supplementary Information is available in the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
References
- 1.Netz DJ, Mascarenhas J, Stehling O, Pierik AJ, Lill R. Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol. 2014;24:303–312. doi: 10.1016/j.tcb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Rouault TA, Tong WH. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol. 2005;6:345–351. doi: 10.1038/nrm1620. [DOI] [PubMed] [Google Scholar]
- 3.Lill R, Muhlenhoff U. Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- 4.Baker PR, 2nd, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS BOLA3 and the novel gene GLRX5. Brain. 2014;137:366–379. doi: 10.1093/brain/awt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, et al. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–495. doi: 10.1016/j.ajhg.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banci L, Brancaccio D, Ciofi-Baffoni S, Del Conte R, Gadepalli R, Mikolajczyk M, et al. [2Fe-2S] cluster transfer in iron-sulfur protein biogenesis. Proc Natl Acad Sci U S A. 2014;111:6203–6208. doi: 10.1073/pnas.1400102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–667. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Hassnan ZN, Al-Dosary M, Alfadhel M, Faqeih EA, Alsagob M, Kenana R, et al. ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet. 2015;52:186–194. doi: 10.1136/jmedgenet-2014-102592. [DOI] [PubMed] [Google Scholar]
- 9.Ajit Bolar N, Vanlander AV, Wilbrecht C, Van der Aa N, Smet J, De Paepe B, et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet. 2013;22:2590–2602. doi: 10.1093/hmg/ddt107. [DOI] [PubMed] [Google Scholar]
- 10.Ahting U, Mayr JA, Vanlander AV, Hardy SA, Santra S, Makowski C, et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Frontiers in genetics. 2015;6:123. doi: 10.3389/fgene.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girisha KM, Shukla A, Trujillano D, Bhavani GS, Hebbar M, Kadavigere R, et al. A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet. 2016 doi: 10.1111/cge.12762. [DOI] [PubMed] [Google Scholar]
- 12.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Y, Ding X, Shen Y, Lyon GJ, Wang K. SeqMule: automated pipeline for analysis of human exome/genome sequencing data. Sci Rep. 2015;5:14283. doi: 10.1038/srep14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vigeland MD, Gjotterud KS, Selmer KK. FILTUS: a desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics (Oxford, England) 2016;32:1592–1594. doi: 10.1093/bioinformatics/btw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang H, Thomas PD. PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics. 2016;32:2230–2232. doi: 10.1093/bioinformatics/btw222. [DOI] [PubMed] [Google Scholar]
- 21.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics. 2015;16(Suppl 8):S1. doi: 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muhlenhoff U, Richter N, Pines O, Pierik AJ, Lill R. Specialized function of yeast Isa1 and Isa2 proteins in the maturation of mitochondrial [4Fe-4S] proteins. J Biol Chem. 2011;286:41205–41216. doi: 10.1074/jbc.M111.296152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsasser HP, Muhlenhoff U, et al. The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol Biol Cell. 2012;23:1157–1166. doi: 10.1091/mbc.E11-09-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brancaccio D, Gallo A, Mikolajczyk M, Zovo K, Palumaa P, Novellino E, et al. Formation of [4Fe-4S] clusters in the mitochondrial iron-sulfur cluster assembly machinery. J Am Chem Soc. 2014;136:16240–16250. doi: 10.1021/ja507822j. [DOI] [PubMed] [Google Scholar]
- 27.Song D, Tu Z, Lee FS. Human ISCA1 interacts with IOP1/NARFL and functions in both cytosolic and mitochondrial iron-sulfur protein biogenesis. J Biol Chem. 2009;284:35297–35307. doi: 10.1074/jbc.M109.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.