

SUMMARY
Over 5% of the world population have varying degrees of hearing loss. Mutations in GJB2 are the most common cause of autosomal recessive non-syndromic hearing loss (NSHL) in many populations. The frequency and type of mutations are influenced by ethnicity. Guatemala is a multi-ethnic country with four major populations: Maya, Ladino, Xinca, and Garifuna. To determine the mutation profile of GJB2 in a NSHL population from Guatemala, we sequenced both exons of GJB2 in 133 unrelated families. A total of six pathogenic variants were detected. The most frequent pathogenic variant is c.131G>A (p.Trp44*) detected in 21 of 266 alleles. We show that c.131G>A is associated with a conserved haplotype in Guatemala suggesting a single founder. The majority of Mayan population lives in the west region of the country from where all c.131G>A carriers originated. Further analysis of genome-wide variation of individuals carrying the c.131G>A mutation compared to those of Native American, European, and African populations shows a close match with the Mayan population.
Keywords: connexin 26, founder effect, GJB2, hearing loss, Maya, mutations
INTRODUCTION
One of every thousand babies born has hearing loss, at least 50% of cases are the result of genetic factors. Genetic hearing loss is divided into non-syndromic and syndromic subgroups. Autosomal recessive non-syndromic hearing loss (ARNHL) is the most common form (1, 2). Pathogenic variants in GJB2 (MIM 121011) have been found in many populations worldwide as the most common cause of ARNHL. GJB2 is located in the first locus defined for recessive deafness (DFNB1) on chromosome 13q11-q12, and codes for the protein Connexin 26. Connexin 26 has an important role in maintaining the high potassium concentration in the endolymph of the inner ear (3). Thus, mutations in GJB2 result in a dysregulation of potassium ion concentration, which subsequently results in hearing impairment (1).
Previous studies have shown a correlation between the ethnicity of a given cohort and the frequency and type of pathogenic variants observed in GJB2 (NM_004004.5). The most common mutation in European, North American, and some Latin American populations is c.35delG (4). In Chinese and Japanese populations, the most common pathogenic variant is c.235delC (5–7), and in India the most prevalent pathogenic variant is c.71G>A (p.Trp24*) (8).
No previous data is available for deafness mutations in Guatemala. The objective of the present study is to determine which GJB2 pathogenic variants are present in the Guatemalan population with non-syndromic deafness.
MATERIAL AND METHODS
Participants
One hundred and thirty three blood samples were collected from unrelated probands with non-syndromic deafness from different regions of Guatemala. Probands were referred from the Center for Hearing and Phonetic training (CEDAF) and the Therapeutic Center of Hearing and Language (CEAL) in Guatemala. Hearing loss was diagnosed with a standard pure tone audiometry. Syndromic findings were excluded with a review of medical history and a thorough physical examination. Each participant signed an informed consent form approved by the local Ethics Committee.
GJB2 variant screening
DNA was extracted by the Gentra Puregen Cell kit (Qiagen, MD, USA) according to the manufacture´s procedures. The primers and PCR conditions used were described previously (9). The PCR products were purified by using the SpinPrep PCR Clean-up kit (EMD Millipore, MA, and USA), and sent to Macrogen (MACROGEN, MD, USA) for Sanger sequencing. The GENEIOUS R6 software (Auckland, New Zealand) was used to analyze the electropherograms.
Genome wide genotyping
We performed genome wide genotyping in seven unrelated probands who were either homozygous (two probands) or heterozygous (five probands) for the GJB2 c.131G>A variant. Samples from other individuals with c.131G>A were not available for the study. 240,000 SNPs were genotyped using Illumina Human Exome v1.0 Bead–chip. Each sample was processed according to standard protocol for Infinium HD ® Ultra assay (Illumina). Illumina ® Genome Studio software was used to extract data created by the Illumina iscan.
Bioinformatics
For the ancestry analysis we used the Exome Chip reference population in 1000 Genomes (10)(Table S1) and the HGDP-CEPH Human Genome Diversity Project panel database version 3.0 (Table S2). We initially used exome chip reference population in 1000 Genomes. The quality control (QC) procedure included confirmation of the origin of samples from unrelated individuals, single nucleotide variant (SNV) call rates > 97%, minor allele frequency (MAF) > 0, Hardy Weinberg Equilibrium (HWE) only for common variants (HWE > 1e-6 in MAF > 5%), and removing SNV duplicates (different SNV name, but the same chromosome and position). After QC checks and merging with the seven GUA (Guatemala) samples, we had 18,316 SNVs in total. In order to add the local ancestry population (Maya) into the reference population, we added more samples from the Americas region of the HGDP database (11). After QC cleaning of both reference data sets, we combined them with our GUA samples resulting in a total number of 7,367 shared SNVs. We subsequently used both EIGENSTRAT (12) and ADMIXTURE (13) to perform ancestry estimation. EIGENSTRAT estimates algorithmic ancestry with the techniques from principal component analysis. We then visually compared the distribution of the GUA samples with the reference populations. ADMIXTURE is a model-based ancestry estimation in unrelated individuals and adopts the likelihood model embedded in structure. Not only do we observe the population structure, but also we can obtain the ancestry fractions for every individual.
RESULTS
Among 133 Guatemalan unrelated families with hearing loss, 110 were simplex and 23 were multiplex with presumably autosomal recessive inheritance based on the presence of multiple affected children born to unaffected parents. Information on consanguinity was not available. Hearing loss was congenital or prelingual onset in all cases with a severity ranging from moderate to profound.
The overall frequency of pathogenic variants found in the GJB2 gene was 12.7% (34/266) (Table 1,2). Pathogenicity of variants was assessed according to ClinVar and American College of Medical Genetics (ACMG) recommendations (14, 15). There were six variants which were predicted to be pathogenic or likely pathogenic. The most common pathogenic variant was c.131G>A (21/34; 62%). This variant results in a stop codon (p.Trp44*). Remarkably, c.35delG was absent in our data set. One variant of uncertain significance was identified, c.380G>T in two probands. The most common alteration was a benign variant, c.79G>A, present on 89 chromosomes.
Table 1.
Identified variant frequencies and pathogenicity.
| c.DNA | Protein notation | dbSNP | Pathogenicity (ClinVar15 and ACMG criteria14) | Present study allele frequency | ExAC# allele frequency in Latinos |
|---|---|---|---|---|---|
| c.79G>A | p.Val27Ile | rs2274084 | Benign | 89/266 | 0.231 |
| c.131G>A | p.Trp44* | rs104894413 | Pathogenic | 21/266 | 0.00008637 |
| c.250G>T | p.Val84Leu | rs104894409 | Likely Pathogenic | 5/266 | 0.0000864 |
| c.439G>A | p.Glu147Lys | n/a | Likely Pathogenic | 3/266 | 0 |
| c.94C>T | p.Arg32Cys | rs371024165 | Likely Pathogenic | 3/266 | 0 |
| c.380G>T | p.Arg127Leu | rs111033196 | Uncertain significance | 2/266 | 0.0008667 |
| c.511G>A | p.Ala171Thr | rs201004645 | Likely Benign | 1/266 | 0 |
| c.101T>C | p.Met34Thr | rs35887622 | Likely Pathogenic | 1/266 | 0.00285 |
| c.617A>G | p.Asn206Ser | rs111033294 | Pathogenic | 1/266 | 0.00008675 |
Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org)
Table 2.
Identified GJB2 variants in 133 Guatemalan probands with non-syndromic hearing loss#
| cDNA | PROTEIN | NUMBER OF PROBANDS |
|---|---|---|
| HOMOZYGOUS & COMPOUND HETEROZYGOUS GENOTYPES | ||
|
| ||
| c.[79G>A]; [79G>A] | p.[Val27Ile];[Val27Ile] | 15 |
| c.[79G>A;131G>A];[79G>A;131G>A] | p.[Val27Ile;Trp44*];[Val27Ile;Trp44*] | 6 |
| c.[79G>A;131G>A(;)439G>A] | p.[Val27Ile;Trp44*(;)Glu147Lys] | 2 |
| c.[94C>T];[94C>T] | p.[Arg32Cys];[Arg32Cys] | 1 |
| c.[250G>T];[250G>T] | p.[Val84Leu];[Val84Leu] | 1 |
| c.[79G>A];[439G>A] | p.[Val27Ile];[Glu147Lys] | 1 |
| c.[101T>C];[511G>A] | p.[Met34Thr];[Ala171Thr] | 1 |
| c.[79G>A];[380G>T] | p.[Val27Ile];[Arg127Leu] | 1 |
|
| ||
| HETEROZYGOUS GENOTYPES | ||
|
| ||
| c.[79G>A];[=] | p.[Val27Ile];[=] | 36 |
| c.[79G>A;131G>A];[=] | p.[Val27Ile;Trp44*];[=] | 7 |
| c.[94C>T];[=] | p.[Arg32Cys];[=] | 1 |
| c.[250G>T];[=] | p.[Val84Leu];[=] | 3 |
| c.[380G>T];[=] | p.[Arg127Leu];[=] | 1 |
| c.[617A>G];[=] | p.[Asn206Ser];[=] | 1 |
|
| ||
| WILD TYPE | ||
|
| ||
| WT | WT | 56 |
Genotypes are annotated according to Human Genome Variation Society Nomenclature (http://www.hgvs.org/mutnomen/)
Evaluation of SNVs flanking the c.131G>A in seven individuals were consistent with a shared haplotype, suggesting a single founder (Table S3). Furthermore, among the 133 probands, all six probands who were homozygous for the c.131G>A variant were also homozygous for the c.79G>A variant and all nine individuals who were heterozygous for the c.131G>A variant were also heterozygous for the c.79G>A variant. The presence of probands with a c.79G>A without a c.131G>A suggests that the c.131G>A variant occurred on a c.79G>A chromosome (Table S4).
Guatemala is divided into four major geographical regions: west, east, north and central. The majority of GJB2 pathogenic variants reported here are from individuals living in the west region, which has a large Mayan population. Evaluation of global ancestry from seven probands who are either homozygous (two) or heterozygous (five) for c.131G>A showed a close match with Mayans and Mexicans as compared to those of European, African, and other Native American populations (Figure 1). Our samples with the c.131G>A variant (GUA) originate from a close geographical region where the Mayan samples were obtained (Fig.1A). Analysis of global ancestry with EIGENSTRAT shows that the GUA samples overlap with Mexican (MXL) and Mayan samples (Fig.1B). Finally, ADMIXTURE analysis shows a very similar distribution of ancestry between the GUA samples and the Mayan and Mexican samples (Fig.1C).
Figure 1.
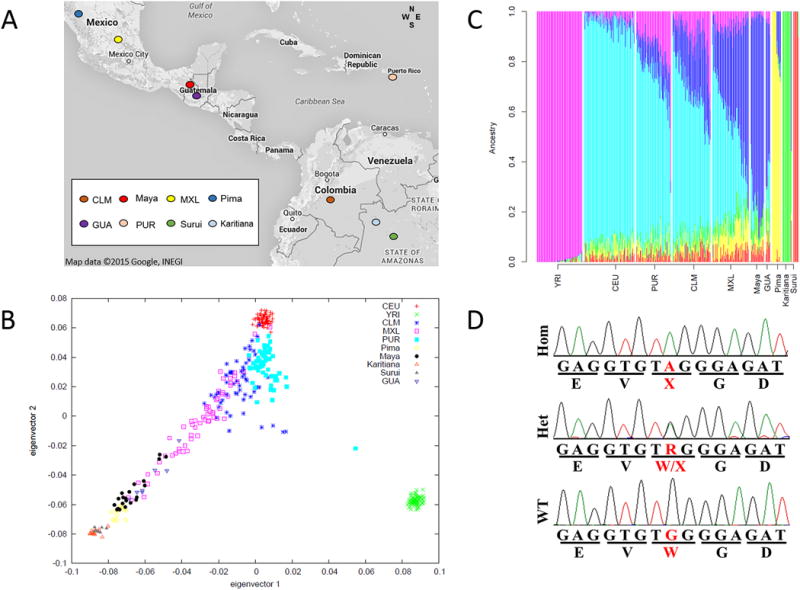
Geographic locations of the studied other populations (Modified from “Map data ©2015 Google, INEGI”) (A). Eigenvector plot analysis of the GUA samples and the compared populations. GUA samples that we studied are clustered closely and they overlap with HDGP Mayan samples (B). Ancestry analysis of the other populations and GUA samples represents identical genetic background between GUA and Mayan population (K=6). Each color represents an ancestral population (C) Electropherogram of the GJB2 c.131G>A pathogenic variant (D).
DISCUSSION
The frequency of variants in GJB2 can vary significantly among different countries or ethnic populations (16). The Mayans are ancient ethnic population which occupied a wide territory in southeastern Mexico to northern Central America and the territory is now incorporated into many modern countries including Guatemala. The largest and most traditional Mayan populations are in the western highlands in Guatemala where the majority of our cohort lives. The indigenous Mayan people share a distinctive set of genetic traits such as ABO blood group markers and mitochondrial and chromosome Y haplotypes; therefore it can be inferred that these people originated from a founder population (17, 18).
In this study we report that the most common GJB2 pathogenic variant in a deaf population from Guatemala is c.131G>A (p.Trp44*). Ancestry analysis of individuals with this pathogenic variant shows close match with Mayans (Figure 1). This variant results in a premature stop codon (p.Trp44*) and has previously been reported in only one study with two cases out of 610 probands from the U.S.; the authors did not specify ethnic origins of these individuals (19). There is single occurrence of this variant in the Exome Aggregation Consortium (ExAC) database, a heterozygous Latino (URL: http://exac.broadinstitute.org). Another variant in this same codon, which also results in a stop codon, has been reported (c.132G>A; rs104894407) (20). Carriers of this latter variant have been identified in Africans and non-Finnish Europeans. While p.Trp44* has been reported in four other studies, a cDNA description is not provided for these cases so that it is impossible to determine the specific base change (4, 21–23).
It is important to note that the allele frequency of the c.79G>A variant in GJB2 is 0.33 in our cohort. This polymorphism is common in East Asians as well as in the studied Latin American populations, supporting an East Asian origin of the early settlers of the Americas (Table S5) (9, 24). While c.79G>A is shared by many Native American populations, c.131G>A likely occurred in a subpopulation within America. Linkage disequilibrium (LD) analysis with PLINK between these two variants shows them to be in extreme LD (D′=1.0, r=0.17), although these values must be interpreted with caution as neither variant is in Hardy Weinburg Equilibrium, a not unexpected finding given the selection bias of the sample (c.79G>A, p=0.01; c.131G>A, p=8.733E-06) (25).
In the remainder of the cohort, repeating pathogenic variants are c.250G>T (5/266), c.94C>T (3/266), and c.439G>A (3/266). These variants were previously reported as rare causes of deafness (20, 23, 26). c.250G>T is reported in only one allele out of 121,362 alleles in the Exome Aggregation Consortium database and that single allele belongs to a Latino individual. (Table S5). It is therefore likely that c.250G>T is another rare variant in Guatemala.
In conclusion, c.131G>A (p.Trp44*) is the most frequent pathogenic variant found in our Guatemalan cohort. This is the first time c.131G>A has been reported as a prevalent variation in a population. In conjunction with the geographical location of the samples studied, the high concentration of living Mayans in that area and the results of ancestry analysis, it is likely that c.131G>A is a Mayan-specific pathogenic variant for hearing loss.
Supplementary Material
Acknowledgments
Support for this research was provided by INVEGEM, The Rozas-Botrán Foundation from Guatemala and supported by National Institutes of Health grant R01DC009645 to MT.
Footnotes
Conflict of interest
Authors declare no conflict of interest.
References
- 1.Morton CC, Nance WE. Newborn hearing screening–a silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Najmabadi H, Kahrizi K. Genetics of non-syndromic hearing loss in the Middle East. Int J Pediatr Otorhinolaryngol. 2014;78:2026–2036. doi: 10.1016/j.ijporl.2014.08.036. [DOI] [PubMed] [Google Scholar]
- 3.Mese G, Londin E, Mui R, et al. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum Genet. 2004;115:191–199. doi: 10.1007/s00439-004-1142-6. [DOI] [PubMed] [Google Scholar]
- 4.Snoeckx RL, Huygen PL, Feldmann D, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi C, Funayama M, Li Y, et al. Prevalence of GJB2 causing recessive profound non-syndromic deafness in Japanese children. Int J Pediatr Otorhinolaryngol. 2011;75:211–214. doi: 10.1016/j.ijporl.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Chai Y, Pang X, Chen D, et al. Molecular etiology of non-dominant, non-syndromic, mild-to-moderate childhood hearing impairment in Chinese Hans. Am J Med Genet A. 2014;164A:3115–3119. doi: 10.1002/ajmg.a.36785. [DOI] [PubMed] [Google Scholar]
- 7.Chen T, Jiang L, Liu C, et al. Update of the spectrum of GJB2 mutations in 107 patients with nonsyndromic hearing loss in the Fujian population of China. Ann Hum Genet. 2014;78:235–242. doi: 10.1111/ahg.12062. [DOI] [PubMed] [Google Scholar]
- 8.Padma G, Ramchander PV, Nandur UV, et al. GJB2 and GJB6 gene mutations found in Indian probands with congenital hearing impairment. J Genet. 2009;88:267–272. doi: 10.1007/s12041-009-0039-5. [DOI] [PubMed] [Google Scholar]
- 9.Tekin M, Xia XJ, Erdenetungalag R, et al. GJB2 mutations in Mongolia: complex alleles, low frequency, and reduced fitness of the deaf. Ann Hum Genet. 2010;74:155–164. doi: 10.1111/j.1469-1809.2010.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genomes Project C. Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavalli-Sforza LL. The Human Genome Diversity Project: past, present and future. Nat Rev Genet. 2005;6:333–340. doi: 10.1038/nrg1596. [DOI] [PubMed] [Google Scholar]
- 12.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 13.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukada K, Nishio SY, Hattori M, et al. Ethnic-Specific Spectrum of GJB2 and SLC26A4 Mutations: Their Origin and a Literature Review. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):61S–76S. doi: 10.1177/0003489415575060. [DOI] [PubMed] [Google Scholar]
- 17.Ibarra-Rivera L, Mirabal S, Regueiro MM, et al. Delineating genetic relationships among the Maya. Am J Phys Anthropol. 2008;135:329–347. doi: 10.1002/ajpa.20746. [DOI] [PubMed] [Google Scholar]
- 18.Sochtig J, Alvarez-Iglesias V, Mosquera-Miguel A, et al. Genomic insights on the ethno-history of the Maya and the ‘Ladinos’ from Guatemala. BMC Genomics. 2015;16:131. doi: 10.1186/s12864-015-1339-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang HY, Fang P, Ward PA, et al. DNA sequence analysis of GJB2, encoding connexin 26: observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet A. 2006;140:2401–2415. doi: 10.1002/ajmg.a.31525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Putcha GV, Bejjani BA, Bleoo S, et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med. 2007;9:413–426. doi: 10.1097/gim.0b013e3180a03276. [DOI] [PubMed] [Google Scholar]
- 21.Lin JW, Chowdhury N, Mody A, et al. Comprehensive diagnostic battery for evaluating sensorineural hearing loss in children. Otol Neurotol. 2011;32:259–264. doi: 10.1097/MAO.0b013e31820160fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipan M, Ouyang X, Yan D, et al. Clinical comparison of hearing-impaired patients with DFNB1 against heterozygote carriers of connexin 26 mutations. Laryngoscope. 2011;121:811–814. doi: 10.1002/lary.21422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasad S, Cucci RA, Green GE, et al. Genetic testing for hereditary hearing loss: connexin 26 (GJB2) allele variants and two novel deafness-causing mutations (R32C and 645-648delTAGA) Hum Mutat. 2000;16:502–508. doi: 10.1002/1098-1004(200012)16:6<502::AID-HUMU7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 24.Paz-y-Mino C, Beaty D, Lopez-Cortes A, et al. Frequency of GJB2 and del(GJB6-D13S1830) mutations among an Ecuadorian mestizo population. Int J Pediatr Otorhinolaryngol. 2014;78:1648–1654. doi: 10.1016/j.ijporl.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frei K, Lucas T, Ramsebner R, et al. A novel connexin 26 mutation associated with autosomal recessive sensorineural deafness. Audiol Neurootol. 2004;9:47–50. doi: 10.1159/000074186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.