

Abstract
Tauopathies are a major group of neurodegenerative proteinopathies characterized by the accumulation of abnormal and hyperphosphorylated tau proteins in the brain. Tau pathology is characterized as 3R (repeat) or 4R predominant or mixed 3R and 4R type. Here we report three cases lacking mutations in the microtubule associated protein tau (MAPT) gene with unusual tau pathology. The age at onset and duration of illness, respectively, were 63 and 20 years (male), 67 and 5 years (female) and 72 and 20 years (female). The clinical presentation was compatible with a diagnosis of progressive supranuclear palsy (PSP) in two subjects and with cognitive decline in all three subjects. Common neuropathological features included neuronal loss in the hippocampus and dentate gyrus associated with spherical basophilic Pick body‐like inclusions showing 4R tau immunoreactivity, which was supported by the detection of predominantly 4R tau species by Western blot examination. In addition, accumulation of tau immunoreactive argyrophilic astrocytes in the hippocampus and amygdala and oligodendroglial coiled bodies in the hippocampal white matter were observed. These morphologies appeared in combination with Alzheimer disease‐related pathology and subcortical tau pathology compatible with PSP. Together with a single case report in the literature, our observations on these three cases expand the spectrum of previously described tauopathies. We suggest that this tauopathy variant with hippocampal 4R tau immunoreactive spherical inclusions might contribute to the cognitive deficits in the patients reported here. The precise definition of the clinicopathological relevance of these unusual tau pathologies merits further study.
Keywords: astroglia, hippocampus, Pick's disease, 4R tau isoform, tau, tauopathy
Introduction
Neurodegenerative diseases are characterized by progressive dysfunction and loss of neurons associated with deposition of misfolded proteins in various cell types or extracellularly 18. Clinical symptoms usually depend on the anatomical involvement of neuronal loss and do not necessarily reflect the molecular pathological background. Due to recent observations suggesting that the progression of many neurodegenerative diseases is driven by template‐directed misfolding, seeded aggregation, cell‐to‐cell transmission and sequential dissemination of pathological proteins 5, the importance of protein‐based classification of neurodegenerative diseases with relevance for biomarker and therapy development has been emphasized 18.
One major group of neurodegenerative proteinopathies is associated with the deposition of pathological tau protein, hence called tauopathies or frontotemporal lobar degeneration (FTLD) with tau pathology (FTLD‐Tau). Tauopathies are characterized by the accumulation of abnormal and hyperphosphorylated tau proteins in the brain and are also classified as primary or secondary 23. Tau pathology is characterized as 3R (repeat) or 4R predominant or mixed 3R and 4R type 15, 17. Primary tauopathies comprise Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), neurofibrillary tangle (NFT) predominant senile dementia (NFT‐dementia or “tangle‐only” dementia; now included in the category of primary age‐related tauopathy, PART), and globular glial tauopathy (GGT) 6, 17. In addition, there are other tau pathologies, such as those seen in chronic traumatic encephalopathy 25, aging‐related tau astrogliopathy (ARTAG) 19, or anti‐IgLON5 syndrome related tauopathy 10, which require further studies to better interpret their frequency and relevance. Moreover, there are still peculiar constellations of tau pathologies, which do not fit into these classifications. Many of them belong to the group of hereditary FTLD‐Tau disorders that are caused by autosomal dominant mutations in the microtubule‐associated protein tau gene (MAPT) 11; however, there are reports of unclassifiable sporadic tauopathies as well, such as a 4R tauopathy clinically presenting as posterior cortical atrophy 16.
In 2009, Miki et al reported a seemingly sporadic case with hippocampal sclerosis and 4R tau‐positive round inclusions in the dentate gyrus, interpreted as a new type of 4R tauopathy 26. A similar case was detected in a large archival series of Pick's disease but was not further characterized 21. During the reevaluation of archival cases we detected three cases with similar 4R tau pathology and provide here a comprehensive description of these cases.
Materials and Methods
Selection of cases
The patients were seen and systematically investigated as part of the routine neurological diagnostic practice and later referred for neuropathological autopsy diagnosis. During the reevaluation of archival cases of the Center for Neurodegenerative Disease Research (CNDR), Philadelphia 36, we found two out of 50 consecutive PSP cases and one out of 240 cases with AD‐related or PART pathology.
Genetic analysis
Genomic DNA was extracted from brain tissues using QIAamp DNA mini kit (QIAGEN Sciences Inc. Qiagen Sciences, Germantown, MD USA) following manufacturer recommendations. Bidirectional Sanger sequencing of MAPT exons 1 and 9–13 with flanking intronic primers (sequences available on request) was performed as previously described 37. Data were analyzed with Mutation Surveyor software (Soft Genetics LLC, State College, PA).
Neuropathology
Formalin fixed, paraffin‐embedded tissue blocks from the investigated cases were evaluated including the following anatomical regions: frontal, anterior cingulate, parietal, temporal, occipital cortex, hippocampus, entorhinal cortex, amygdala, basal ganglia, thalamus, mesencephalon, pons, medulla oblongata and cerebellum. In addition to hematoxylin and eosin (HE) and Gallyas silver staining, the following mouse monoclonal antibodies were used for immunohistochemistry: anti‐p62 (1:1000 + MW; Abnova Abnova, Walnut, CA USA EMD Millipore, Billerica, MA USA), anti‐tau PHF‐1 (Ser396/Ser404, 1:2000; Gift of Peter Davies), anti 4R (RD4, 1:10000 + MW; Millipore) and 3R (RD3, 1:1000 + FA; Millipore) tau isoforms; anti‐phospho‐TDP‐43 (rat monoclonal TAR5P‐1D3; pS409/410 TDP‐43; Ascenion, Munich, Germany; gift of Manuela Neumann) 33, anti‐α‐synuclein (Syn303, 1:30000 + FA; CNDR), anti‐Aβ (Nab228, 1:20000; CNDR) and anti‐FUS (fused in sarcoma; 1:8000 + MW; ProteinTech Proteintech, Rosemont, IL USA). The Vectashield ABC detection kit, peroxidase/DAB, rabbit/mouse/rat (BA1000/BA2000/BA4001, 1:1000; Vector Laboratories Vector Laboratories, Inc, Burlingame, CA USA) was used for the visualization of antibody reactions. Neuropathological alterations (astrogliosis/neuronal loss, degree of various protein depositions) were semi‐quantitatively (none, mild, moderate and severe) evaluated in different anatomical regions.
Brain extraction and immunoblot analysis
Frozen postmortem brain tissue samples of the hippocampus and putamen from cases 1 and 2 was dissected, weighed and sequentially extracted at 5 mL/g tissue with high salt buffer (50 mM Tris‐HCl, pH 7.4, 10 mM NaF, 5 mM EDTA and 750 mM NaCl) followed by high salt buffer containing 1% Triton X‐100 (HS‐TX). Each extraction step was done twice at 4°C and both buffers contained a cocktail of protease and phosphatase inhibitors as described 38. After removal of myelin, the HS‐TX insoluble pellet was washed in 5 mL/g tissue PBS and re‐suspended followed by sonication in 0.5 mL/g tissue PBS. Proteins were resolved in Tris‐glycine 7.5% SDS‐PAGE, transferred to nitrocellulose and probed with primary antibody (rabbit polyclonal phospho‐independent antibody 17025 (1:2000; CNDR); mouse monoclonal phospho‐dependent antibody PHF‐1 (1:1000; Gift of Peter Davies), rabbit polyclonal 4R‐tau specific antibody (1:3000; Cosmo Bio Cosmo Bio USA, Inc, Carlsbad, CA USA) and mouse monoclonal 3R‐tau specific antibody clone 8E6/C11 (1:1000; Millipore). Signals were visualized using goat polyclonal anti‐mouse or anti‐rabbit IgG IRDye‐680 or IRDye‐800 conjugated secondary antibody (Li‐Cor) with a Li‐Cor Odyssey imaging system.
Results
Case reports
Case 1
This gentleman presented with falling and decline in balance and walking at the age of 63, which progressed slowly. Later he experienced difficulties with gaze and his family noticed decreased short‐term memory (described as he “lives in the moment”). One year before his death he was examined by a movement disorder specialist who found mild bradykinesia, mildly slurred speech, marked supranuclear palsy and unsteady gait. Mini mental examination revealed a score of 28/30. Hoehn‐Yahr score was III. In the last years of his life, he lived in an assisted living facility. Seven months before death, he had two grand mal seizures, which were observed by the staff at the assisted living facility. From this time onwards, he remained unconscious and died at the age of 83 years. The duration of illness was 20 years, and for 15 years, he was without noticeable cognitive impairment. There was no family history of neurological disorders or dementia. The clinical diagnosis was PSP and mild cognitive impairment (Table 1).
Table 1.
Summary of clinicopathological data. ABC score indicates the score provide for Aβ deposition (A), for Braak stage of neurofibrillary degeneration (B) and neuritic plaque score (C) 28. Abbreviations: PSP, progressive supranuclear palsy; MCI, mild cognitive impairment.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Age at death (years) | 83 | 72 | 91 |
| Sex | Male | Female | Female |
| Duration of illness (years) | 20 | 5 | 20 |
| Clinical diagnosis | PSP+ MCI | PSP+ dementia | Unclassified dementia |
| Symptoms at early stage | Imbalance and gait disorder | Incoordination | Not available |
| Symptoms at late stage | Cognitive decline | Cognitive decline | Cognitive decline |
| Supranuclear gaze palsy | Supranuclear gaze palsy | ||
| 2 grand mal seizures | Parkinsonism | ||
| Motor–dementia interval | 15 years | 0 years | 0 years |
| Family history | No | No | Yes |
| ApoE | E2/E3 | E3/E3 | E2/E3 |
| H1/H2 haplotype | H1/H1 | H1/H1 | H2/H2 |
| MAPT mutation | None | None | None |
| Brain weight | 1378 | 1147 | 1066 |
| ABC score | A0B2C0 | A1B1C0 | A3B3C3 |
| pTDP‐43 | No | No | Yes |
| α‐Synuclein | No | No | No |
| Hippocampal sclerosis | no | yes | Yes |
| Others | – | Focal encephalitis in brainstem | – |
| Lacunar infarcts in Globus pallidus and brainstem |
Case 2
This woman presented at the age of 67 years with incoordination and falls. Roughly 2 years prior to presentation she was involved in several car accidents that were thought to be related to changes in coordination. A few months later she developed frequent falls. In addition, she complained of forgetfulness, loss of smell and smaller handwriting. Her first clinical evaluation found executive and visuospatial dysfunction with mild hypophonia, bilateral bradykinesia, increased tone in axial and limb muscles with a mild intention tremor but no resting tremor. Extraocular movements were full but square‐wave jerks were present. Possible Parkinson disease was diagnosed; however, a trial of L‐DOPA treatment (25/100 tablets up to 6 times daily) did not improve her state. The clinical picture soon evolved into what was regarded as a typical PSP‐syndrome with prominent axial rigidity, postural instability and supranuclear gaze palsy. Mini mental examination revealed a score of 17/30 three years before death. DAT scan at this time revealed depressed tracer uptake in the striatum with minimal sparing of the left caudate. Social withdrawal and mild apathy were also noted. She developed severe dementia and was completely dependent, living in a hospice in the last months of her life. Hoehn–Yahr score at the end of life was V. She died at the age of 72 after five years duration of her illness. There was no family history of neurological disorders or dementia. In summary, the clinical phenotype was consistent with a diagnosis of PSP (Table 1).
Case 3
This patient presented with a slowly progressive cognitive decline thought to be consistent with Alzheimer's disease dementia. The clinical course was more than 20 years and she died at the age of 92 years. Documentation of detailed neurological examination is not available. Her daughter had been diagnosed with FTLD at the age of 69 years. Furthermore, there was a history for mental illnesses including panic attacks and bipolar disorder in her family (Table 1).
Neuropathology
Macroscopic and histopathological findings
Case 1
The brain was examined 20 h postmortem, and it weighed 1378 g. Only mild atherosclerosis was noted in the basal arteries. Macroscopic inspection revealed mild atrophy of the frontal lobes. The ventricles were symmetrical and moderately dilated. There was no evidence of infarction, hemorrhage, or tumor mass. The nuclei of the brain, including the thalami, the caudate, the lentiform, the lateral geniculate bodies and the subthalamic nuclei were unremarkable. The hippocampus was moderately atrophic. The amygdala was normal. The substantia nigra and the locus coeruleus showed mild depigmentation.
Histopathological findings comprised neuronal loss in subcortical areas, including substantia nigra, dentate nucleus and globus pallidus, as well as in the hippocampus, particularly the CA4 and CA2/3 subregion. Basophilic or eosinophilic neuronal cytoplasmic inclusions were seen in the dentate gyrus and less in the hippocampus (Figure 1A). Although mild gliosis was present in the CA1 and subiculum, this was not compatible with the definition of hippocampal sclerosis (Figure 1D). Prominent hyalinosis and fibrosis was noted in subcortical vessels. Ballooned neurons were observed in the amygdala. There was a lack of Aβ plaques, as well as α‐synuclein, FUS, or pTDP‐43 pathology (Table 1).
Figure 1.
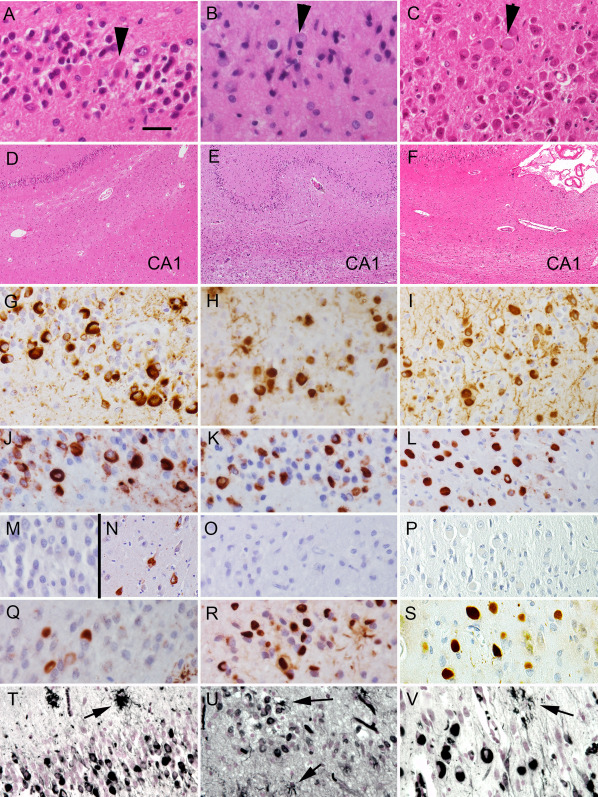
Basophilic inclusions in the dentate gyrus (A–C; indicated by arrowheads) and variable cell loss in the CA1 subregion (D–F) in the three cases (A and D: case 1; B and E: case 2; and C and F: case 3) reported in this study. Immunostaining for PHF‐1 (G–I), 4R tau (J–L), 3R tau (M–P), p62 (Q–S) and for Gallyas silver staining (T–V) in cases 1 (G, J, M, N, Q and T), 2 (H, K, O, R and U) and 3 (I, L, Q, P, S and V) demonstrating the spherical inclusions in the dentate gyrus. Immunostaining for 3R tau in the CA1 subregion (N) demonstrates neurofibrillary tangles in case 1 corresponding to the immunoblot results. Note the argyrophilic astrocytes in (T)–(V) (arrows). The bar in (A) represents 25 μm for (A)–(C) and (G)–(V) and 100 μm for (D)–(F).
Tau pathology (Figures 1G and 2A,D,G,J,M and Table 2) was characterized by NFTs in the hippocampus (Figure 2A,D), entorhinal and inferior temporal cortex and amygdala (Figure 2G) corresponding to stage III according to Braak. NFTs and tufted astrocytes were present in cortical areas (Figure 2J), the basal ganglia (Figure 2M), thalamus and brainstem nuclei and dentate nucleus. Abundant ring‐like and spherical tau positive neuronal cytoplasmic inclusions were seen in the granule cells of the dentate gyrus and some also were present in the CA regions of the hippocampus (Figure 1G). These were strongly immunoreactive for 4R tau (Figure 1J) but not for 3R tau (Figure 1M) and they also showed immunoreactivity for p62 (Figure 1Q) and they were Gallyas positive as well (Figure 1T). However, immunostaining for 3R tau labeled the NFTs in CA subregions of the hippocampus, but we interpret these NFTs as aging or PART related (Figure 1N). In addition, 4R tau positive oligodendroglial coiled bodies were seen in the white matter in the hippocampus and subcortical areas. Astrocytic 4R tau positive pathology was mostly compatible with tufted astrocytes appearing not only in the striatum but also in the cornu ammonis, dentate gyrus and particularly in the amygdala, where they were associated with numerous granular/fuzzy astrocytes as described in ARTAG (Figure 2G) 19, which is unusual in PSP. Immunostaining for p62 detected grain‐like pathology in the gyrus ambiens, which is compatible with an early stage of AGD 35. Biochemical analyses of the Triton X‐100 insoluble fraction of the brain of this patient showed abundant tau pathology in the hippocampus, but not in the putamen (Figure 3). Similar to the immunohistochemical results, this tau pathology was immuno‐positive for both 4R and 3R‐tau.
Figure 2.
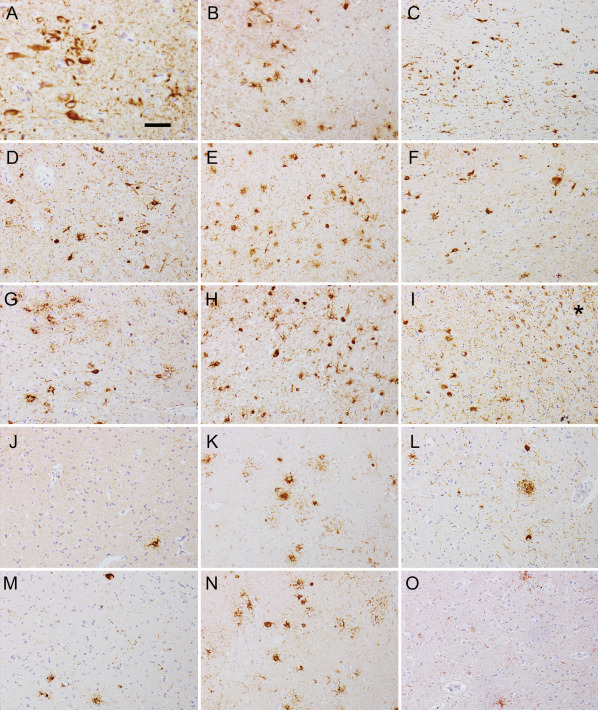
Immunostaining for PHF‐1 in the CA1 (A–C), CA4 (D–F) subregions, amygdala (G–I), frontal cortex (J–L) and putamen (M–O) in cases 1 (A, D, G, J and M), 2 (B, E, H, K and N) and 3 (C, F, I, L and O). Note the numerous tau positive astrocytes in case 2 in all subregions, while less are seen in case 1 together with neurofibrillary tangles in the CA1 subregion. Case 3 exhibits neurofibrillary tangles and dystrophic neuritic plaques together with astrocytes in CA subregions and amygdala (* in image I indicates white matter in amygdala with thorn shaped astrocytes compatible with ARTAG). The bar in (A) represents 40 μm for (A) and 100 μm for (B)–(O).
Table 2.
Anatomical distribution of neuronal. Abbreviations: NFT, neurofibrillary tangle; PT, pretangle; SB, spherical body; TA, tufted astrocytes; RA, ramified astrocytes; GFA, granular/fuzzy astrocytes; subthal, subthalamic nucleus; AG, astroglial; OG, oligodendroglial (in the adjacent white matter) tau pathologies. −: none; +: occasional; ++: moderate number; +++: abundant/many. *Thorn‐shaped astrocytes (TSA) in the white matter.
| Case 1 | Case 2 | Case 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region/pathology | NFT/PT | SB | AG | OG | NFT/PT | SB | AG | OG | NFT/PT | SB | AG | OG |
| Frontal | + | − | +TA | + | + | − | +++TA | + | ++ | − | − | + |
| Temporal | + | − | − | − | + | − | +++TA | ++ | +++ | − | − | + |
| Parietal | − | − | − | − | − | − | +++TA | ++ | ++ | − | +* | + |
| Anterior cingulate | ++ | − | +TA | + | + | − | +++TA | ++ | ++ | − | − | + |
| Entorhinal | +++ | − | +TA | + | + | − | +++TA | + | +++ | − | − | − |
| Hippocampus | ||||||||||||
| CA1/subiculum | +++ | + | ++TA | +++ | + | + | +++TA | ++ | +++ | + | +TA/RA | + |
| CA2/3 | +++ | + | +++TA | +++ | + | + | +++TA | − | +++ | + | ++TA/RA | + |
| CA4 | ++ | − | +++TA | − | + | − | +++TA | − | + | − | ++TA/RA | − |
| Dentate gyrus | + | +++ | ++TA/TSA | − | + | ++ | ++TA | − | + | +++ | +TA/RA | − |
| Amygdala | +++ | − | +++TA/GFA | +++ | +++ | + | +++TA | ++ | +++ | − | ++GFA/TSA | + |
| Striatum | ++ | − | +++TA | + | + | − | +++TA | + | + | − | − | − |
| Globus pallidus | + | − | ++TA | +++ | ++ | − | +TA | +++ | − | − | − | − |
| Thalamus + subthal | ++ | − | − | + | ++ | − | +++TA | +++ | ++ | − | − | − |
| Substantia nigra | ++ | − | − | − | +++ | − | +TA | +++ | + | − | − | − |
| Locus coeuruleus | +++ | − | − | − | + | − | − | + | ++ | − | − | − |
| Pontine base | − | − | − | + | + | − | − | +++ | − | − | − | + |
| Inferior olives | +++ | − | − | − | + | − | − | + | − | − | − | − |
| Hypoglossal nucleus | − | − | − | − | − | − | − | − | − | − | − | − |
| Dorsal vagus nucleus | ++ | − | − | − | − | − | − | − | − | − | − | − |
| Dentate nucleus | ++ | − | − | − | +++ | − | − | + | − | − | − | − |
Figure 3.
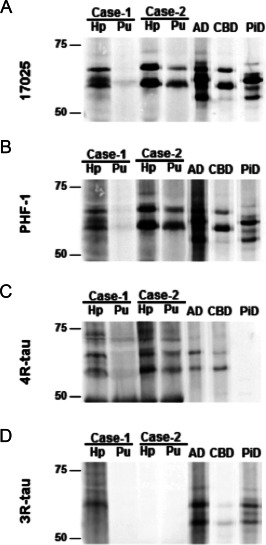
Biochemical analysis of tau in Triton X‐100 insoluble fraction extracted from frozen brain tissues. (A) Total‐tau (17025) and (B) phosphorylated‐tau (PHF‐1) immunoblot showed abundance of tau pathology in case 1 and case 2 Hp but not in case 1 Pu. Isoform‐specific immunoblot revealed predominant 4R‐tau pathology in both cases (C) consistent with IHC finding. Furthermore, case 1 Hp was also immune‐reactive to 3R‐tau antibody (d). Hp = hippocampus, Pu = putamen; Alzheimer disease (AD), corticobasal degeneration (CBD) and Pick's disease (PiD) control samples are also shown.
Case 2
The brain was examined 14 h postmortem and weighed 1147 g. Severe atherosclerosis was noted in the basal arteries. The nuclei of the brain, including the thalami, the caudate, the lateral geniculate bodies and the subthalamic nuclei were unremarkable. The hippocampus and amygdala were severely atrophic. The substantia nigra was depigmented while the locus coeruleus was well pigmented. The ventricles were symmetrical and severely dilated. Multiple lacunar infarcts were observed, including in both sides in the globus pallidus and also at the level of the left upper pons.
Major histopathological findings included loss of pigmented neurons in the substantia nigra, and neuronal loss and gliosis in the amygdala, hippocampus (all CA subregions) and entorhinal cortex. Macrophages, including siderophages were seen in the lentiform nucleus, amygdala, entorhinal cortex, hippocampus and midbrain. Basophilic neuronal cytoplasmic inclusions were seen in the granule cells of the dentate gyrus (Figure 1B) and occasionally in pyramidal cells of CA subregions. Although the CA subregions showed macrophages, however, gliotic segments of the CA1 without macrophages were suggestive of hippocampal sclerosis (Figure 1E). Moderate to prominent perivascular lymphocytic infiltrates were present in the medulla, cerebellum and midbrain, with microglial nodules in the medulla. Diffuse Aβ plaques were present in the frontal, temporal and occipital cortex; neurofibrillary tangles were restricted to the entorhinal cortex. The ABC score 28 was A1B1C0; no Lewy bodies, Lewy neurites, FUS or pTDP‐43 inclusions were seen (Table 1).
Tau pathology included abundant tau‐positive tufted astrocytes, NFTs, coiled bodies and gliosis in the lentiform nucleus, thalamus, subthalamus, dentate nucleus, hippocampus with prominent tau inclusions in the dentate neurons, amygdala, cingulate gyrus and midbrain (Figures 1H and 2B,E,H,K,N and Table 2). The frontal and temporal cortex as well as the subcortical white matter, the angular gyrus, the lentiform nucleus, the thalamus and the occipital cortex showed tau positive tufted astrocytes and oligodendrocytes with coiled bodies. Hippocampal tau pathology comprised abundant argyrophilic tufted astrocytes in all CA subregions and the dentate gyrus. The granule cells of the dentate gyrus exhibited many spherical and ring‐like inclusions, which showed p62, 4R (but not 3R) tau immunoreactivity and Gallyas positivity (Figure 1H,K,O,R,U). A few spherical inclusions were detected in the CA subregion of the hippocampus as well. 3R positive NFTs were not seen in the hippocampus. Immunoblot analysis of Triton X‐100 insoluble tau from the hippocampus and putamen showed abundant phosphorylated tau as evidenced by PHF‐1 positivity in both regions (Figure 3). Moreover, the 4R‐tau predominant pathology seen by immunohistochemistry was confirmed by Western blots.
Case 3
The brain was examined 14 h postmortem and weighed 1066 g. Severe atherosclerosis was noted in the basal arteries. The ventricles were symmetrical and mildly dilated. There was no evidence of infarction, hemorrhage, or tumor mass. The nuclei of the brain, including the thalami, the caudate, the lentiform nucleus, the lateral geniculate bodies and the subthalamic nuclei were all unremarkable. The hippocampus and amygdala were severely atrophic. The substantia nigra and the locus coeruleus were well pigmented.
Histopathological findings comprised neuronal loss predominantly in the amygdala, hippocampus (all CA subregions) and dentate gyrus. In addition to NFTs visible in H&E staining, spherical basophilic inclusions as well as ring‐like swelling of the cytoplasm were noted in the granule cells of the dentate gyrus (Figure 1C). Gliosis in the CA1 and subiculum was compatible with hippocampal sclerosis (Figure 1F). There was a lack of α‐synuclein and FUS pathology. High degree of AD‐related pathology including high density of neuritic plaques, deposition of Aβ together with NFTs in neocortical areas were noted (A3B3C3). In the entorhinal cortex pTDP‐43 immunoreactive neuronal cytoplasmic inclusions and small and thin neurites were observed.
Tau pathology (Figures 1I and 2C,F,I,L,O and Table 2) was characterized by AD‐related changes including NFTs, pretangles, neuropil threads and dystrophic neurites surrounding plaques in the entorhinal cortex, hippocampus and neocortical areas. Spherical neuronal cytoplasmic inclusions larger than the size of the nucleus were noted in the granule cell layer of the dentate gyrus (Figure 1I,L,P,S,V) and occasionally in hippocampal CA subregions. These were strongly immunoreactive for 4R tau but not for 3R tau and they also showed immunoreactivity for p62 and were Gallyas positive. In the pyramidal layers of the cornu ammonis abundant NFTs, including ghost tangles were seen. The astrocytic tau pathology was Gallyas positive and resembled partly ramified astrocytes while tufted astrocytes also were seen in the cornu ammonis and dentate gyrus. Furthermore, thorn‐shaped astrocytes were noted in the white matter of the amygdala (Figure 2I), parietal lobe and midline of the medulla oblongata as well as subpial in several locations as described in ARTAG 19. Granular/fuzzy astrocytes were observed in the amygdala. No grain‐like tau pathology was seen but small numbers of tau positive oligodendroglial coiled‐bodies were seen in several regions (Table 2). The glial tau pathology in this case was consistently 4R tau positive.
Discussion
We expand the spectrum of 4R tauopathies by presenting three cases with a peculiar constellation of tau pathology. Common features comprise neuronal loss in the hippocampus and dentate gyrus associated with spherical, mostly basophilic inclusions in the granule cell layer of the dentate gyrus showing 4R tau and p62 immunoreactivity and accumulations of tau immunoreactive argyrophilic astrocytes in the hippocampus and amygdala together with oligodendroglial coiled bodies in the hippocampal white matter. We detected combinations of additional tau pathologies in these cases, such as various degrees of AD/PART‐ and AGD‐related pathology and subcortical tau pathology compatible with PSP, as well as presence of ARTAG in the two elderly cases and ischemic/hypoxic lesions and inflammatory infiltrates in one.
In 2009 Miki et al reported similar 4R tau pathology in the dentate gyrus associated with hippocampal sclerosis, while other anatomical regions did not show significant tau pathologies 26. The patient died at the age of 78 years due to subarachnoid hemorrhage and clinically presented with depression three years prior to death. Cognitive decline was not documented 26. A 93‐year‐old female patient presenting with eight years of cognitive decline and with similar morphology has been reported in a study on archival Pick's disease cases focusing only on hippocampal tau pathology 21. In the present study, two of our cases showed additional features of PSP in subcortical areas and the third case showed prominent AD/PART‐related tau pathology. Two of these cases (cases 2 and 3) showed hippocampal sclerosis and both of these two cases had a clinical history of dementia. One patient with mild cognitive impairment (case 1) showed fewer inclusions in the dentate gyrus and only mild neuronal loss in the CA1 subregion.
Regarding the 4R tau positive spherical neuronal inclusion bodies, the early vulnerability of the dentate gyrus is similar to the 3R tauopathology in Pick's disease 14, 21. However, in contrast to Pick's disease and other neurodegenerative FTLD‐Tau variants 5, 14 the spherical neuronal inclusions in the present 4R tauopathy cases seem to show an anatomically restricted distribution and do not show a hierarchical involvement of further anatomical regions. Interestingly, the morphology of inclusions seen in H&E and Gallyas staining is strongly reminiscent of that seen in the α‐synucleinopathy multiple system atrophy with frontotemporal lobar involvement (atypical multiple system atrophy), where the neuronal inclusions in the dentate gyrus are composed of α‐synuclein and not tau as in our cases 2, 34. These observations suggest a peculiar involvement of the dentate gyrus cells by compact inclusion bodies, which is different from the hippocampal vulnerability patterns of other tauopathies and AD/PART 27. Indeed, the dentate gyrus shows diffuse neuronal cytoplasmic tau immunoreactivity (i.e., pretangles) in AGD 17, 27, which frequently associates with PSP 9; however, in AGD these are not visible in HE staining and do not show p62 immunoreactivity as the basophilic structures in our cases. The lack of comparative neuropsychological studies on the reported cases does not allow the definition of the precise clinical phenotype associated with the dentate gyrus pathology. This would be particularly important, since PSP 4R tau pathology (i.e.`, seen also in two cases) itself might also associate with cognitive decline 24. Since the case reported by Miki et al did not present with cognitive decline 26 and one of our case showed only mild cognitive impairment, it might be that these conspicuous 4R tau inclusions in the dentate gyrus, even with the involvement of the pyramidal cells of CA subregions, are not sufficient to cause clinical symptoms or alternatively a threshold of the number of inclusion bodies or combined pathologies must be reached in order to be associated with neuronal dysfunction and clinical symptoms.
Spherical neuronal cytoplasmic inclusions in non‐hippocampal cells have been described in CBD 7, 13 and also in GGT cases with prominent white matter tau pathology 20. Furthermore, in MAPT mutation cases a mixture of 4R‐ and 3R‐positive spherical inclusions can be detected 11. Importantly, in 4R tauopathies, including the present form, the Pick‐body‐like inclusions are Gallyas positive contrasting 3R tau positive Pick bodies of Pick's disease 13, 26. The distribution of tau pathology and the presence of basophilic inclusions distinguish our cases from the neuropathology of chronic traumatic encephalopathy 25 and the tauopathy associated with IgLON5 antibodies, where the hippocampus and brainstem shows prominent involvement 10. Basophilic neuronal inclusions can be observed also in basophilic inclusion body disease; however, the anatomical distribution of those inclusions is different, involving mostly subcortical structures 22 and not the dentate gyrus; moreover, they are immunoreactive for fused in FUS and not hyperphosphorylated tau 29. However, in our cases, we excluded immunoreactivity for FUS in these basophilic inclusions.
Prominent astrocytic tau pathology, representing a mixture of tufted astrocytes, ramified astrocytes and granular/fuzzy in the hippocampus and amygdala expanding beyond that seen in PSP cases were seen in all our cases. One of our cases showed additional prominent white matter and subpial thorn shaped astrocytes in the frame of ARTAG pathology 19. Astrocytic tau pathology was not mentioned by Miki et al 26. Another study reported astroglial tau pathology in the hippocampus associated with hippocampal sclerosis and tauopathy but did not mention basophilic spherical inclusions in the dentate gyrus 3. Prominent astroglial tau pathology in the hippocampus is not a typical finding in PSP 17. Moreover, in case 1, the hippocampal astroglial tau pathology expanded far beyond the degree seen in early stage AGD 35, since in AGD it does not involve prominently the hippocampal CA subregions, especially not the CA4 subregion 4, as seen in our case 1. Furthermore, astroglial tau pathology appeared in the hippocampus in the case without PSP (case 3); accordingly, tau positive and argyrophilic astrocytes in CA subregions of the hippocampus seem to be consistently present and therefore are additional features of the 4R tauopathy with spherical hippocampal neuronal inclusions. Similarly to tufted astrocytes in PSP and astrocytic plaques in CBD 17, the astrocytes seen in the hippocampus in our cases showed strong Gallyas positivity, which is distinct from the non‐argyrophilic nature of the globular astroglial inclusions seen in GGT 1, 17 and granular/fuzzy astrocytes of gray matter ARTAG 17, 19.
Interestingly, hippocampal sclerosis was detected in the case reported by Miki et al 26, in two of our cases with prominent dementia, but not in the case with mild cognitive impairment. Hippocampal sclerosis in aging is thought to be a separate entity with AD type clinical symptoms 31. Although not part of current consensus‐based diagnostic criteria, TAR‐DNA binding protein 43 (TDP‐43) pathology is strongly linked to hippocampal sclerosis in aging 12, 31. TDP‐43 immunoreactivity has been reported in two published cases 21, 26 and was seen in one in our cohort. Interestingly, hippocampal sclerosis in GGT with white matter predominant tau pathology also does not associate with TDP‐43 pathology 20. This suggests that the pathogenesis of hippocampal sclerosis in a subset of tauopathies might be different than in hippocampal sclerosis of the aging. It is of note that we observed gliosis also in all CA subregions contrasting age‐related hippocampal sclerosis where CA1 is more affected. We observed TDP‐43 pathology only in the eldest (92 years‐old) patient. Indeed, due to the fact that hippocampal sclerosis of aging affects the oldest‐old with arteriolosclerosis and TDP‐43 pathologies that extend well beyond the hippocampus, the term cerebral age‐related TDP‐43 and sclerosis (CARTS) has been recently recommended 32. However, in our case (Nr.3) we observed also severe degree of hippocampal AD‐related pathology, thus most likely combined pathogenesis led to the severe neuronal loss.
Our biochemical studies confirm that in one case the 4R isoform was predominant, while in the second examined case 3R tau isoforms also were present in the hippocampus sample. Differential solubility of tau in AD and related tauopathies is well described 38 and this sample contained AD‐related NFT pathology, which is most likely responsible for the 3R component. Genetic analysis excluded mutations in MAPT and showed that two patients carried the H1/H1 haplotype. Importantly, these two showed also subcortical tauopathy compatible with PSP, which is known to associate strongly with this haplotype 8, 30. The presence of H2/H2 in the third case without PSP pathology could suggest that the present hippocampal tauopathy is not exclusively linked to the presence of H1/H1 and that atypical tauopathies might be expected in individuals carrying the H2/H2 haplotype.
In summary, based on our report here and prior observations, the novel 4R tauopathy with hippocampal spherical inclusions (i) affects mostly elderly individuals, (ii) may associate with features of other tauopathies or AD/PART, (iii) associate frequently with tau positive astrocytes in the hippocampus and amygdala and (iv) frequently, but not inevitably, combines with hippocampal sclerosis with or without TDP‐43 immunoreactivity. Our aim is to raise awareness of this peculiar tauopathy, which can be screened for in archival material also. Our study suggests that this tau pathology may be a morphological substrate of neuropsychiatric symptoms, however, due to the overlap with other tau pathologies in our cohort, the precise definition of its clinicopathological relevance merits further study.
Conflict of Interest
Authors report no conflict of interest.
Acknowledgments
Support for this work was provided by grants from the National Institute on Aging of the National Institutes of Health (P30‐AG10124, PO1‐AG17586 and NS088341). We thank the members of the Center for Neurodegenerative Disease Research at the University of Pennsylvania, Philadelphia, PA, who contributed to the work, Manuela Neumann and Elisabeth Kremmer for providing the phosphorylation specific TDP‐43 antibody, and the many patients studied and their families for making the research described here possible.
Contributor Information
Gabor G. Kovacs, Email: gabor.kovacs@meduniwien.ac.at.
John Q. Trojanowski, Email: trojanow@mail.med.upenn.edu.
References
- 1. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA et al (2015) Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha‐synuclein. Acta Neuropathol 130:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beach TG, Sue L, Scott S, Layne K, Newell A, Walker D et al (2003) Hippocampal sclerosis dementia with tauopathy. Brain Pathol 13:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Botez G, Probst A, Ipsen S, Tolnay M (1999) Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol 98:251–256. [DOI] [PubMed] [Google Scholar]
- 5. Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathologica 128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K et al (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946. [DOI] [PubMed] [Google Scholar]
- 8. Dickson DW, Rademakers R, Hutton ML (2007) Progressive supranuclear palsy: pathology and genetics. Brain Pathol 17:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dugger BN, Adler CH, Shill HA, Caviness J, Jacobson S, Driver‐Dunckley E et al (2014) Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord 20:525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gelpi E, Hoftberger R, Graus F, Ling H, Holton JL, Dawson T et al (2016) Neuropathological criteria of anti‐IgLON5‐related tauopathy. Acta Neuropathol. doi: 10.1007/s00401-016-1591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M (2015) Invited review: frontotemporal dementia caused by microtubule‐associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 41:24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ighodaro ET, Jicha GA, Schmitt FA, Neltner JH, Abner EL, Kryscio RJ et al (2015) Hippocampal sclerosis of aging can be segmental: two cases and review of the literature. J Neuropathol Exp Neurol 74:642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikeda K, Akiyama H, Arai T, Tsuchiya K (2002) Pick‐body‐like inclusions in corticobasal degeneration differ from Pick bodies in Pick's disease. Acta Neuropathol 103:115–118. [DOI] [PubMed] [Google Scholar]
- 14. Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE et al (2016) Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 79:272–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM et al (2015) Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 129:469–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jellinger KA, Grazer A, Petrovic K, Ropele S, Alpi G, Kapeller P et al (2011) Four‐repeat tauopathy clinically presenting as posterior cortical atrophy: atypical corticobasal degeneration?. Acta Neuropathol 121:267–277. [DOI] [PubMed] [Google Scholar]
- 17. Kovacs GG (2015) Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 41:3–23. [DOI] [PubMed] [Google Scholar]
- 18. Kovacs GG (2016) Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci 17:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al (2016) Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R et al (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovacs GG, Rozemuller AJ, van Swieten JC, Gelpi E, Majtenyi K, Al‐Sarraj S et al (2013) Neuropathology of the hippocampus in FTLD‐Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol Appl Neurobiol 39:166–178. [DOI] [PubMed] [Google Scholar]
- 22. Lashley T, Rohrer JD, Mead S, Revesz T (2015) Review: an update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol 41:858–881. [DOI] [PubMed] [Google Scholar]
- 23. Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 24. Ling H (2016) Clinical approach to progressive supranuclear palsy. J Mov Disord 9:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miki Y, Mori F, Hori E, Kaimori M, Wakabayashi K (2009) Hippocampal sclerosis with four‐repeat tau‐positive round inclusions in the dentate gyrus: a new type of four‐repeat tauopathy. Acta Neuropathol 117:713–718. [DOI] [PubMed] [Google Scholar]
- 27. Milenkovic I, Petrov T, Kovacs GG (2014) Patterns of hippocampal tau pathology differentiate neurodegenerative dementias. Dement Geriatr Cogn Disord 38:375–388. [DOI] [PubMed] [Google Scholar]
- 28. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S et al (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627. [DOI] [PubMed] [Google Scholar]
- 30. Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L et al (2007) The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 25:561–570. [DOI] [PubMed] [Google Scholar]
- 31. Nelson PT, Smith CD, Abner EL, Wilfred BJ, Wang WX, Neltner JH et al (2013) Hippocampal sclerosis of aging, a prevalent and high‐morbidity brain disease. Acta Neuropathol 126:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nelson PT, Trojanowski JQ, Abner EL, Al‐Janabi OM, Jicha GA, Schmitt FA et al (2016) “New Old Pathologies”: AD, PART, and cerebral age‐related TDP‐43 with sclerosis (CARTS). J Neuropathol Exp Neurol 75:482–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y et al (2009) Phosphorylation of S409/410 of TDP‐43 is a consistent feature in all sporadic and familial forms of TDP‐43 proteinopathies. Acta Neuropathol 117:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rohan Z, Rahimi J, Weis S, Kapas I, Auff E, Mitrovic N et al (2015) Screening for alpha‐synuclein immunoreactive neuronal inclusions in the hippocampus allows identification of atypical MSA (FTLD‐synuclein). Acta Neuropathol 130:299–301. [DOI] [PubMed] [Google Scholar]
- 35. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y et al (2004) Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol 63:911–918. [DOI] [PubMed] [Google Scholar]
- 36. Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL et al (2014) A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 10:477–484 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Deerlin VM, Gill LH, Farmer JM, Trojanowski JQ, Lee VM (2003) Familial frontotemporal dementia: from gene discovery to clinical molecular diagnostics. Clin Chem 49:1717–1725. [DOI] [PubMed] [Google Scholar]
- 38. Zhukareva V, Shah K, Uryu K, Braak H, Del Tredici K, Sundarraj S et al (2002) Biochemical analysis of tau proteins in argyrophilic grain disease, Alzheimer's disease, and Pick's disease: a comparative study. Am J Pathol 161:1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]