

Abstract
Background/Purpose
Ethanol co-administered with immediate-release dl-methylphenidate (dl-MPH) or dexmethylphenidate (d-MPH) significantly increases the geomean maximum plasma concentration (Cmax) of d-MPH 22% and 15%, respectively, and elevates overall drug exposure and psychostimulant effects. We asked the question: Are these ethanol-MPH interactions based more fundamentally on: (1) inhibition of post-absorption d-MPH metabolism or (2) acceleration of MPH formulation gastric dissolution by ethanol in the stomach? This was investigated using the pulsatile, distinctly biphasic, Spheroidal Oral Drug Absorption Systems of dl-MPH and d-MPH.
Methods/Procedures
In a randomized, 4-way crossover study, fourteen healthy subjects received pulsatile dl-MPH (40 mg) or d-MPH (20 mg), with or without ethanol (0.6 g/kg), dosed 4 hours later. These 4 hours allowed the delayed-release second MPH pulse to reach a more distal region of the gut to preclude gastric biopharmaceutical influences. Plasma was analyzed using a highly sensitive chiral method. Subjective/physiological effects were recorded.
Findings/Results
Ethanol increased the second pulse of d-MPH Cmax for dl-MPH by 35% (P < 0.01), and the partial area under the curve (pAUC4–8h) by 25% (P < 0.05). The respective values for enantiopure d-MPH were 27% (P = 0.001) and 20% (P < 0.01). The carboxylesterase 1-mediated transesterification metabolite ethylphenidate served as a biomarker for co-exposure. Ethanol significantly potentiated stimulant responses to either formulation.
Implications/Conclusions
These findings support drug dispositional interactions between ethanol and MPH as dominant over potential biopharmaceutical considerations. Understanding the pharmacology underlying the frequent co-abuse of MPH-ethanol provides rational guidance in the selection of first-line pharmacotherapy for comorbid ADHD-alcohol use disorder.
INTRODUCTION
Racemic methylphenidate (dl-MPH), a 50:50 mixture of d-threo-(R:R)-MPH and l-threo-(S:S)-MPH isomers, continues to be one of the most widely used psychostimulant to treat attention-deficit/hyperactivity disorder (ADHD) throughout the world. However, abuse of this controlled substance is well documented and patterns of misused or diverted dl-MPH very commonly involve concomitant alcohol (ethanol).1–10 Compounding the societal cost of MPH-ethanol co-abuse, alcohol use disorder (AUD) is overrepresented in adolescent11 and adult12,13 ADHD patients, especially in women.14 In addition, comorbid ADHD-AUD patients may be at special risk of AUD relapse.15
Twice or three times daily immediate-release (IR)-dl-MPH regimens had served for decades as the “gold standard” for ADHD pharmacotherapy.16 A potential therapeutic advantage of IR-MPH over the subsequent once daily formulation, a wax matrix sustained-release dl-MPH product, has been attributed to the more rapid absorption rate of onset for IR-MPH.17 This pharmacokinetic (PK) distinction has also been implicated in heightened stimulant euphoria and abuse liability of IR-dl-MPH relative to the sustained-release product.4,18–22 More recently, once daily second generation pulsatile/biphasic, modified-release (MR)-MPH products have largely supplanted IR-MPH regimens. These newer formulations incorporate both an IR-MPH component to facilitate rapid onset and a delayed-release component to mitigate compliance, diversion and peer ridicule concerns – especially pertinent to school time IR-MPH dosing.
In 2001, the pure active23–27 IR-d-MPH isomer, dexmethylphenidate (d-MPH; Focalin, Novartis Pharmaceuticals, East Hanover, NJ), was approved for the treatment of ADHD,28,29 followed by approval of a distinctly pulsatile (relative to other MR-MPH products17) MR capsule in 2005. This enantiopure MR-d-MPH formulation contains 50% IR beads and 50% MR beads with the MR beads developed to dissolve approximately 4 hours after dosing using the spheroidal oral drug absorption system (SODAS) formulation technology (Focalin XR; Novartis Pharmaceuticals, East Hanover, NJ).30
For conversion from a maintenance dose of once daily SODAS dl-MPH (Ritalin LA; Novartis Pharmaceuticals, East Hanover, NJ) to a once daily regimen of the SODAS d-MPH product, the labeling recommends a reduction to one-half the total daily mg drug strength of MR-dl-MPH owing to the inactivity of the l-isomer.23–27 SODAS d-MPH was preceded by a corresponding racemic SODAS dl-MPH formulation
Use of MR-MPH products in general have been reported to reduce MPH-ethanol co-abuse liability,31 as attributed to less rapid rate of drug absorption following most once daily MR-MPH formulations compared to IR-dl-MPH.21,32,33 However, the 50% IR-MPH component in the SODAS formulations represents the highest IR percent of any of the several existing MR-MPH products34 and consequently results in a PK profile closely resembling the distinct dual plasma d-MPH concentration peaks of a typical twice daily IR-MPH regimen.35,36
dl-MPH interacts with ethanol to potentiate a range of positive subjective effects, as consistent with the illicit popularity of concomitant MPH-ethanol co-abuse.1–10 This pharmacodynamic (PD) interaction has correlated with ethanol-induced increases in d-MPH exposure.20,22 Ethanol appears to inhibit d-MPH hydrolysis by carboxylesterase 1 (CES1) in the course of serving as a CES1 transesterification substrate, enantioselectively forming l-ethylphenidate (EPH). This metabolite serves as a biomarker for concomitant dl-MPH – ethanol drug exposure.37–39
Following oral dosing with dl-MPH, approximately 25% of the active23–27 d-MPH, while only 1–5% of l-MPH reaches the systemic circulation.40–43 In addition to ethanol influencing d-MPH PK, formulation differences between IR-dl-MPH and IR-d-MPH have also been reported to markedly alter early exposure to d-MPH.44
In the present study, we extend the characterization of dl-MPH-ethanol compared to d-MPH – ethanol PK/PD interactions to the MR-MPH SODAS formulations described above. These are the only MR-MPH products available as both racemic and enantiopure isomeric forms, and both use the same MR pharmaceutical technology, providing virtually indistinguishable rates and extents of d-MPH absorption in the absence of ethanol.36 Accordingly, these formulations were selected for the present comparative study of ethanol interactions.
Ethanol administration commenced at the beginning of the reported 4 hour mean plasma d-MPH trough time,36 then consumed at a constant rate over the course of 0.25 hours. Four hours corresponds to both the trough time of the typical twice daily IR-MPH regimen as well as to the uniquely distinct d-MPH trough time characterizing these SODAS products; just in advance of the steep absorption phase of the second drug release pulse after 4 hours of small gut transit time. This study design allowed avoidance of any potential influence of ethanol on the dissolution rate of the formulations, unlike the previous IR-MPH-ethanol studies where the racemate20, or the racemate compared to the pure d-enantiomer22,39 were taken within the same 0.5–0.75 hours ethanol.
We asked the question: Is the significant elevation in overall d-MPH exposure resulting from concomitant ethanol primarily due to a hypothetical pharmaceutics/formulation dissolution rate acceleration, or due to an influence of ethanol on MPH metabolism. Data analysis included partial area under the plasma concentration curve (pAUC) comparisons, this being a timely extension of a new PK metric used by the FDA in accessing bioequivalence of other MR-dl-MPH products designed to be bioequivalent to the MR osmotic release oral system.46–48 Our findings extend PK-PD relationships pertaining to the apparent increase in MPH abuse liability when combined with ethanol.1–10 These findings offer guidance in the rational first-line drug selection for the treatment of patients with comorbid ADHD and substance use disorder.
MATERIALS AND METHODS
Research subjects
Each subject provided a written informed consent approved by the Medical University of South Carolina’s Office of Research Integrity. The study was conducted in the Clinical & Translational Research Center located at the Medical University of South Carolina. The study population consisted of 14 volunteers 22–42 years of age who were healthy as assessed by medical history, physical examination, 12-lead electrocardiogram, and routine laboratory tests. All subjects were within 15% of ideal body weight, were non-smokers and were asked to abstain from the use of caffeine containing beverages beginning at 7:30 p.m. the evening before and continuing through each active study day. The subjects consented to being medication free (with the exception of allowed FDA-approved oral contraceptives for female subjects) from 3 days before initiation of the first active study day and through the duration of the study, including the use of vitamins, dietary supplements and OTC medications.
Study design
An open label, randomized, 4-way crossover study design was used. There were four treatment schedules: Oral MR-dl-MPH (40 mg) or MR-d-MPH (20 mg), with or without ethanol (0.6g/kg) 4.0–4.25 hours later. All subjects were admitted to the research center on the morning of each active study day. One hour prior to dosing, the subjects consumed a light breakfast consisting of a plain bagel (36 g total: fat 1 g, carbohydrate 29 g, protein 6 g) with cream cheese (30 g total: fat 9.2 g, protein 4.4 g, carbohydrate 0.03 g) and skim milk (240 ml total: fat 9.2 g, carbohydrate 11.5 g, protein 8.4 g), finished over a period of 10 min… Following a urine drug screen and pregnancy test (females), an indwelling venous catheter was placed in each subject’s arm for serial blood sampling at 0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 10, 24 hours post-dosing. MR-dl-MPH (40 mg) or MR-d-MPH (20 mg) was administered with 240 ml of room temperature water. Subjects remained seated, with feet on the floor, for 2 hours after MPH administration.
Depending on the randomization assignment, an ethanol or non-ethanol orange juice drink was consumed 4.0–4.25 hours after MPH dosing. The ethanol drink was administered as 0.6 g/kg ethanol (0.66 ml/kg 95% ethanol) in 180 ml of orange juice and 60 ml of soda water, with additional water added to provide a total volume of 450 ml. For alcohol-free treatments, the alcohol volume was replaced with water. The drinks were consumed over 15 min at a rate of approximately 30 ml/min. Subjects received a standard lunch consumed in its entirety over the 3.25–3.75 hour period after MPH dosing. Lunch consisted of a turkey sandwich on whole wheat bread (2 slices bread, 3 slices turkey, 31 g), 1 1/8 oz potato chips (Baked Lay’s, 32 g), 1 canned fruit in light syrup (Dole mixed fruit cups, 120 g), and 360 ml of water. A standardized dinner was provided 10.5 hours after MPH dosing. There was at least a 6-day washout period between treatments and negative urine drug screens and urine pregnancy results (females) were obtained at the beginning of each active study session.
Blood collection tubes (Vacutainers®, Becton Dickinson, Rutherford, NJ) were previously stored in an ice bath and contained sodium oxalate to minimize post-sampling MPH and ethylphenidate hydrolysis. Green stoppered tubes (2 ml) were used to collect whole blood for blood ethanol analysis by the hospital clinical laboratory per standard procedures (< 10 mg/dl lower limit of quantitation). Venous catheter lines were flushed of residual heparin solution prior to sampling. Samples were promptly centrifuged at 4°C for 5 min and the plasma immediately aspirated into separate labeled polypropylene vials and stored at −70°C until analysis.
Vital signs
Blood pressure, heart rate, temperature and respiratory rate were obtained at the screening visit and recorded at the beginning and end of each of the four active sessions. Blood pressure and heart rate were recorded before dosing and again at the following intervals after receiving MR-MPH: 0.75, 1.75, 2.75, 3.75, 4.75, 5.75 and 11.75 hours.
Visual Analog Scales
A visual analog drug subjective effect questionnaire was used with nine subscales: (1) Do you feel any drug effect?; (2) How high are you?; (3) Do the drugs have any good effects?; (4) Do the drugs have any bad effects?; (5) Do you like the drugs?; (6) Do you feel depressed?; (7) How anxious are you?; (8) How stimulated do you feel?; (9) How intoxicated do you feel? This questionnaire was administered before (baseline) dosing with MR-MPH, then repeated at 0.75, 1.25, 2.25, 3.25, 4.25, 5.25, 6.25, 7.25, 8.25, 9.25 and 11.75 hours after MR-MPH dosing. The subscales allowed rating of the degree to which the subject was experiencing each effect by making a vertical mark on a horizontal solid line ranging in intensity of drug effect from “not at all” (0) to “extremely” (100), as previously used.20,22,39
Recovery Period
Following each study period, subjects remained on the study unit site until blood ethanol concentrations were below 10 mg% (mg/dl), as determined by a breathalyzer measurement (AlcoHAWK®, Independence, IA).
d-MPH, l-MPH, d-EPH and l-EPH plasma analysis
Plasma analyses were conducted by a robustly validated and enantiospecific method using liquid chromatography-tandem mass spectrometry and a vancomycin based chiral stationary phase (Astec Chirobiotic V2 column, 5 μm, 250×2.1 mm, Sigma–Aldrich, St. Louis, MO). Analytical control was provided by incorporation of a deuterated internal standard and byanalyzing a range of spiked plasma calibrators run in parallel to the unknowns. The following transitions were monitored in the multiple reaction monitoring (MRM) mode: both isomers of dl-MPH, m/z 234 > 84; both isomers of dl-EPH, m/z 248 > 84; both isomers of 2H3-dl-MPH, m/z 237 > 84. The lower limit of quantitation was 0.025 ng/ml for each isomer. The analysis used 0.5 ml of plasma.49
Pharmacokinetic analysis
PK parameters were calculated by standard methods.50 The non-compartmental analysis of enantiospecific MPH and EPH plasma concentrations was performed using WinNonlin v 5.1® software (Pharsight, Cary, NC).
Statistical analysis
The mean and the least square geometric means of the two test treatments (MR-d-MPH with/without ethanol) and the reference (MR-dl-MPH with/without ethanol) were calculated for the Cmax and AUC values. Ratios of the test geometric means to the reference, as well as the 90% confidence intervals about the reference, were determined. Correlations between parameters for individuals were assessed by linear regression analysis (Instat® 3.01, GraphPad software, San Diego, CA). The primary endpoint variables were compared using analysis of variance (ANOVA) with the four treatments as a between (repeated measures) factor using the Latin square design to take into account sequence (carry-over) effect as described by Winer.51 The pAUC4.25–8.25 hours VAS subscales between formulations, with ethanol and without ethanol, were analyzed using a paired, parametric, one-tailed T-test (GraphPad 6® software, San Diego, CA). The level of significance was set at P = 0.05.
RESULTS
Human subjects
Fourteen healthy volunteers completed the study; eight men (7 Caucasian; 1 Asian; mean weight 82.2 kg, range 71.9 kg – 93.6 kg ; mean age 25.7 years, range 22 – 30 years), and six women (all Caucasian; mean weight 59.6 kg, range 47.9 kg– 82.2 kg; mean age 28.1 years, range 21 – 42 years). One woman withdrew from the study, completing only the two dl-MPH-MR treatment groups, due to trouble with intravenous cannulation, and these data were excluded from analysis. Four occurrences of headaches were reported and were treated with ibuprofen (400 mg). One subject reported taking acetaminophen the day before the dl-MPH-MR only treatment and was allowed to continue. No subject had any clinically significant findings on post-study “exit” laboratory tests. No subject exhibited a phenotype suggestive of CES1 poor metabolizer status.20,37
Influence of Ethanol on d-MPH PK: MR-dl-MPH versus MR-d-MPH
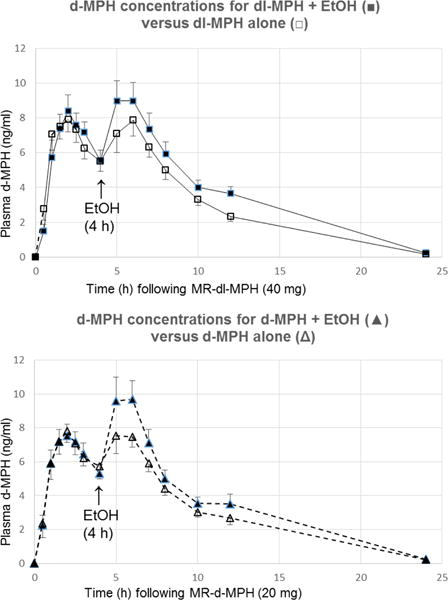
The mean plasma concentration (+/− SD) time profiles for the active d-MPH isomer following oral MR-dl-MPH (40 mg/kg; comparator) versus MR-d-MPH (20 mg; test), with or without ethanol (0.6 g/kg) consumed during 4–4.25 hours after MR-MPH administration, are shown in Figure 1 (Top) and 1 (Bottom), respectively. The associated PK parameters are found in Table 1. The statistical comparisons of treatments by geometric mean ratios are presented in Table 2. Following a dose of MR-dl-MPH, ethanol significantly elevated the mean d-MPH Cmax2 (P = 0.001) and AUC4–8h (P < 0.001) values. Ethanol also significantly elevated these corresponding parameters after dosing with the pure isomer MR-d-MPH (P < 0.001 and P < 0.05, respectively).
FIGURE 1.

(Top): Arithmetic mean (+/− SD) d-MPH plasma concentrations after dosing with MR-dl-MPH (40 mg) alone (□) or dosing with racemic MR-dl-MPH (40 mg MR) followed by ethanol (0.6 g/kg) 4–4.25 h later (▪); (Bottom): Arithmetic mean d-MPH plasma concentrations after dosing with enantiopure MR-d-MPH (20 mg) alone (Δ) or dosing with MR-d-MPH (20 mg) followed by ethanol (0.6 g/kg;) 4–4.25 h later. Values only include those at or above the lower limit of detection (≥ 0.025 ng/ml).
TABLE 1. d-Methylphenidate Pharmacokinetic Parameters.
Subjects (n = 14) received either racemic MR-dl-MPH (40 mg/kg) or enantiopure MR-d-MPH (20 mg/kg), then orange juice, with or without ethanol (0.6 g/kg), 4.0–4.25 hours later.
| Parameter | dl-MPH + EtOH | dl-MPH alone | d-MPH + EtOH | d-MPH alone |
|---|---|---|---|---|
| K (h−1) | 0.214 (21) | 0.221 (14) | 0.218 (12) | 0.215 (15) |
| T1/2 (h) | 3.4 (26) | 3.2 (14) | 3.2 (12) | 3.3 (15) |
| Cmax1 (ng/ml) | 9.5 (32) | 9.1 (31) | 12.4 (23) | 10.7 (21) |
| Cmax2 (ng/ml) | 10.3 (42) | 7.9 (44) | 10.8 (46) | 8.5 (43) |
| Tmax1 (h) | 1.5 (1–3) | 1.5 (1–3) | 2 (1–2.5) | 2 (1–3) |
| Tmax2 (h) | 6 (5–12) | 6 (5–10) | 6 (4–6) | 6 (4–7) |
| AUC0-last (ng*h/ml) | 90.8 (33) | 79.1 (30) | 88.4 (33) | 78.4 (29) |
| AUC0-inf (ng*h/ml) | 97.1 (29) | 80.0 (30) | 90.0 (32) | 79.8 (29) |
| AUC0–4h (ng*h/ml | 23.6 (32) | 24.0 (32) | 22.6 (31) | 22.7 (34) |
| AUC4–8h (ng*h/ml) | 31.2 (42) | 25.1 (39) | 31.6 (40) | 26.0 (35) |
dl-MPH, methylphenidate; d-MPH, dexmethylphenidate; MR, modified-release; EtOH, ethanol; AUC0-inf, area under the plasma concentration-time curve to infinity (arithmetic mean (CV%); AUC0-last, area under the plasma d-MPH concentration-time curve from 0 hour to the time of the last concentration (arithmetic mean (CV%); Cmax, maximum plasma concentration (arithmetic mean (CV%); Tmax, time to Cmax [median (range)]; K (h−1), first-order rate constant. Only plasma d-MPH concentrations at or above the lower limit of quantitation (≥ 0.025 ng/ml) were included in the data set.
TABLE 2.
Statistical Comparisons between treatment groups by GeoMean ratios: d-Methylphenidate least square geometric mean (90% CI) pharmacokinetic parameter ratios for the 14 subjects. Subjects received racemic MR-dl-MPH (40 mg MR) or enantiopure MR- d-MPH (20 mg) with or without EtOH (0.6 g/kg) 4–4.25 h later.
| Parameter d-MPH |
Statistical Test | MR-dl-MPH + Ethanol to MR-dl-MPH | MR-dl-MPH + Ethanol to MR-d-MPH + Ethanol | MR-dl-MPH to MR-d-MPH | MR-d-MPH + Ethanol to MR-d-MPH |
|---|---|---|---|---|---|
| Cmax | GeoMean Ratio | 1.15 | 0.96 | 1.04 | 1.24 |
| 90% CI | 1.03–1.27 | 0.87–1.07 | 0.94–1.16 | 1.11–1.38 | |
| P value | 0.038 | 0.545 | 0.518 | 0.002 | |
| Tmax (h) | GeoMean Ratio | 2.18 | 1.12 | 0.58 | 1.14 |
| 90% CI | 1.57–3.03 | 0.80–1.55 | 0.42–0.81 | 0.82–1.58 | |
| P value | 0.000 | 0.573 | 0.009 | 0.501 | |
| AUC0-last | GeoMean Ratio | 1.14 | 1.03 | 1.00 | 1.10 |
| 90% CI | 1.04–1.24 | 0.94–1.13 | 0.92–1.10 | 1.10–1.21 | |
| P value | 0.022 | 0.538 | 0.968 | 0.077 | |
| AUC0-inf | GeoMean Ratio | 1.23 | 1.10 | 1.00 | 1.12 |
| 90% CI | 1.14–1.33 | 1.02–1.19 | 0.92–1.08 | 1.03–1.21 | |
| P value | 0.000 | 0.043 | 0.949 | 0.024 | |
| K d-MPH | GeoMean Ratio | 0.95 | 0.95 | 1.02 | 1.02 |
| 90% CI | 0.88–1.02 | 0.88–1.02 | 0.94–1.10 | 0.95–1.10 | |
| P value | 0.226 | 0.220 | 0.699 | 0.685 | |
| T1/2 (h) | GeoMean Ratio | 1.06 | 1.06 | 0.98 | 0.98 |
| 90% CI | 0.98–1.14 | 0.98–1.14 | 0.91–1.06 | 0.91–1.06 | |
| P value | 0.226 | 0.220 | 0.699 | 0.685 | |
| Cmax1 (0–4h) | GeoMean Ratio | 1.06 | 1.07 | 1.03 | 1.02 |
| 90% CI | 0.94–1.19 | 0.95–1.21 | 0.91–1.16 | 0.90–1.15 | |
| P value | 0.434 | 0.356 | 0.697 | 0.806 | |
| Tmax1 (0–4h) | GeoMean Ratio | 1.12 | 1.08 | 0.98 | 1.01 |
| 90% CI | 0.92–1.37 | 0.88–1.32 | 0.80–1.19 | 0.83–1.24 | |
| P value | 0.353 | 0.528 | 0.836 | 0.924 | |
| AUC1 (0–4h) | GeoMean Ratio | 0.99 | 1.04 | 1.06 | 1.00 |
| 90% CI | 0.89–1.11 | 0.93–1.16 | 0.95–1.18 | 0.90–1.12 | |
| P value | 0.893 | 0.522 | 0.413 | 0.964 | |
| Cmax2 (4–8h) | GeoMean Ratio | 1.35 | 0.99 | 0.93 | 1.27 |
| 90% CI | 1.17–1.55 | 0.86–1.14 | 0.81–1.07 | 1.10–1.46 | |
| P value | 0.001 | 0.884 | 0.384 | 0.008 | |
| Tmax2 (4–8h) | GeoMean Ratio | 0.98 | 1.11 | 1.07 | 0.94 |
| 90% CI | 0.90–1.07 | 1.02–1.22 | 0.98–1.17 | 0.87–1.03 | |
| P value | 0.737 | 0.042 | 0.196 | 0.268 | |
| AUC2 (4–8h) | GeoMean Ratio | 1.25 | 0.99 | 0.95 | 1.20 |
| 90% CI | 1.10–1.43 | 0.87–1.13 | 0.83–1.08 | 1.05–1.36 | |
| P value | 0.006 | 0.931 | 0.500 | 0.026 |
dl-MPH, methylphenidate; d-MPH, dexmethylphenidate; MR, modified-release; EtOH, ethanol; AUCinf, area under the serum concentration-time curve to infinity; AUClast, area under the plasma d-MPH concentration-time curve from 0 hours to the time of the last concentration; Cmax, maximum plasma concentration; Tmax, time to Cmax.
The mean plasma Cmax for the enantioselectively formed ethanol transesterification metabolite, l-EPH, was 0.33 ng/ml (highest single value: 0.47 ng/ml; T = 5 hours). The mean pharmacologically active26 d-EPH plasma concentration following the MR-dl-MPH-ethanol treatment was 0.03 ng/ml (highest single value: 0.069 ng/ml; T = 7 hours); that for the MR-d-MPH-ethanol treatment was 0.027 ng/ml (highest single value: 0.068 ng/ml; T = 7 hours).
Compared to racemic MR-dl-MPH alone, ethanol increased the mean d-MPH Cmax2 (CV%) from 7.9 ng/ml (44) to 10.3 (46), and the pAUC4–8h value from 25.1 ng·h/ml (39) to 31.2 ng·h/ml (41). In the corresponding comparisons using the pure isomer MR-d-MPH, ethanol increased the plasma d-MPH Cmax from 8.5 (44) to 10.8 (46), and the pAUC4–8h from 26.0 (35) to 31.6 (40) – see Table 1. Statistical comparisons between treatment groups by geometric mean ratios revealed that the influence of ethanol on MR-dl-MPH elevated the Cmax2 by 35% and by 27% after MR-d-MPH dosing. Ethanol increased d-MPH AUC4–8h in the MR-dl-MPH treatment group by 25% and 20%, respectively, for the MR-d-MPH treatment (Table 2).
Regarding initial exposure to these drug combinations, the 5 hour plasma concentration (SEM) following MR-dl-MPH-ethanol was 9.0 ng (1.2), compared to 7.1 ng/ml (1.1) (P < 0.05) without ethanol. The corresponding values for MR-d-MPH with/without ethanol were 9.8 (1.4) and 7.5 (1.1) (P < 0.05).
The geometric mean ethanol Cmax values resulting from this experimental design did not significantly differ between the two drug combination treatments: 53 mg% (0.053%) and 56 mg% for MR-d-MPH and MR-dl-MPH, respectively (90% CI; 83.4–107.5). Similarly, the AUC4–8h values were within 95% of one another (90% CI; 81–110).
PK of d-MPH following MR-dl-MPH versus MR-d-MPH without ethanol
In the evaluation of MR-dl-MPH versus MR-d-MPH Cmax1 and pAUC0–4h values for all four treatment groups, as well as Cmax2 and pAUC4–8h with the MR products without ethanol, all parameters were comparable (Table 1), i.e., the 90% confidence intervals for the geometric mean ratios for both the Cmax and AUC0-inf of d-MPH demonstrated bioequivalence (i.e., CI’s within 80–125; Table 2).52
Statistical comparisons of the pAUC0–4h, Cmax and Tmax geometric mean ratios (i.e., prior to ethanol) for both MR-dl-MPH and MR-d-MPH were not significant different. Ratios for these parameters were 1.04, 1.06 and 0.98, respectively. The corresponding unitary values (CV%) were 24.0 (32) versus 22.7 (43) ng•h/ml; 9.1 (31) versus 8.7 (35) ng/ml; and 1.8 (48) versus 1.8 (34) – Table 2.
Influence of Ethanol on MR-MPH PD: MR-dl-MPH versus MR-d-MPH
The combination of ethanol with racemic MR-dl-MPH resulted in a significant potentiation of the cumulative subjective effects pAUC4.25h–8.25h values for the “stimulated” subscale (P < 0.005); as well as for the potentiation of the “high” (P = 0.013) and “any effect” rating (P = 0.017). Ethanol-induced potentiation of enantiopure MR-d-MPH cumulative subjective effects pAUC4.25h–8.25h was significant for “stimulated” (P < 0.05), while approaching statistical significance for “good” and “any effect” – Table 3.
TABLE 3.
Cumulative potentiation of positive subjective effects (pAUC4.25–8.25h) of racemic MR-dl-MPH (40 mg; T = 0) or enantiopure MR-d-MPH (20 mg; T = 0) by EtOH (T = 4–4.25 hours).
| Racemate | dl-MPH+EtOH | dl-MPH alone | Potentiation by EtOH | |
|---|---|---|---|---|
| Mean (+/−SEM) | Mean (+/−SEM) | Mean | P Value | |
| “Stimulated” | 41 (12) | 25 (9) | 16 | 0.0046 |
| “High” | 16 (6) | 11 (5) | 5 | 0.0134 |
| “Good” | 52 (16) | 49 (23) | 3 | 0.3307 |
| “Any effect” | 41 (12) | 33 (11) | 8 | 0.0168 |
| d-Isomer | dexMPH+EtOH | dexMPH | Potentiation by EtOH | |
| Mean (+/−SEM) | Mean | P Value | ||
| “Stimulated” | 33 (15) | 19 (8) | 14 | 0.0447 |
| “High” | 21 (9) | 7 (3) | 14 | 0.0884 |
| “Good” | 59 (24) | 45 (22) | 8 | 0.0687 |
| “Any effect” | 38 (14) | 25 (8) | 13 | 0.0634 |
pAUC4.25–8.25h, partial area under the subjective effect curves from 4.25–8.25 hours; EtOH, ethanol; MR, modified-release; dl-MPH, dl-methylphenidate; d-MPH, dexmethylphenidate.
The VAS subscale potentiation induced by combining ethanol consumed over the 4.0–4.25 hour period following MR-MPH reached a maximum at 4.25 hours for MR-dl-MPH and at 5.25 hours for MR-d-MPH.
The effects of ethanol on the VAS subscales “bad,” “depressed,” “anxious” and “intoxicated” were unremarkable for either the MR-dl-MPH-MR or MR-d-MPH-MR treatment groups. While not found to be statistically significant, the subscale of “Like” trended to be potentiated by ethanol.
Blood pressures and pulse rates at times 12:45 and 13:45 were analyzed to assess the influence of ethanol. The ethanol combination caused a statistically significant increase in pulse rate when given concomitantly with either MR-dl-MPH or MR-d-MPH (P < 0.05). For these two time points, the mean increase induced by concomitant ethanol for dl-MPH-MR and d-MPH-MR were 6.8 bpm and 11.2 bpm, respectively. There were no significant changes in blood pressure upon combining ethanol with either MR-MPH formulation.
DISCUSSION
We hypothesized that ethanol-induced elevation in plasma d-MPH concentrations20,39 results primarily from post-absorption drug dispositional factors rather than simply the presence of ethanol in the stomach accelerating the dissolution of MPH formulations. Further, we asked the question: Does the presence of l-MPH in combination with ethanol increase the exposure to d-MPH as CES1 enantioselectively transesterifies l-MPH to l-ethylphenidate? Most of an oral dose of MPH is hydrolyzed by CES1 presystemically and eliminated in the urine as the corresponding amino acid. Our ethanol interaction findings are consistent with elevated d-MPH plasma concentrations being primarily dependent on a general inhibition of CES1 by ethanol. Ethanol induced inhibition of CES1-mediated drug hydrolysis finds precedent in the elevation of drug exposure in cases where their structures contain metabolically labile small esters, e.g., methyl or ethyl esters.53–57
Relative to other MR-MPH formulations, the SODAS MR-dl-MPH or SODAS MR-d-MPH formulations provide the most distinctly pulsatile plasma d-MPH concentration profiles, very much resembling the time course of a twice daily IR-MPH regimen with doses separated by 4 hours.17,34 The experimental design used in the present study precluded the possibility of a biopharmaceutics-based dissolution rate component influencing the MPH-ethanol interaction. The ethanol drink was consumed beginning 4 hours after either of these SODAS MPH formulations, allowing the necessary time for gastric dissolution of the IR portion of the dose as well as the gastric emptying of the delayed-release MPH beads.
Following a dose of racemic MR-dl-MPH, ethanol increased the geomean d-MPH Cmax2 by 35% (P < 0.01) and the pAUC4–8h by 25% (P < 0.05). The ethanol-induced increases in d-MPH following the enantiopure MR-d-MPH product were 27% (P = 0.001) and 20% (P < 0.01), respectively.
Accordingly, the mechanism of this PK MPH-ethanol interaction appears more related to a more general inhibition of CES1 by ethanol rather than based on formulation dissolution effects by ethanol. Any substantive significance of the transesterification pathway competitively inhibiting d-MPH hydrolysis to further elevate the drug concentration seems unlikely.
These ethanol influences on d-MPH Cmax2 and AUC4–8h are illustrated in Figure 1 and quantified in Tables 1 and 2. Table 1 shows the arithmetic mean PK parameters, as altered by ethanol-MPH interactions, while Table 2 delineates the comparative statistical analysis of the geometric mean ratios of these parameters. It is noted that ethanol also significantly increased the rate of plasma d-MPH ascent as the concentrations reached Cmax for both MR-dl-MPH and MR-d-MPH, a temporal relationship potentially pertinent to MPH abuse liability.18,19,58–61
Assessing the 0–4 hour time periods for all 4 treatment groups, i.e., all initial periods in the absence of ethanol, the d-MPH Cmax1 and pAUC0–4h values for both MR-d-MPH and MR-dl-MPH met the regulatory standards of bioequivalence. However, racemic dl-MPH and enantiopure d-MPH are classified as separate drug entities, and these comparisons are not technically subject to conventional bioequivalence evaluation. The entire d-MPH plasma concentration-time course for the two MR-d-MPH and MR-dl-MPH treatment groups, without ethanol, produced essentially identical profiles as consistent with an earlier comparison.36
Fundamental studies of MPH-ethanol pharmacological interactions were initially conducted with IR-dl-MPH,20,45 followed by IR-dl-MPH compared against IR-d-MPH20,22 formulations, to gain an understanding of the mechanisms underlying metabolic and psychopharmacological interactions uncomplicated by the range of PK characteristics differentially distinguishing the MR-MPH products.
Since the early 2000s, a range of once daily dosed pulsatile MR-MPH products have largely supplanted multiple daily dose IR-MPH regimens in an effort to mitigate issues of compliance, peer ridicule and diversion.17,34 These various pulsatile (biphasic) MR-MPH treatment options include IR compositions from 20%,63 22%, 30%, 40% and 50% of the total dose.34 In keeping with these more contemporary MPH regimens, the results reported here extend the characterization of MPH-ethanol interactions to pulsatile MR-MPH formulations.
In our study design, the delayed-release component of the MR-MPH dose enters the portal vein from a more the distal region of the gut, with ethanol primarily entering the portal vein from the duodenum when administered 4 hours later.64 This delayed exposure to ethanol offers the advantage of circumventing theoretical confounds potentially associated with enteric first-pass effects of concomitant MPH-ethanol consumption. The present study design: (1) avoids the potential for ethanol to delay in gastric emptying time64 which would extend the period of possible acid catalyzed interactions of the 2 drugs; (2) precludes any proximal digestive and bacterial enzyme actions between the drugs following concomitant gastric emptying; and (3) obviates any simultaneous MPH-ethanol gut wall absorption/metabolism influences.
Ethanol can potentially influence gastric dissolution properties of some MPH dosage forms,65 Drawing from our own experience, the plasma d-MPH concentration-time profiles from IR-d-MPH tablets compared to IR-dl-MPH tablets (administered as equimolar d-MPH doses) resulted in virtually identical rates of absorption when co-administered with an ethanol drink,22 while in the absence of ethanol the absorption rate of d-MPH was significantly lower for dl-MPH when compared to d-MPH tablets.44 Guidance from the Food and Drug Administration explicitly addresses concern over the potential influence of concomitant ethanol on the gastric formulation dissolution rate,66 in the context of dose dumping.67 Accordingly, the present study avoided any such potential confounds to the interpretation of results in view of ethanol being administered 4 hours after MR-MPH dosing. However, in more broadly considering the potential for extra-hepatic ethanol-MPH interactions, it is recognized that ethanol can increase portal venous blood flow and influence the rate of drug absorption.68
Cocaine exhibits neuropharmacological26 and structural69 similarities to d-MPH. Both drugs contain a metabolically labile methyl ester whose CES1-mediated hydrolysis is inhibited by ethanol.34 In controlled human studies, a one g/kg oral dose of ethanol increased intranasal cocaine exposure by over 50% in the course of CES1 transesterifing cocaine with ethanol to yield cocaethylene70 in a biotransformation pathway analogous to that of CES1 transesterifying l-MPH with ethanol to yield l-ethylphenidate.34
Ethanol dosed at the 4 hour trough time following racemic MR-dl-MPH significantly potentiated VAS subscales of “Stimulated?”, “High?” and “Any effect?” over the cumulative AUC4.25–8.25h period which followed concomitant drug exposure. The corresponding AUC4.25–8.25h responses using the pure isomer MR-d-MPH product reached significance only for the subscale of “Stimulated?” (Table 3). In addition, there was a tendency toward a delay in the time to maximal subjective effect potentiation for the pure isomer MR-d-MPH which could relate to first-pass l-MPH-ethanol metabolic factors (Figure 2).
FIGURE 2.
Visual analog subscale positive subjective drug effects of “Stimulated; “High”; “Good”; and “Any effect” (0 – 12.5 hours): Racemic MR-dl-MPH (40 mg) was administered alone (□) or followed by ethanol (0.6 g/kg) 4–4.25 hours later (▪). Enantiopure MR-d-MPH (20 mg) was administered alone (Δ) or followed by ethanol (0.6 g/kg) 4–4.25 hours later (■). MPH administration was at T = 0 hours. Effect ratings were from 0 = “Not at all” to 100 = “Extremely”; Values represent the arithmetic mean (+/− SD); *P < 0.05; **P < 0.01; aP = 0.071; bP = 0.078.
Several limitations of this study regarding the interpretation of subjective responses to the MPH-ethanol combination compared to MPH alone are conspicuous. (1) No ethanol alone treatment group was included. (2) Accordingly, the study design cannot parse out the ethanol effect from the ethanol-MPH interaction. The study participants were carefully screened (Michigan scale) for only light ethanol use. Literature studies of ethanol alone, at doses of 0.4 g/kg or 0.8 g/kg, determined that only heavy drinkers felt stimulated, or liked the effects of ethanol; and then only at 0.5 and 1 h following the drink. Light drinkers reported not “liking” ethanol using the lower 0.5 g/kg dose at 1 h, and otherwise their reported effects did not differ from placebo.71 (3) This notwithstanding, a biphasic stimulant-depressant ethanol dose-response relationship is certainly recognized.72,73 (4) Another limitation of this study was the lack of inclusion of a placebo group. Practical considerations factored into not increasing our 4-way crossover study design to a 6-way crossover, with the inclusion of ethanol alone and placebo treatment groups. A 6-way design introduces ethical and health concerns regarding the total amount of blood needed to be drawn, and the likely increase in dropout rates of participants, as has been our experience as we have increased the number of treatment groups from 145 to 322 to 439 disproportionately leads to a higher number of subjects discontinuing the study before completing all treatment groups. (5) Additional limitations include the study being performed open-labelled, (6) as well as the problematic blinding of the alcohol-orange juice treatment, versus the orange juice without ethanol treatment groups. Unequivocally disguising a 0.6 g/kg ethanol drink represents a daunting challenge given the taste and olfactory ques of ethanol.
Placing this investigation in the context of the current clinical landscape, the number of prescriptions for MPH exceeds that of any other drug regardless of therapeutic class within the adolescent age group.74 Further, the high prevalence of underage ethanol consumption, and the frequent binge drinking pattern within this age group,75 adds to the importance of better understanding concomitant MPH-ethanol use and abuse.
Correlations between an increase in d-MPH absorption rate/exposure, and enhanced positive subjective effects, have previously served as surrogates for gauging abuse liability.22,39 Increasing the rate of d-MPH absorption,32,33,58,59 and consequently the rate of delivery to the central nervous system,19,60,61 strongly influences the stimulatory effects underlying the reward value of MPH-ethanol co-abuse.1–10 Accordingly, the PK and psychopharmacological findings reported here provide guidance for rational drug selection and individualization when treating ADHD with or without comorbid AUD.
Acknowledgments
This publication was supported by NIH grant R01AA016707 (K.S.P.) with additional support from the South Carolina Clinical & Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina, via use of the Clinical & Translational Research Center, NIH UL1 TR000062, UL1 RR029882, support from the Southeastern Pre-doctoral Training in Clinical Research Program TL1 RR029881 (OTR, KSP), as well as from NIH grant R01 DA022475–01A1 (J.S.M.). The authors appreciate the contributions of Michael Calos in editing this manuscript.
Funding support for this work was solely received from the National Institutes of Health (K.S.P.). No other funding could be construed as a conflict of interest. K.S.P. has been a consultant for Janssen, ALZA Corporation, Shire and Noven within the last 36 months. J.S.M., K.S.P., H-J. Z. received a provisional patent for isopropylphenidate (ritalinic acid isopropyl ester) as a novel psychotropic agent through the MUSC Foundation for Research Development, with a Notice of Abandonment Jan 2014. A.B.S. is a member of the Board of Directors for Cingulate Therapeutics and has been a consultant for Watson. R.J.M. is funded through NIH/NIDA and has research funding from Janssen. J.S.M. has been a consultant for Janssen.
Footnotes
AUTHOR DISLOSURE INFORMATION: No disclosures were declared for the remaining authors.
References
- 1.Levin FR, Kleber HD. Attention-deficit hyperactivity disorder and substance abuse: relationships and implications for treatment. Harv Rev Psychiatry. 1995;2:246–58. doi: 10.3109/10673229509017144. [DOI] [PubMed] [Google Scholar]
- 2.Barrett SP, Pihl RO. Oral methylphenidate-alcohol co-abuse. J Clin Psychopharmacol. 2002;22:33–34. doi: 10.1097/00004714-200212000-00020. [DOI] [PubMed] [Google Scholar]
- 3.Teter CJ, McCabe SE, Boyd CJ, et al. Illicit methylphenidate use in an undergraduate student sample: prevalence and risk factors. Pharmacotherapy. 2003;23:609–17. doi: 10.1592/phco.23.5.609.34187. [DOI] [PubMed] [Google Scholar]
- 4.Barrett SP, Darredeau C, Bordy LE, et al. Characteristics of methylphenidate misuse in a university student sample. Can J Psychiatry. 2005;50:457–61. doi: 10.1177/070674370505000805. [DOI] [PubMed] [Google Scholar]
- 5.Barrett SP, Darredeau C, Pihl RO. Patterns of simultaneous polysubstance use in drug using university students. Hum Psychopharmacol. 2006;21:255–263. doi: 10.1002/hup.766. [DOI] [PubMed] [Google Scholar]
- 6.McCabe SE, Cranford JA, Morales M. Simultaneous and concurrent polydrug use of alcohol and prescription drugs: Prevalence, correlates, and consequences. J Stud Alcohol. 2006;6:529–537. doi: 10.15288/jsa.2006.67.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darredeau C, Barrett SP, Jardin B, et al. Patterns and predictors of medication compliance, diversion, and misuse in adult prescribed methylphenidate users. Hum Psychopharmacol. 2007;22:529–536. doi: 10.1002/hup.883. [DOI] [PubMed] [Google Scholar]
- 8.Novak SP, Kroutil LA, Williams, et al. The nonmedical use of prescription ADHD medications: results from a national Internet panel. Subst Abuse Treat Prev Policy. 2007;2:32. doi: 10.1186/1747-597X-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilens TE. The nature of the relationship between attention-deficit/hyperactivity disorder and substance abuse. J Clin Psychiatry. 2007;(suppl 11):4–8. [PubMed] [Google Scholar]
- 10.Wilens TE, Adler LA, Adams J, et al. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. J Am Acad Child Adolesc Psychiatry. 2008;47:21–31. doi: 10.1097/chi.0b013e31815a56f1. [DOI] [PubMed] [Google Scholar]
- 11.Molina BS, Pelham WE, Gnagy EM, et al. Attention-deficit/hyperactivity disorder risk for heavy drinking and alcohol use disoder is age specific. Alcohol Clin Exp Res. 2007;31:643–654. doi: 10.1111/j.1530-0277.2007.00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kessler RC, Adler L, Barkley R. The prevalence and corrates of adult ADHD in the United States: Results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163:716–723. doi: 10.1176/appi.ajp.163.4.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards AC, Kendler KS. Twin studies of the relationship between adolescent attention-deficit/hyperactivity disorder and adult alcohol dependence. J Stud Alcohol Drugs. 2012;3:185–194. doi: 10.15288/jsad.2012.73.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schubiner H, Tzelepis A, Milberger S, et al. Prevalence of attention-deficit/hyperactivity disorder and conduct disorder among substance abusers. J Clin Psychiatry. 2000;61:244–251. doi: 10.4088/jcp.v61n0402. [DOI] [PubMed] [Google Scholar]
- 15.Ercan ES, Coskunol H, Varan A, et al. Childhood attention deficit/hyperactivity disorder and alcohol dependence: A 1-year follow-up. Alcohol Alcol. 2003;38:352–356. doi: 10.1093/alcalc/agg084. [DOI] [PubMed] [Google Scholar]
- 16.Wilens TE, Dodson W. A clinical perspective of attention-deficit/hyperactivity disorder into adulthood. J Clin Psychiatry. 2004;65:1301–1313. doi: 10.4088/jcp.v65n1003. [DOI] [PubMed] [Google Scholar]
- 17.Patrick KS, Straughn AB, Perkins JS, et al. Evolution of stimulants to treat ADHD: Transdermal methylphenidate. Human Psychopharmacol Clin Exper. 2009;24:1–17. doi: 10.1002/hup.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volkow ND, Swanson JM. Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry. 2003;160:1909–1918. doi: 10.1176/appi.ajp.160.11.1909. [DOI] [PubMed] [Google Scholar]
- 19.Spencer TJ, Biederman J, Ciccone PE, et al. PET study examining pharmacokinetics, detection and likeability, and dopamine transporter receptor occupancy of short- and long-acting oral methylphenidate. Am J Psychiatry. 2006;163:387–95. doi: 10.1176/appi.ajp.163.3.387. [DOI] [PubMed] [Google Scholar]
- 20.Patrick KS, Straughn AB, Minhinnett RR, et al. Influence of ethanol and gender on methylphenidate pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2007;81:346–353. doi: 10.1038/sj.clpt.6100082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szobot CM, Bukstein O. Attention deficit hyperactivity disorder and substance use disorders. Child Adolesc Psychiatric Clin North Am. 2008;1:309–323. doi: 10.1016/j.chc.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Patrick KS, Straughn AB, Reeves OT, III, et al. Comparative ethanol-induced stimulatory responses to dexmethylphenidate versus methylphenidate. J Clin Psychopharmacol. 2015;35:464–467. doi: 10.1097/JCP.0000000000000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patrick KS, Caldwell RW, Ferris RM, et al. Pharmacology of the enantiomers of threo-methylphenidate. J Pharmacol Exp Ther. 1987;241:152–158. [PubMed] [Google Scholar]
- 24.Srinivas NR, Hubbard JW, Quinn D, et al. Enantioselective pharmacokinetics and pharmacodynamics of dl-threo-methylphenidate in children with attention deficit hyperactivity disorder. Clin Pharmacol Ther. 1992;52:561–8. doi: 10.1038/clpt.1992.185. [DOI] [PubMed] [Google Scholar]
- 25.Ding Y-S, Fowler JS, Volkow ND, et al. Chiral drugs: comparison of the pharmacokinetics of [11C]d-threo and l-threo-methylphenidate in the human and baboon brain. Psychopharacology. 1997;131:71–78. doi: 10.1007/s002130050267. [DOI] [PubMed] [Google Scholar]
- 26.Williard RL, Middaugh LD, Zhu HJ, et al. Methylphenidate and its ethanol transesterification metabolite ethylphenidate: brain disposition, monoamine transporters and motor activity. Behav Pharmacol. 2007;18:39–51. doi: 10.1097/FBP.0b013e3280143226. [DOI] [PubMed] [Google Scholar]
- 27.Markowitz JS, Patrick KS. Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter? J Clin Psychopharmacol. 2008;28:54–61. doi: 10.1097/JCP.0b013e3181733560. [DOI] [PubMed] [Google Scholar]
- 28.Wigal S, Swanson JM, Feifel D, et al. A double-blind, placebo-controlled trial of dexmethylphenidate hydrochloride and d,l-threo-methylphenidate hydrochloride in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2004;43:1406–1414. doi: 10.1097/01.chi.0000138351.98604.92. [DOI] [PubMed] [Google Scholar]
- 29.Weiss M, Wasdell M, Patin J. A post hoc analysis of d-threo-methylphenidate hydrochloride (Focalin) versus d,l-threo-methylphenidate hydrochloride (Ritalin) J Am Acad Child Adolesc Psychiatry. 2004;43:1415–1421. doi: 10.1097/01.chi.0000138352.06229.b0. [DOI] [PubMed] [Google Scholar]
- 30.Robinson DM, Ketting GM. Dexmethylphenidate extended-release in attention deficit hyperativity disorder. Drugs. 2006;66:661–668. doi: 10.2165/00003495-200666050-00006. [DOI] [PubMed] [Google Scholar]
- 31.Simon N, Rolland B, Karila L. Methylphenidate in adults with attention deficit disorder and substance abuse disorders. Curr Pharm Design. 2015;21:3359–3366. doi: 10.2174/1381612821666150619093254. [DOI] [PubMed] [Google Scholar]
- 32.Kollins SH, Rush CR, Pazzaglia PJ, Ali JA. (1998) Comparison of acute behavioral effects of sustained-release and immediate-release methylphenidate. Exp Clin Psychopharmacol. 1998;6:367–374. doi: 10.1037//1064-1297.6.4.367. [DOI] [PubMed] [Google Scholar]
- 33.Parasrampuria DA, Schoedel KA, Sculler R, et al. Do formulation differences alter abuse liability of methylphenidate?: A placecebo-controlled, randomized, double-blind, crossover study in recreational drug users. J Clin Psychopharmacol. 2007;27:459–467. doi: 10.1097/jcp.0b013e3181515205. [DOI] [PubMed] [Google Scholar]
- 34.Markowitz JS, Straughn AB, Patrick KS. Advances in the pharmacotherapy of attention- deficit hyperactivity disorder: Focus on methylphenidate formulations. Pharmacotherapy. 2003;23:1281–1299. doi: 10.1592/phco.23.12.1281.32697. [DOI] [PubMed] [Google Scholar]
- 35.Patrick KS, Straughn AB, Jarvi EJ, et al. The absorption of sustained-release methylphenidate formulations compared to an immediate-release formulation. Biopharm Drug Dispos. 1989;10:165–171. doi: 10.1002/bdd.2510100206. [DOI] [PubMed] [Google Scholar]
- 36.Tuerck D, Wang Y, Maboudian M, et al. Similar bioavailability of dexmethylphenidate extended (bimodal) release, dexmethyl-phenidate immediate release and racemic methylphenidate extended (bimodal) release formulations in man. Int J Clin Pharmacol Ther. 2007;45:662–668. doi: 10.5414/cpp45662. [DOI] [PubMed] [Google Scholar]
- 37.Zhu HJ, Patrick KS, Yuan HJ, et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am J Hum Genet. 2008;82:1241–1248. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markowitz JS, Zhu HJ, Patrick KS. Isopropylphenidate: An ester homolog of methylphenidate with sustained and selective dopaminergic activity and reduced drug interaction liability. J Child Adolesc Psychopharmacol. 2013;23:648–654. doi: 10.1089/cap.2013.0074. [DOI] [PubMed] [Google Scholar]
- 39.Patrick KS, Straughn AB, Reeves OT, et al. Differential influences of ethanol on early exposure to racemic methylphenidate compared to dexmethylphenidate in humans. Drug Metab Dispos. 2013;41:197–205. doi: 10.1124/dmd.112.048595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srinivas NR, Hubbard JW, Quinn D, et al. Enantioselective pharmacokinetics and pharmacodynamics of dl-threo-methylphenidate in children with attention deficit hyperactivity disorder. Clin Pharmacol Ther. 1992;52:561–568. doi: 10.1038/clpt.1992.185. [DOI] [PubMed] [Google Scholar]
- 41.Srinivas NR, Hubbard JW, Korchinski ED, et al. Enantioselective pharmacokinetics of dl-threo-methylphenidate in humans. Pharm Res. 1993;10:14–21. doi: 10.1023/a:1018956526016. [DOI] [PubMed] [Google Scholar]
- 42.Aoyama T, Sasaki T, Kotaki J. Pharmacokinetics and pharmacodynamics of (R)-threo-methylphenidate enantiomer in patients with hypersomnia. Clin Pharmacol Ther. 1994;55:270–276. doi: 10.1038/clpt.1994.27. [DOI] [PubMed] [Google Scholar]
- 43.Modi NB, Lindemulder G, Gupta SK. Single-and multiple-dose pharmacokinetics of oral once-a-day osmotic controlled-release OROS™ (methylphenidate HCl) formulation. J Clin Pharmacol. 2000;40:379–388. doi: 10.1177/00912700022009080. [DOI] [PubMed] [Google Scholar]
- 44.Patrick KS, Straughn AB. Absorption differences between immediate-release dexmethylphenidate and dl-methylphenidate. Drug Met Dispos. 2016;44:418–421. doi: 10.1124/dmd.115.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markowitz JS, DeVane CL, Boulton DV, et al. Ethylphenidate formation in human subjects after the administration of a single dose of methylphenidate and ethanol. Drug Metab Disp. 2000;26:620–624. [PubMed] [Google Scholar]
- 46.Chen ML, Shah VP, Ganes D, et al. Challenges and opportunities in establishing scientific and regulatory standards for assuring therapeutic equivalence of modified-release products: workshop summary report. Eur J Pharm Sci. 2010;40:148–153. doi: 10.1016/j.ejps.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 47.Zirkelbach JF, Jackson AJ, Wang Y, et al. Use of partial AUC (PAUC) to evaluate bioequivalence – a case study with complex absorption: Methylphenidate. Pharm Res. 2013;30:191–202. doi: 10.1007/s11095-012-0862-x. [DOI] [PubMed] [Google Scholar]
- 48.Jackson A. Impact of release mechanism on the pharmacokinetic performance of PAUC metrics for three methylphenidate products with complex absorption. Pharm Res. 2014;31:173–181. doi: 10.1007/s11095-013-1150-0. [DOI] [PubMed] [Google Scholar]
- 49.Zhu HJ, Patrick KS, Markowitz JS. Enantiospecific determination of DL-methylphenidate and DL-ethylphenidate in plasma by liquid chromatography-tandem mass spectrometry: application to human ethanol interactions. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:783–788. doi: 10.1016/j.jchromb.2011.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowland M, Tozer TN. Clinical Pharmacokinetics: Concepts and Applications. Philadelphia: Lea & Febiger; 1989. [Google Scholar]
- 51.Winer B. Book Statistical Principles in Experimental Design. 1st. McGraw-Hill; New York: 1962. [Google Scholar]
- 52.Metzler CM. Sample sizes for bioequivalence studies. Statistics in medicine. 1991;10:961–970. doi: 10.1002/sim.4780100617. [DOI] [PubMed] [Google Scholar]
- 53.Zhu H-J, Xinwen G, Brinda BE, et al. Carboxylesterase 1 as a determinant of clopidogel metabolism and activation. J Pharmacol Exp Ther. 2013;344:665–672. doi: 10.1124/jpet.112.201640. [DOI] [PubMed] [Google Scholar]
- 54.Tang M, Mukundan M, Yang Jian, et al. Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel Is transesterificated in the presence of ethyl alcohol. J Pharmacol Exper Ther. 2006:1467–1476. doi: 10.1124/jpet.106.110577. [DOI] [PubMed] [Google Scholar]
- 55.Parker RB, Laizure SC. The effect of ethanol on oral cocaine pharmacokinetics reveals an unrecognized class of ethanol-mediated drug interactions. Drug Metab Dispos. 2010;28:317–22. doi: 10.1124/dmd.109.030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu Z-Y, Edginton AN, Laizure SC, Parker RB. Physiologically based pharmacokinetic modeling of impaired carboxylesterase-1 activity: effects on oseltamivir disposition. Clin Pharmacokinet. 2014;53:825–836. doi: 10.1007/s40262-014-0160-3. [DOI] [PubMed] [Google Scholar]
- 57.Parker RB, Hu Z-Y, Melbohm B, et al. Effect of alcohol on human carboxylesterase drug metabolism. Clin Pharmacokin. 2015;54:627–638. doi: 10.1007/s40262-014-0226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spencer TJ, Biederman J, Martin JM, et al. Importance of pharmacokinetic profile and timing of coadministration of short- and long-acting formulations of methylphenidate on patterns of subjective responses and abuse potential. Postgrad med. 2012;124:166–73. doi: 10.3810/pgm.2012.01.2529. [DOI] [PubMed] [Google Scholar]
- 59.Stoops WW, Glaser PE, Fillmore MT, et al. Reinforcing, subject-rated, performance and physiological effects of methylphenidate and d-amphetamine in stimulant abusing humans. J Psychopharmacol. 2004;18:534–43. doi: 10.1177/0269881104047281. [DOI] [PubMed] [Google Scholar]
- 60.Swanson JM, Volkow ND. Serum and brain concentrations of methylphenidate: implications for use and abuse. Neurosci Biobehav Rev. 2003;27:615–21. doi: 10.1016/j.neubiorev.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 61.Volkow ND, Swanson JM. Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry. 2003;160:1909–18. doi: 10.1176/appi.ajp.160.11.1909. [DOI] [PubMed] [Google Scholar]
- 62.Patrick KS, Gonzalez MA, Straughn, et al. New methylphenidateformulations for the treatment of attention-deficit/hyperactivity disorder. Expert Opin Drug Deliv. 2005;2:121–143. doi: 10.1517/17425247.2.1.121. [DOI] [PubMed] [Google Scholar]
- 63.Childress A, Sallee FR. The use of methylphenidate hydrochloride extended-release oral suspension for the treatment of ADHD. Exp Rev Neurother. 2013;13:979–988. doi: 10.1586/14737175.2013.833002. [DOI] [PubMed] [Google Scholar]
- 64.McConnell EL, Fadda HM, Basit AW. Gut instincts: Exploration in intestinal physiology and drug delivery. Inter J Pharm. 2008;364:213–226. doi: 10.1016/j.ijpharm.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Lennernas H. Ethanol – drug absorption interaction: Potential for a significant effect on the plasma pharmacokinetics of ethanol vulnerable formulations. Mol Pharmaceutics. 2009;6:1429–1440. doi: 10.1021/mp9000876. [DOI] [PubMed] [Google Scholar]
- 66.Anand O, Yu LX, Conner DP, et al. Dissolution testing for generic drugs: An FDA perspective. AAPS J. 2011;13:328–335. doi: 10.1208/s12248-011-9272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fribe TP, Asgaradeh F, Gray A, et al. Regulatory considerations for alcohol-induced dose dumping of oral modified-release formulations. Pharmaceut Tech. 2015;38:40–46. [Google Scholar]
- 68.Yoshihara H, Sato N, Sasaki Y, et al. Effect of alcohol ingestion on portal venous blood flow in healthy volunteers: Comparison between the subjects with and without ALDH I isozyme. Alcohol. 1985;2:463–468. doi: 10.1016/0741-8329(85)90116-8. [DOI] [PubMed] [Google Scholar]
- 69.Froimowitz M, Patrick K, Cody V. Conformational analysis of methylphenidate and its structural relationship to other dopamine reuptake blockers such as CFT. Pharm Res. 1995;12:1430–1434. doi: 10.1023/a:1016262815984. [DOI] [PubMed] [Google Scholar]
- 70.Farre M, de la Torre R, Gonzalez ML, et al. Cocaine and alcohol interactions in humans: neuroendocrine effects and cocaethylene metabolism. J Pharmacol Exp Ther. 1997;283:164–76. [PubMed] [Google Scholar]
- 71.King AC, de Wit H, McNamara PJ, et al. Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch Gen Psychiatry. 2011;68:389–99. doi: 10.1001/archgenpsychiatry.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morean ME, de Wit H, King AC, et al. The drug effects questionnaire: psychometric support across three drug types. Psychopharmacology. 2013;227:177–92. doi: 10.1007/s00213-012-2954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rueger SY, McNamara PJ, King AC. Expanding the utility of the Biphasic Alcohol Effects Scale (BAES) and initial psychometric support for the Brief-BAES (B-BAES) Alcohol Clinical Exper Res. 2009;33:916–24. doi: 10.1111/j.1530-0277.2009.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chai G, Governale L, McMahon AW, et al. Trends of outpatient prescription drug utilization in US children, 2002–2010. Pediatrics. 2012;130:23–31. doi: 10.1542/peds.2011-2879. [DOI] [PubMed] [Google Scholar]
- 75.Monti PM, Miranda R, et al. Adolescence: Booze, Brains, and Behavior. Alcoh Clin Exper Res. 2005;29:207–220. doi: 10.1097/01.alc.0000153551.11000.f3. [DOI] [PubMed] [Google Scholar]