

Abstract
Abnormal activity of various N-methyl-d-aspartate receptor (NMDAR) subtypes has been implicated in a wide variety of neurological disorders such as Alzheimer's disease, schizophrenia, and epilepsy. Imaging agents for PET and SPECT that target NMDARs in a subtype-selective fashion may enable better characterization of those disorders and enhance drug development. On the basis of a pyrazoline derivative that demonstrated neuroprotective effects in vivo, we synthesized a series of para-substituted analogues and measured their affinities to various NMDAR subtypes. Compounds 4a–c and 4e showed greater, nanomolar affinity for the GluN1/2A subtype versus GluN1/2B. Dicarbomethoxy (pro-drug) analogues of [124/125I]4d and [11C] 4e (i.e., [124/125I]11d and [11C]11e) were generated and tested for NMDAR binding specificity in ex vivo autoradiography and brain biodistribution studies. Although NMDAR-specific binding could be demonstrated for [125I]11d and [11C]11e through autoradiography and biodistribution studies, imaging of neither [124I]11d nor [11C]11e could demonstrate brain penetration sufficient for detection by PET.
Graphical abstract
Introduction
Glutamate-activated receptors belong to two families: ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs). Within the class of ionotropic glutamate receptors, N-methyl-d-aspartate receptors (NMDARs) have been widely investigated for their involvement in neurodegenerative processes.1 The majority of NMDA receptors are heterotetrameric assemblies of two glycine-binding GluN1 subunits and two glutamate binding GluN2 subunits, of which four types exist (GluN2A, -B, -C, -D).2 The various GluN2 subunits show differential temporal and spatial distribution in the central nervous system (CNS) and provide an opportunity to develop region-specific pharmacological regulation of NMDA receptor function using compounds selective for one individual GluN2 subunit.3
Unlike dopaminergic and serotonergic transmission, glutamatergic transmission has proven to be an elusive target for brain imaging, although several promising agents exist.4,5 Positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging agents for glutamate receptors (GluRs) could be powerful tools for the evaluation of excitatory neurotransmission, the study of the pathophysiology of related neurological disorders, and the quantification of GluR drug occupancy in vivo. The strategy for the development of PET and SPECT probes for NMDA receptors involves the structural modification of NMDA receptor antagonists, including channel blockers, e.g., [11C]-ketamine, [18F]fluoromemanthine, and N-(1-naphthyl)-N-(3-[123I]iodophenyl)-N-methylguanidine ([123I]CNS-1261, 1),6 glycine site antagonists, e.g., 3-[3-(4-[11C]methoxybenzyl)-phenyl]-4-hydroxy-7-chloroquinolin-2(1H)-one ([11C]L-703,717),6 and GluN2B negative modulators, e.g., compound 26 (Figure 1).
Figure 1.
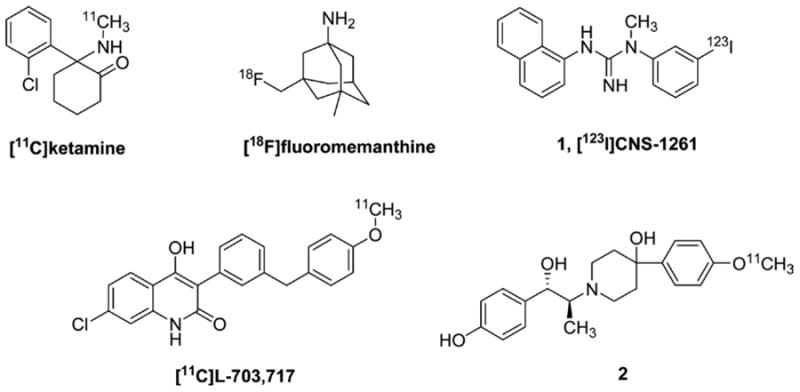
Structures of imaging agents for NMDA receptors.
For example, 1 is a channel-binding NMDA receptor radioligand for SPECT that has been used to study NMDA receptor function in vivo in humans.7 While of high affinity and having been tested in patients with schizophrenia, changes in 1 signal noted in affected individuals relative to controls, including those on clozapine, were small.8 The results obtained with 1 proved sufficiently encouraging to pursue other compounds of its class for PET.5 Nevertheless, most competitive antagonists of the glutamate binding site have shown low selectivity for NMDARs and poor blood–brain barrier (BBB) permeability, and at present there are no validated radioligands interacting with this region of the NMDARs.
In the past decade, we developed several new competitive NMDA receptor antagonists as potential neuroprotective agents.9 In particular, compound 3 (Figure 2) was highlighted for its promising pharmacological activity as a neuroprotective agent.10 In binding assays, compound (5S,αR)-3 showed nanomolar affinity for native receptors. Furthermore, preliminary evaluation of GluN2 subunit selectivity suggested that the racemic mixture (±)-3 displayed a preference for inhibition of NMDA receptors containing GluN2A or GluN2B subunits compared to receptors with GluN2C or GluN2D subunits.10
Figure 2.
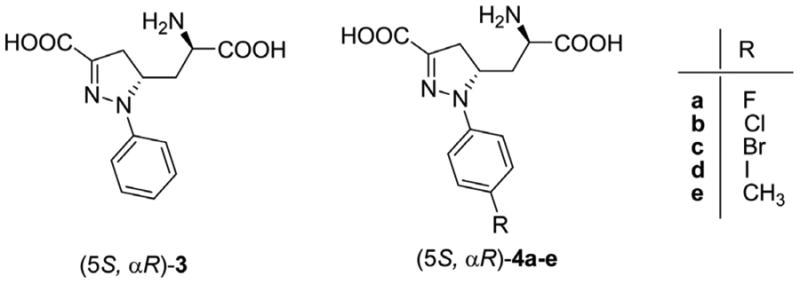
Structures of model and target compounds.
Despite being a charged molecule, (±)-3 displayed anticonvulsant activity after intraperitoneal (ip) administration to mice, indicating the capacity to cross the intact BBB at the doses employed.10 Therefore, (5S,αR)-3 appeared to us as a suitable tool to generate radiolabeled analogues for PET (or SPECT) imaging within the CNS.
Considering the molecular structure of (5S,αR)-3, we identified the para position of the aromatic ring as a suitable position for the insertion of a radiolabeled atom or group. We synthesized and pharmacologically tested several p-substituted-analogues, i.e., (5S,αR) 4a–e (Figure 2), to measure how para substitution would affect the binding properties in terms of affinity and subtype selectivity. The para substituents included either a halogen (F, Br, I, compounds 4a,c,d) or a methyl group (compound 4e), that could be subsequently replaced by their radioactive counterpart, namely, 18F, 76Br, 124/125I, or 11C. For a more thorough description of the structure–activity relationships (SARs), we included in this series also the p-Cl derivative (compound 4b). The clogD and polar surface area (PSA) values of compounds 4a-e were calculated as shown in Table 1. The negative clogD and high PSA values caution that, although the (5S,αR)-3 possesses CNS activity, 4a–e may not have sufficient BBB penetration when administered at the radiotracer level. Converting 4a–e to the prodrugs 11a–e, containing biologically cleavable methyl esters groups, significantly increased the clogD to positive values and also decreased PSA values, which may improve BBB penetration.
Table 1. Calculated CLogD and Polar Surface Areasa.
| compd | CLogD (pH 7.4) | PSA |
|---|---|---|
| 4a | −3.9 | 116.2 |
| 4b | −3.4 | 116.2 |
| 4c | −3.2 | 116.2 |
| 4d | −3.0 | 116.2 |
| 4e | −3.8 | 116.2 |
| 11a | 0.1 | 94.2 |
| 11b | 0.7 | 94.2 |
| 11c | 0.9 | 94.2 |
| 11d | 1.1 | 94.2 |
| 11e | 0.3 | 94.2 |
ACD/LogD Software, Advanced Chemistry Development, ACD/ Laboratories Release 9.0, product version 9.1.
Compounds 4d, 11d (prodrug of 4d), and 11e (prodrug of 4e) were then prepared in radioactive form and submitted for biological investigation.
Results and Discussion
Chemistry
Chloro-hydrazones 7a–e were prepared by reacting the p-substituted anilines 5a–e with methyl 2-chloro-3-oxobutanoate 6, in the presence of sodium nitrite, 6 N HCl, sodium acetate, and MeOH at 0 °C.11 Chloro-hydrazones 7a–e were treated with base to generate in situ the corresponding N-substituted-methoxycarbonylformonitrilimines that underwent 1,3-dipolar cycloaddition with suitably protected allylglycine 8 (Scheme 1). The cycloaddition reaction produced a mixture of stereoisomers 9 and 10 in an approximate 1:1 ratio, as estimated by 1H NMR spectroscopy. Chromatographic separation of the two stereoisomers was unsuccessful at this stage; therefore, the mixture was treated with a 30% solution of trifluoroacetic acid (TFA) in dichloromethane to remove the N-Boc protecting group. The couples of amines 11a–e and 12a–e were easily separated by flash chromatography. The desired stereoisomers 11a–e were then converted into the final amino acids 4a–e through a three-step protection/deprotection strategy including (a) N-Boc protection, (b) hydrolysis of the two ester functions, and (c) N-Boc deprotection, using standard procedures.
Scheme 1.
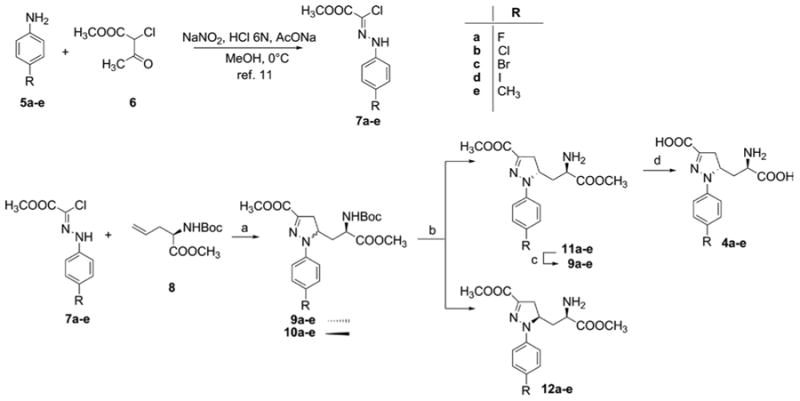
a(a) NaHCO3, EtOAc, Δ, 70–90% yield; (b) (i) 30% TFA, CH2Cl2, 78–87% yield, (ii) flash chromatography; (c) Boc2O, TEA, CH2Cl2, quantitative yield; (d) (i) 1N NaOH, MeOH, (ii) 30% TFA, CH2Cl2, 58–63% yield after two steps.
Pharmacology
Final amino acids 4a–e were evaluated using two-electrode voltage-clamp electrophysiology at recombinant NMDA receptors expressed in Xenopus oocytes (Supporting Information, Figure 1 and Table 2).
Table 2. IC50 Values for Inhibition of Current Responses Activated by 100 μM Glycine and l-Glutamate in Oocytes Expressing Recombinant Rat GluN1/2A, GluN1/2B, GluN1/2C, and GluN1/2D Receptorsa.
| compd | GluN1/2A | GluN1/2B | GluN1/2C | GluN1/2D | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| IC50 (nM) | est Ki (nM) | IC50 (nM) | est Ki (nM) | IC50 (nM) | est Ki (nM) | IC50 (nM) | est Ki (nM) | |
| 3 | 320 ± 10 | 87 ± 4 | 210 ± 40 | 80 ± 15 | 200 ± 10 | 49 ± 2 | 390 ± 80 | 120 ± 30 |
| 4a | 160 ± 10 | 43 ± 2 | 860 ± 10 | 330 ± 40 | 920 ± 20 | 230 ± 10 | 920 ± 20 | 290 ± 10 |
| 4b | 98 ± 4 | 27 ± 1 | 280 ± 20 | 110 ± 10 | 180 ± 10 | 44 ± 2 | 520 ± 20 | 160 ± 10 |
| 4c | 91 ± 4 | 25 ± 1 | 250 ± 20 | 94 ± 7 | 190 ± 10 | 47 ± 3 | 630 ± 40 | 200 ± 10 |
| 4d | 910 ± 80 | 250 ± 20 | 700 ± 40 | 270 ± 20 | 560 ± 20 | 140 ± 10 | 1.310 ± 90 | 110 ± 30 |
| 4e | 180 ± 10 | 48 ± 3 | 690 ± 50 | 250 ± 20 | 380 ± 20 | 96 ± 5 | 1.420 ± 60 | 440 ± 20 |
10 μM l-glutamate was used for GluN1/2A, 3 μM l-glutamate for GluN1/2B and GluN1/2C, and 1 μM l-glutamate for GluN1/2D receptors. Ki values were estimated using the Cheng–Prusoff relationship with the following l-glutamate EC50 values: GluN1/2A 3.8 μM, GluN1/2B 1.9 μM, GluN1/2C 1.0 μM, and GluN1/2D 0.45 μM.12,13 Hill slopes for the concentration–inhibition curves were 1.1 to 1.4, and 5–8 oocytes were recorded for each IC50 value using two-electrode voltage-clamp electrophysiology.
The estimated Ki values for the model compound (5S,αR)-3 at GluN1/2A and GluN1/2B receptor subtypes were 87 and 80 nM, respectively. Interestingly, we observed that the insertion of a substituent in the para position of compound (5S,αR)-3 gave rise to compounds endowed with an increased affinity and selectivity for the GluN1/2A subtype versus GluN1/2B. In detail, by increasing the size of the substituent, the affinity for GluN1/2A increased up to a certain size (H < F, CH3 < Cl, Br) and then it dropped with larger substituents such as I (compound 4d). Notably, the best selectivity GluN1/2A over GluN1/2B was obtained with the insertion of F (compound 4a) or CH3 (compound 4e).
The estimated Ki values for compounds 4a and 4e were 7.6-fold and 5.3-fold higher, respectively, at GluN1/2B compared to at GluN1/2A (namely, the binding affinities were higher at GluN1/2A). Insertion of Cl (compound 4b) resulted in an estimated Ki value that was 4.0-fold higher at GluN1/2B compared to at GluN1/2A. Among glutamate site competitive antagonists described to date, [(R)-[(S)-1-(4-bromophenyl)-ethylamino]-(2,3-dioxo-1,2,3,4 -tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077)14 is generally considered to have the highest selectivity for GluN1/2A over GluN1/2B. However, its binding affinity is only modestly 5.2-fold higher for GluN1/2A (15 nM) compared to GluN1/2B (78 nM).15 Thus, the 7.6-fold selectivity of compound 4a for GluN1/2A over GluN1/2B represents a useful starting point for the development of a PET or SPECT ligand to image GluN1/2A receptors in the CNS. The 18F-labeled analogue of 4a proved synthetically challenging, requiring us to focus our radiosynthetic efforts on [11C]4e and the corresponding radioiodinated analogues for SPECT (125I) and PET (124I), [125/124I] 4d.
Radiochemistry
The synthesis of the stannyl precursor 14 for the radioligand [125I]4d is shown in Scheme 2. Aryl bromide 13, prepared starting from 9c, was treated with hexabutylditin in the presence of tetrakis(triphenylphosphine)palladium in toluene to give 14. Radioiodination of 14 was carried out by treatment with [125I]NaI in the presence of N-chlorosuccinimide, followed by deprotection with TFA/H2O to give [125I] 4d.
Scheme 2.
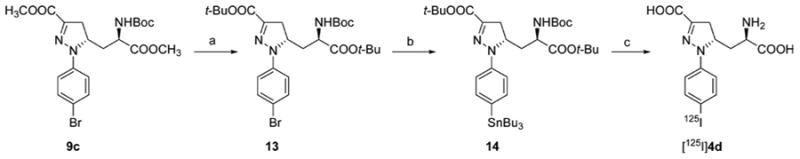
a(a) (i) 1N NaOH, MeOH, 95% yield, (ii) TBTA, dry THF/CH2Cl2, 42% yield; (b) Bu6Sn2, Pd(PPh3)4, toluene, 55% yield; (c) (i) AcOH, [125]NaI, NCS, MeOH, (ii) TFA/H2O, 54% radiochemical yield.
The synthesis of the stannyl precursor 15 for the radioligand [125I]11d, [124I]11d, and [11C]11e is shown in Scheme 3. Aryl bromide 9c was treated with hexabutylditin in the presence of tetrakis(triphenylphosphine)palladium in toluene to give 15. Radioiodination of 15 was carried out by treatment with [125I]NaI or [124I]NaI in the presence of N-chlorosuccinimide, followed by deprotection with TFA/H2O to give [125I]11d or [124I]11d. [11C]11e was prepared by Stille coupling reaction of 15 with [11C]methyl iodide, followed by deprotection with TFA/H2O.
Scheme 3.
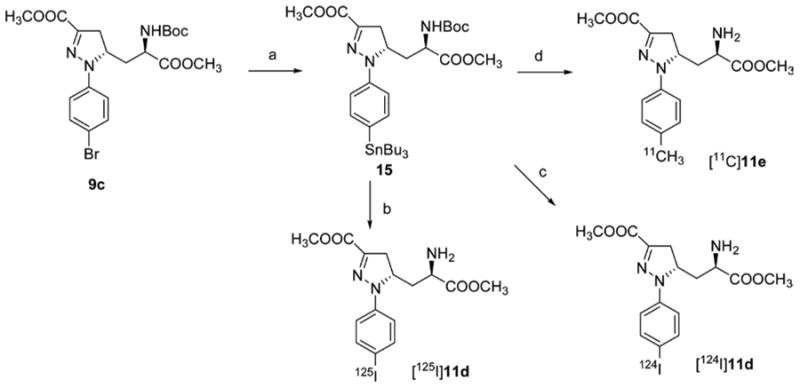
a(a) Bu6Sn2, Pd(PPh3)4, toluene, 33% yield; (b) (i) AcOH, [125]NaI, NCS, MeOH, radiochemical yield 58%, (ii) TFA/H2O; (c) (i) AcOH, [124]NaI, NCS, MeOH, (ii) TFA/H2O, radiochemical yield 49%; (d) (i) [11C]CH3I, Pd2(dba)3, P(o-tol)3, DMF, (ii) TFA/H2O, radiochemical yield 2–5%.
Autoradiography
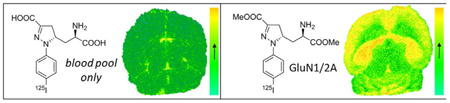
Ex vivo autoradiography studies were conducted in female athymic nude mice with radiolabeled compounds [125I]4d and prodrug [125I]11d to qualitatively assess both the ability to cross the BBB as well as brain subregion selectivity. Each mouse was injected with radiotracer, allowed to undergo the indicated uptake time (Figure 3), and sacrificed, and then sections were cut and exposed to X-ray film. Compound [125I]4d displayed no evidence of brain uptake (Figure 3A), while prodrug compound [125I]11d displayed brain region-selective uptake (Figure 3B), particularly in cortex, hippocampus, thalamus (Supporting Information, Figure 2), and cerebellum, with absence of uptake in caudate putamen, which is consistent with the distribution of GluN1/2A subtype receptor mRNA expression.16 Brain uptake of [125I]11d exhibited high-contrast and region-specific retention but was low in absolute value as estimated by exposure time.
Figure 3.
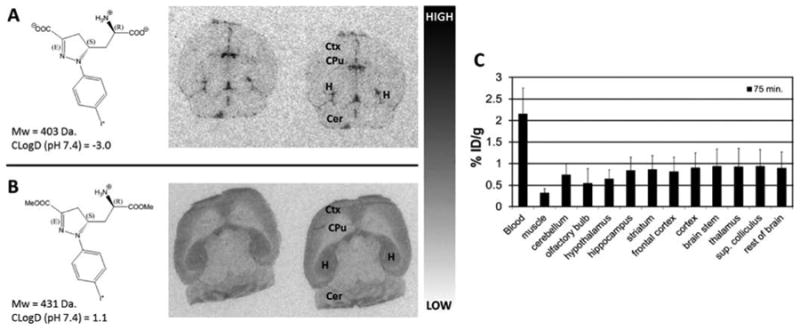
In vivo and ex vivo brain uptake and distribution of [125I]4d and [125I]11d prodrug in mice. (A) Ex vivo autoradiography using [125I]4d in serial brain sections following a 60 min conscious uptake. No evidence of radiotracer penetration into brain tissue is apparent. (B) Ex vivo autoradiography using [125I]11d prodrug in serial brain sections following a 40 min conscious uptake. Evidence of selective uptake in GluN2A subunit-rich regions is apparent including cortex (Ctx), hippocampus (H)m, and cerebellum (Cer). (C) Ex vivo biodistribution of [124I]11d in mice reflecting a 75 min uptake period confirms low overall brain uptake in the indicated tissue regions, indicating unsuitability for in vivo imaging.
Biodistribution
Ex vivo biodistribution in CD-1 mice was undertaken with prodrug compounds [124I]11d and [11C]11e to ascertain and quantify region-selective uptake. Compound [124I]11d was injected into mice and allowed to undergo a 75 min biological uptake period to maximize in vivo prodrug cleavage and receptor binding. Figure 3C shows a graph of those uptake values as a percentage of injected dose per gram of wet tissue (% ID/g) in selected regions. Uptake across regions was low and never exceeded 1% ID/g. In vivo PET imaging with this compound was also undertaken through 90 min and verified that brain uptake was below the threshold of detection for this modality (Supporting Information).
Dimethyl ester [11C]11e, for which GluN1/2A affinity of the free acid analogue 4e was found to be higher than that for 4d, was also assayed by ex vivo biodistribution. Figure 4 shows the graph of uptake values from 15 min through 60 min of uptake (Figure 4A) as well as autoblockade at 60 min (Figure 4B) to verify radiotracer specificity. Figure 4A demonstrates radiotracer wash-in at 15 min followed by reduction at 30 min and then steady accumulation through 60 min, suggesting in vivo cleavage of the methyl esters and subsequent receptor binding over time. Figure 4B shows the effect of coadministration of 4.5 mg/kg of unlabeled compound 11e with radiotracer at 60 min of uptake time as well as increasing uptake in radiotracer only mice from 60 to 75 min, continuing the trend seen in Figure 4A.
Figure 4.
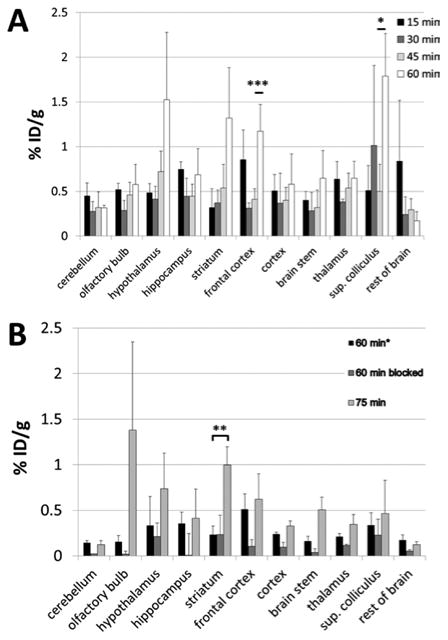
Ex vivo brain biodistribution of [11C]11e prodrug in mice. (A) Mice were injected with [11C]11e and sacrificed at the indicated uptake times. The pattern of uptake in indicated brain tissues shows increasing radiotracer uptake over time in all regions except cerebellum and the remaining brain tissue following dissection. Significant radiotracer uptake was observed in frontal cortex (P = 0.004) and superior colliculus (P = 0.04) between 45 and 60 min of uptake. (B) Autoblockade at 60 min after radiotracer injection compared with radiotracer alone at 60 and 75 min postinjection. Blockade was observed in cerebellum (85%), olfactory bulb (88%), hippocampus (98%), frontal cortex (80%), cortex (60%), brainstem (77%), thalamus (45%), and in the remaining tissue (71%). Radiotracer only uptake increased from 60 to 75 min in the same regions as in (A) where significance was only reached in striatum between 60 and 75 min of uptake (P = 0.009). Note: Specific radiotracer activity in (A) was 1400 Ci/mmol, while in (B) it was 11500 Ci/mmol at the time of injection, which may account for differences in absolute % ID/g values.
Conclusion
Here we report the development of radiolabeled ligands [125I] 4d, [124/125I]11d, and [11C]11e for in vivo targeting of NMDA receptors. [125I]4d was not able to cross BBB when administered intravenously as a radiotracer. Applying a prodrug approach, both [124/125I]11d and [11C]11e, containing cleavable methyl ester groups, did demonstrate BBB penetration following intravenous administration and exhibited differentially selective brain subregional binding. [124/125I]11d displayed an autoradiographic uptake profile consistent with the distribution of GluN2A subunits,16 while ex vivo biodistribution of [11C]11e suggested binding to both GluN1/2B and GluN1/2C, which accounts for both significant measured uptake in superior colliculus (GluN2C profile) and caudate putamen (GluN2B profile). Compounds 11d and 11e differ only in the radiolabeled atom present in the same para position on the aryl ring, where 11d contains a large iodine atom and 11e contains a smaller, less lipophilic methyl group. These differences may account for different murine in vivo GluN2 subunit binding selectivity, contrasting with the trends measured in Xenopus oocytes in Table 2. It is also possible that the murine brain contains disparately distributed methyl esterase activity and/or each analogue has a significantly different affinity for the esterase, efficient cleavage of which is necessary to convert prodrug analogues into the dicarboxylic acid species necessary to bind the targeted receptors (Supporting Information, Table 1).
Aside from the ex vivo evidence showing selective brain uptake for 11d and 11e, in vivo PET-CT imaging with [124I] 11d revealed no observable brain uptake (Supporting Information). That was not unexpected when measured uptake values from ex vivo biodistribution studies revealed % ID/g values consistently below 1% ([124I]11d, Figure 3C) or below 1.5% ID/g ([11C]11e, Figure 4A,B). Although we previously showed that 3 could demonstrate neuroprotective properties,10 further structure–activity relationships expanding upon this scaffold are necessary to identify compounds with sufficient affinity and suitable lipophilicity to serve as imaging agents for NMDARs in CNS. SAR studies will also be aimed at improving even further the specificity for the different subtypes of NMDA receptors. A potential utility for free-acids [124I]4d and [11C]4e would include imaging of NMDARs in peripheral neuroendocrine and neuroendocrine-like tumors, including melanomas, breast, small-cell lung cancer, and castrate-resistant prostate cancers.17 These free acids possess high affinity for their receptor subtypes (Table 2) and favorable clogD values (Table 1) for peripheral imaging.
Experimental Section
Solvents and chemicals purchased from commercial sources were of analytical grade or better and used without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker Ultrashield 400 MHz or on a Varian Mercury 300 (300 MHz) spectrometer. Chemical shifts (δ) are expressed in ppm, and coupling constants (J) are expressed in Hz. Rotary power determinations were carried out using a Jasco P-1010 spectropolarimeter, coupled with a Haake N3-B thermostat. TLC analyses were performed on commercial silica gel 60 F254 aluminum sheets; spots were further evidenced by spraying with a dilute alkaline potassium permanganate solution. Melting points were determined on a model B 540 Büchi apparatus and are uncorrected. MS analyses were performed on a Varian 320-MS triple quadrupole mass spectrometer with ESI source. Microanalyses (C, H, N) of new compounds were within ±0.4% of theoretical values. Chloro-hydrazones 7a–e have been synthesized following a literature procedure.11 HPLC purification of radiolabeled compounds was performed on Varian Prostar systems (Palo Alto, CA), equipped with a Varian ProStar 325 UV–vis variable wavelength detector and a Bioscan Flow-count in-line radioactivity detector (Washington, DC). The specific radioactivity was calculated as the ratio of the radioactivity eluting at the retention time of product during the preparative HPLC purification to the mass corresponding to the area under the curve of the UV absorption.
Pharmacological Studies
Two-Electrode Voltage-Clamp Recordings
Rat cDNAs for GluN1-1a (GenBank IDs U11418 and U08261), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) were provided by Drs. S. Heinemann (Salk Institute, La Jolla, CA), S. Nakanishi (Kyoto University, Kyoto, Japan), and P. Seeburg (University of Heidelberg, Heidelberg, Germany). The cDNA encoding rat GluN2B was modified without changing the amino acid sequence to remove a T7 RNA polymerase termination site located in the intracellular C-terminal domain as previously described.18 For expression of NMDA receptors, cRNA for rat GluN1 (GluN1-1a splice variant), GluN2A, GluN2B, GluN2C, and GluN2D were prepared and injected into Xenopus oocytes as previously described.19 The oocytes were surgically removed from mature female Xenopus laevis and provided by Xenopus1 (Dexter, MI). Recordings were performed 2–4 days after injection using two-electrode voltage-clamp electrophysiology in extracellular solution containing 115 mM NaCl, 2.5 mM KCl, 1.9 mM BaCl2, and 10 mM HEPES (pH 7.6). The membrane potential was clamped at −40 mV. During recordings, 100 μM glycine was included in all agonist and/or antagonist applications. Electrophysiological data were acquired and analyzed as previously described.19
Native NMDA Receptor Binding Assays
Affinities for native NMDA receptors in rat cortical synaptosomes were determined using 2 nM [3H]CGP 39653, as previously described.20
Chemistry
General Procedure for the Synthesis of Compound 4a–e
(a) Derivative 9a–e (1.20 mmol, 1.0 equiv) was dissolved in MeOH (3.6 mL) and treated with 1 N aqueous NaOH (3.6 mL, 3.0 equiv). The disappearance of the starting material was monitored by TLC (cyclohexane/EtOAc 8:2). After evaporation of MeOH, the aqueous layer was washed with Et2O (1 × 5 mL), made acidic with 2 N aqueous HCl and extracted with EtOAc (3 × 5 mL). The organic layer was dried over anhydrous Na2SO4 and, after evaporation of the solvent, the diacid derivative was obtained as a yellow solid. (b) The diacid derivative (1.18 mmol, 1.0 equiv) was treated with a 30% CH2Cl2 solution of trifluoroacetic acid (11.8 mmol, 10 equiv) at 0 °C. The solution was stirred at room temperature for 3 h, and the reaction was followed by TLC (CH2Cl2/MeOH 9:1 + 1% AcOH). The volatiles were removed under reduced pressure, and the residue was crystallized to yield the final amino acid 4a–e.
(S)-5-((R)-2-Amino-2-carboxyethyl)-1-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylic Acid (4a)
Yield: 60%; yellow solid; mp = dec T > 145 °C; Rf = 0.53 (BuOH/H2O/AcOH 4:2:1); [α]D20 = +220.00 (c = 0.25; H2O). 1H NMR (300 MHz, DMSO-d6): 1.75–1.85 (1H, m), 1.98–2.10 (1H, m), 2.98 (1H, dd, J = 3.8, 17.8), 3.20 (1H, dd, J = 12.2, 17.8), 3.62–3.75 (1H, m), 4.62–4.78 (1H, m), 7.08–7.25 (4H, m). 13C NMR (75 MHz, DMSO-d6): 32.69, 37.35, 50.78, 58.79, 116.46, 116.49 (d, J = 22.2), 139.35, 141.30, 157.67 (d, J = 236), 164.39, 170.89. [M + H]+ = 296.1. Anal. calcd for C13H14FN3O4: C, 52.88; H, 4.78; N, 14.23. Found: C, 53.02; H, 4.90; N, 14.11.
(S)-5-((R)-2-Amino-2-carboxyethyl)-1-(4-chlorophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylic Acid (4b)
Yield: 63%; yellow solid; mp = dec T > 175 °C; Rf = 0.58 (BuOH/H2O/AcOH 4:2:1); [α]D20 = +393.2 (c = 0.20; DMSO). 1H NMR (300 MHz, DMSO-d6): 1.75– 1.85 (1H, m), 1.95–2.10 (1H, m), 3.00 (1H, dd, J = 3.9, 18.3), 3.10 (1H, dd, J = 11.7, 18.3), 3.60–3.70 (1H, m), 4.60–4.78 (1H, m), 7.20 (2H, d, J = 7.8), 7.35 (2H, d, J = 7.8). 13C NMR (75 MHz, DMSO-d6): 32.70, 37.38, 50.87, 58.49, 116.36, 124.79, 129.69, 141.62, 142.35, 164.41, 170.70. [M + H]+ = 312.1. Anal. calcd for C13H14ClN3O4: C, 50.09; H, 4.53; N, 13.48. Found: C, 50.30; H, 4.62; N, 13.30.
(S)-5-((R)-2-Amino-2-carboxyethyl)-1-(4-bromophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylic Acid (4c)
Yield: 60%; yellow solid; mp = dec T > 167 °C; Rf = 0.43 (BuOH/H2O/AcOH 4:2:1); [α]D20 = +285.00 (c = 0.20; DMSO). 1H NMR (300 MHz, DMSO-d6): 1.70– 1.85 (1H, m), 1.92–2.02 (1H, m), 2.99 (1H, dd, J = 4.1, 18.1), 3.19 (1H, dd, J = 11.6, 18.1), 3.55–3.65 (1H, m), 4.60–4.75 (1H, m), 7.15 (2H, d, J = 9.1), 7.41 (2H, d, J = 9.1). 13C NMR (75 MHz, DMSO-d6): 32.74, 37.45, 50.94, 58.51, 112.65, 116.87, 132.55, 141.95, 142.26, 164.29, 170.57. [M + H]+ = 355.8. Anal. calcd for C13H14BrN3O4: C, 43.84; H, 3.96; N, 11.80. Found: C, 43.67; H, 3.84; N, 11.92.
(S)-5-((R)-2-Amino-2-carboxyethyl)-1-(4-iodophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylic Acid (4d)
Yield: 58%; yellow solid; mp = dec T > 183 °C; Rf = 0.55 (BuOH/H2O/AcOH 4:2:1); [α]D20 = +336.6 (c = 0.20; DMSO). 1H NMR (300 MHz, DMSO-d6): 1.70– 1.82 (1H, m), 1.92–2.06 (1H, m), 2.97 (1H, dd, J = 3.8, 18.5), 3.18 (1H, dd, J = 11.1, 18.5), 3.58–3.70 (1H, m), 4.60–4.72 (1H, m), 7.02 (2H, J = 8.8), 7.57 (2H, J = 8.8). 13C NMR (75 MHz, DMSO-d6): 32.66, 37.39, 50.77, 58.37, 83.74, 117.30, 138.29, 142.26, 142.39, 164.31, 170.59. [M + H]+ = 404.1. Anal. calcd for C13H14IN3O4: C, 38.73; H, 3.50; N, 10.42. Found: C, 38.98; H, 3.60; N, 10.30.
(S)-5-((R)-2-Amino-2-carboxyethyl)-1-(4-[125I]iodophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylic Acid ([125I]4d)
To a solution of 14 (0.5 mg, 0.64 μmol) in 0.1 mL of methanol was added 0.001 mL of acetic acid and 4.6 mCi [125I]NaI solution, followed by N-chlorosuccinimide (0.005 mg, 0.037 μmol) in methanol. The reaction mixture was kept at room temperature for 20 min and then isolated by radio-HPLC (Econosil C18 10 μ, 250 mm × 4.6 mm, H2O/CH3CN/ TFA = 70/30/0.1, 1 mL/min). The HPLC eluate at 26 min was collected and evaporated under vacuum. To it TFA/H2O (95:5, 0.2 mL) was added. The mixture was heated at 40 °C for 10 min and isolated by radio-HPLC (Econosil C18 10 μ, 250 mm × 4.6 mm, H2O/CH3CN/TFA = 78/22/0.1, 1 mL/min, retention time: 17 min) to give [125I]4d. The radiochemical yield was 54% and the specific activity was 1940 Ci/mmol.
(S)-5-((R)-2-Amino-2-carboxyethyl)-1-p-tolyl-4,5-dihydro-1H-pyrazole-3-carboxylic Acid (4e)
Yield: 62%; yellow solid; mp = dec T > 198 °C; Rf = 0.60 (BuOH/H2O/AcOH 4:2:1); [α]D20 = +335.00 (c = 0.10; H2O). 1H NMR (300 MHz, DMSO-d6): 1.65–1.85 (1H, m), 1.90–2.10 (1H, m), 2.20 (3H, s), 2.95 (1H, dd, J = 4.4, 17.9), 3.15 (1H, dd, J = 11.3, 17.9), 3.35–3.45 (1H, m), 4.50–4.65 (1H, m), 7.02–7.18 (4H, m). 13C NMR (75 MHz, DMSO-d6): 20.85, 33.18, 37.56, 51.53, 58.66, 114.89, 129.43, 130.22, 141.04, 142.78, 165.08, 170.67. [M + H]+ = 292.1. Anal. calcd for C14H17N3O4: C, 57.72; H, 5.88; N, 14.42. Found: C, 58.00; H, 6.01; N, 14.23.
General Procedure for the Cycloaddition Reaction
To a solution of (R)-methyl 2-(tert-butoxycarbonylamino)pent-4-enoate 8 (1.50 g, 6.54 mmol, 1.0 equiv) in EtOAc (25 mL), the appropriate chloro-hydrazone 7a–e (9.81 mmol, 1.5 equiv) and solid NaHCO3 (2.75 g, 32.7 mmol, 5 equiv) were added. The mixture was vigorously stirred for 24 h at 80 °C, and the progress of the reaction was followed by TLC (cyclohexane/EtOAc 8:2). Water (5 mL) was added, and the organic layer was separated and dried over anhydrous Na2SO4. The crude material obtained after evaporation of the solvent was purified by column chromatography on silica gel to give an inseparable mixture of the two diastereoisomers 9a–e and 10a–e (yield: 70–90%).
General Procedure for the Synthesis of Compound 9a–e
(a) The mixture of diastereoisomers 9a–e and 10a–e (4.57 mmol, 1.0 equiv) was treated with a 30% CH2Cl2 solution of trifluoroacetic acid (3.5 mL, 10 equiv) at 0 °C. The solution was stirred at room temperature for 3 h, and the reaction was followed by TLC (cyclohexane/EtOAc 8:2). A saturated solution of NaHCO3 (50 mL) and CH2Cl2 (35 mL) were then added, and the organic layer was separated, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude material was purified on silica gel (cyclohexane/EtOAc 3:7) to give the amines 11a–e and 12a–e (yield: 78–87%). (b) To a stirred solution of the amine 11a–e (1.50 mmol, 1.0 equiv) in CH2Cl2 (10 mL) cooled at 0 °C, triethylamine (2.25 mmol, 1.5 equiv) and a solution of di-tert-butyl dicarbonate (1.95 mmol, 1.3 equiv) in CH2Cl2 (3 mL) were added and the reaction mixture was stirred at room temperature overnight. The reaction was monitored by TLC (EtOAc). The solvent was evaporated under reduced pressure, and the crude material was purified by column chromatography on silica gel (cyclohexane/EtOAc 8:2) to give compound 9a–e in quantitative yield.
(S)-Methyl 5-((R)-2-(tert-Butoxycarbonylamino)-3-methoxy-3-oxopropyl)-1-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (9a)
Yield: 87%; yellow oil; Rf = 0.32 (cyclohexane/EtOAc 7:3); [α]D20 = +242.56 (c = 1.00; CHCl3). 1H NMR (300 MHz, CDCl3): 1.45 (9H, s), 1.82–1.95 (1H, m), 1.95–2.05 (1H, m), 3.10 (1H, dd, J = 4.7, 17.8), 3.40 (1H, dd, J = 11.3, 17.8), 3.75 (3H, s), 3.90 (3H, s), 4.30–4.42 (1H, m), 4.62 (1H, dddd, J = 1.9, 4.7, 11.3, 11.3), 5.20 (1H, bd, J = 8.4), 6.98–7.06 (2H, m), 7.07–7.15 (2H, m). 13C NMR (75 MHz, CDCl3): 28.38, 35.09, 37.34, 50.50, 52.32, 52.86, 59.08, 80.73, 116.14 (d, J = 28.8), 116.24, 138.15, 138.68, 156.65, 157.85 (d, J = 300), 163.31, 172.35. [M + H]+ = 424.2. Anal. calcd for C20H26FN3O6: C, 56.73; H, 6.19; N, 9.92. Found: C, 56.98; H, 6.31; N, 9.78.
(S)-Methyl 5-((R)-2-(tert-Butoxycarbonylamino)-3-methoxy-3-oxopropyl)-1-(4-chlorophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (9b)
Yellow oil; Rf = 0.15 (cyclohexane/EtOAc 8:2); [α]D20 = +230.37 (c = 1.00; CHCl3). 1H NMR (300 MHz, CDCl3): 1.48 (9H, s), 1.80–1.95 (1H, m), 1.95–2.08 (1H, m), 3.10 (1H, dd, J = 4.7, 17.8), 3.40 (1H, dd, J = 11.3, 17.8), 3.75 (3H, s), 3.87 (3H, s), 4.30– 4.42 (1H, m), 4.62 (1H, dddd, J = 2.8, 4.7, 11.3, 11.3), 5.22 (1H, bd, J = 7.5), 7.02–7.12 (2H, m), 7.20–7.28 (2H, m). 13C NMR (75 MHz, CDCl3): 28.50, 35.00, 37.48, 50.58, 52.49, 52.98, 58.76, 80.79, 116.07, 126.64, 129.47, 139.40, 140.49, 156.04, 163.31, 172.47. [M + H]+ = 440.2. Anal. calcd for C20H26ClN3O6: C, 54.61; H, 5.96; N, 9.55. Found: C, 54.87; H, 6.12; N, 9.38.
(S)-Methyl 1-(4-Bromophenyl)-5-((R)-2-(tert-butoxycarbonylamino)-3-methoxy-3-oxopropyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (9c)
Yellow foam; crystallyzed from n-hexane; mp = 70–72 °C; Rf = 0.25 (cyclohexane/EtOAc 8:2); [α]D20 = +268.31 (c = 1.0; CHCl3). 1H NMR (300 MHz, CDCl3): 1.48 (9H, s), 1.80–1.92 (1H, m), 2.02 (1H, ddd, J = 2.4, 10.6, 13.5), 3.09 (1H, dd, J = 4.7, 17.9), 3.38 (1H, dd, J = 11.7, 17.9), 3.73 (3H, s), 3.87 (3H, s), 4.30–4.42 (1H, m), 4.61 (1H, dddd, J = 2.4, 4.7, 11.7, 11.7), 5.26 (1H, bd, J = 8.5), 6.98–7.04 (2H, m), 7.34–7.41 (2H, m). 13C NMR (75 MHz, CDCl3): 28.53, 35.30, 37.58, 50.56, 52.53, 53.03, 58.69, 80.93, 114.12, 116.43, 132.42, 139.56, 140.91, 155.96, 163.28, 172.33. [M + H]+ = 484.1. Anal. calcd for C20H26BrN3O6: C, 49.60; H, 5.41; N, 8.68. Found: C, 49.83; H, 5.58; N, 8.52.
(S)-Methyl 5-((R)-2-(tert-Butoxycarbonylamino)-3-methoxy-3-oxopropyl)-1-(4-iodophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (9d)
Yellow oil; Rf = 0.24 (cyclohexane/EtOAc 8:2); [α]D20 = +262.92 (c = 0.25; CHCl3). 1H NMR (300 MHz, CDCl3): 1.48 (9H, s), 1.87 (1H, ddd, J = 3.5, 10.6, 11.1), 2.02 (1H, ddd, J = 2.4, 10.6, 13.5), 3.09 (1H, dd, J = 4.7, 17.9), 3.38 (1H, dd, J = 11.1, 17.9), 3.73 (3H, s), 3.87 (3H, s), 4.30–4.42 (1H, m), 4.61 (1H, dddd, J = 2.4, 4.7, 11.1, 11.1), 5.20 (1H, bd, J = 8.5), 6.85–6.95 (2H, m), 7.50–7.60 (2H, m). 13C NMR (75 MHz, CDCl3): 28.50, 35.25, 37.57, 50.67, 52.50, 52.99, 58.56, 80.90, 83.97, 116.89, 138.28, 139.69, 141.57, 155.97, 163.24, 172.32. [M + H]+ = 532.2. Anal. calcd for C20H26IN3O6: C, 45.21; H, 4.93; N, 7.91. Found: C, 45.01; H, 4.9381; N, 8.01.
(S)-Methyl 5-((R)-2-(tert-Butoxycarbonylamino)-3-methoxy-3-oxopropyl)-1-p-tolyl-4,5-dihydro-1H-pyrazole-3-carboxylate (9e)
Yellow foam; Rf = 0.42 (cyclohexane/EtOAc 7:3); [α]D20 = +326.90 (c = 1.00; CHCl3). 1H NMR (300 MHz, CDCl3): 1.46 (9H, s), 1.80–1.93 (1H, m), 2.05 (1H, ddd, J = 2.2, 11.2, 13.5), 2.28 (3H, s), 3.05 (1H, dd, J = 4.9, 17.9), 3.36 (1H, dd, J = 11.2, 17.9), 3.70 (3H, s), 3.86 (3H, s), 4.30–4.42 (1H, m), 4.62 (1H, dddd, J = 2.2, 4.7, 11.2, 11.2), 5.20 (1H, bd, J = 8.5), 7.00–7.12 (4H, m). 13C NMR (75 MHz, CDCl3): 20.80, 28.50, 35.11, 37.19, 50.63, 52.30, 52.85, 58.92, 80.69, 115.19, 130.04, 131.36, 137.99, 139.52, 155.44, 163.55, 172.54. [M + H]+ = 520.2. Anal. calcd for C21H29N3O6: C, 60.13; H, 6.97; N, 10.02. Found: C, 59.89; H, 6.77; N, 10.20.
Methyl (S)-5-((R)-2-Amino-3-methoxy-3-oxopropyl)-1-(4-[124I]-iodophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate ([124I]11d)
To a solution of 15 (0.01 mg, 0.014 μmol) in 0.1 mL of methanol was added 0.001 mL of acetic acid and 4.6 mCi [124I]NaI solution, followed by N-chlorosuccinimide (0.02 mg, 0.15 μmol) in methanol. The reaction mixture was kept at room temperature for 20 min and then dried under nitrogen. To it TFA/H2O (95:5, 0.2 mL) was added. After 20 min at 45 °C, the mixture was isolated by radio-HPLC (Phenomenex C18, 10 μ, 250 mm × 4.6 mm, H2O/CH3CN/TFA = 70/30/0.1, 1 mL/min, retention time 16 min) to give [124I]11d. The radiochemical yield was 49% and the specific activity was 13000 Ci/ mmol.
Methyl (S)-5-((R)-2-Amino-3-methoxy-3-oxopropyl)-1-(4-[125I]-iodophenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate ([125I]11d)
To a solution of 15 (0.5 mg, 0.72 μmol) in 0.1 mL of methanol was added 0.002 mL of acetic acid and 1 mCi [125I]NaI solution, followed by N-chlorosuccinimide (0.02 mg, 0.15 μmol) in methanol. The reaction mixture was kept at room temperature for 20 min and then dried under vacuum. To it TFA/H2O (95:5, 0.2 mL) was added. The mixture was kept at room temperature for 20 min and isolated by radio-HPLC (Econosil C18 10 μ, 250 mm × 4.6 mm, H2O/CH3CN/ TFA = 70/30/0.1, 1 mL/min, retention time 16 min) to give [125I] 11d. The radiochemical yield was 58% and the specific activity was 1000 Ci/mmol.
Methyl (S)-5-((R)-2-((tert-Butoxycarbonyl)amino)-3-methoxy-3-oxopropyl)-1-(4-[11C]methylphenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate ([11C]11e)
A reaction vial containing a solution of Pd2(dba)3 (1 mg) and tri(o-tolyl)phosphine (1.3 mg) in 0.1 mL of DMF was purged with nitrogen for 2 min. To it compound 15 (1 mg in 0.1 mL DMF) was added. [11C]MeI (prepared with GE 11C-methyl iodide radiosynthesis module and GE PETtrace cyclotron at Johns Hopkins PET center) was swept by argon flow into the reaction vial. After the radioactivity reached the plateau, the vial was heated at 90 °C for 7 min, 0.25 mL 95% TFA was added, and mixture was heated at 90 °C for another 3 min. The reaction was diluted with 0.3 mL of water and filtered through a 13 mm Teflon filter (0.45 μm) and purified by HPLC (Phenomenex Luna C18, 10 μ, 250 mm × 10 mm. H2O/ CH3CN/TFA = 70/30/0.1, 8 mL/min, retention time 9–10 min) to give [11C]11e. The radiochemical yield was 8–20 mCi starting from 400 to 500 mCi [11C]MeI. Radiochemical purity was >99% and specific radioactivity was 6000–23000 Ci/mmol.
(S)-tert-Butyl 1-(4-Bromophenyl)-5-((R)-3-tert-butoxy-2-(tert-butoxycarbonylamino)-3-oxopropyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (13)
(a) Derivative 9c (290 mg, 0.60 mmol) was dissolved in MeOH (1.8 mL) and treated with 1 N aqueous NaOH (1.8 mL, 3.0 equiv). The disappearance of the starting material was monitored by TLC (cyclohexane/EtOAc 8:2). After evaporation of MeOH, the aqueous layer was washed with Et2O (1 × 5 mL), made acidic with 2 N aqueous HCl, and extracted with EtOAc (3 × 5 mL). The organic layer was dried over anhydrous Na2SO4 and, after evaporation of the solvent, the diacid derivative was obtained as a yellow solid (95% yield). (b) The diacid intermediate (260 mg, 0.57 mmol) was dissolved in a mixture of dry THF (640 μL) and dry CH2Cl2 (2.40 mL), and tert-butyl 2,2,2-trichloroacetimidate (5.7 mmol, 1.02 mL) was added. The mixture was stirred overnight at room temperature. A 5% solution of NaHCO3 (50 mL) was added, and the organic layer was separated. The aqueous phase was extracted with EtOAc (3 × 10 mL). The organic layer was dried over anhydrous Na2SO4 and, after evaporation of the solvent, the crude was purified by column chromatography (cyclohexane/EtOAc 9:1). Compound 13 was further purified by crystallization from n-hexane. Yield: 42%; Rf = 0.59 (cyclohexane/EtOAc 8:2). 1H NMR (300 MHz, CDCl3): 1.42 (9H, s), 1.48 (9H, s), 1.58 (9H, s), 1.78–2.05 (2H, m), 3.02 (1H, dd, J = 4.8, 17.8), 3.38 (1H, dd, J = 11.9, 17.8), 4.12–4.25 (1H, m), 4.52– 4.65 (1H, m), 5.25 (1H, bd, J = 8.5), 6.98–7.05 (2H, m), 7.32–7.42 (2H, m). Anal. calcd for C26H38BrN3O6: C, 54.93; H, 6.74; N, 7.39. Found: C, 55.16; H, 6.91; N, 7.27.
tert-Butyl (S)-5-((R)-3-(tert-Butoxy)-2-((tert-butoxycarbonyl)-amino)-3-oxopropyl)-1-(4-(tributylstannyl)phenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (14)
Compound 13 (0.028 g, 0.049 mmol) and tetrakis(triphenylphosphine)palladium (0.010 g, 0.009 mmol) were placed in an oven-dried, resealable Schlenk tube. Toluene (1 mL) was added via syringe, followed by hexabutylditin (0.1 mL, 0.2 mmol). The tube was purged with nitrogen for 2 min and then sealed. Its contents were stirred for 4 h at 100 °C, then cooled to room temperature and purified by flash chromatography (silica gel, EtOAc:hexanes = 1:4) to afford 14 (0.021 g, 55%). 1H NMR (400 MHz, CDCl3) δ 7.34 (2H, d, J = 8.0 Hz), 7.11 (2H, d, J = 8.0 Hz), 5.30 (1H, m), 4.59 (1H, m), 4.21 (1H, m), 3.31 (1H, m), 3.01 (1H, m), 2.01 (1H, m), 1.85 (1H, m), 1.43–1.54 (33H, m), 1.23–1.29 (6H, m), 0.97–1.02 (6H, m), 0.84–0.88 (9H, m).
Methyl (S)-5-((R)-2-((tert-Butoxycarbonyl)amino)-3-methoxy-3-oxopropyl)-1-(4-(tributylstannyl)phenyl)-4,5-dihydro-1H-pyrazole-3-carboxylate (15)
Compound 9c (0.050 g, 0.103 mmol) and tetrakis(triphenylphosphine)palladium (0.010 g, 0.009 mmol) were placed in an oven-dried, resealable Schlenk tube. Toluene (2 mL) was added via syringe, followed by hexabutylditin (0.3 mL, 0.6 mmol). The tube was purged with nitrogen for 2 min and then sealed. Its contents were stirred overnight at 95 °C, then cooled to room temperature and purified by flash chromatography (silica gel, EtOAc:hexanes = 1:3) to afford 15 (0.024 g, 33%). 1H NMR (400 MHz, CDCl3) δ 7.34 (2H, d, J = 8.0 Hz), 7.10 (2H, d, J = 8.0 Hz), 5.20 (1H, m), 4.64 (1H, m), 4.37 (1H, m), 3.85 (3H, s), 3.71 (3H, s), 3.36 (1H, m), 3.07 (1H, m), 2.06 (1H, m), 1.85 (1H, m), 1.44–1.58 (15H, m), 1.29–1.39 (6H, m), 0.98–1.02 (6H, m), 0.84–0.88 (9H, m).
Animals
All animal studies were performed in compliance with a Johns Hopkins Animal Care and Use Committee approved protocol. Male CD-1 mice (7 weeks of age) were purchased from Charles River Laboratories (Frederick, MD) and were housed for several weeks in a specific pathogen free facility under a 12/12 light/dark cycle with free access to food and water ad libitum. Female athymic nude mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed under the same conditions. Rodent chow is Teklad 8604 (Harlan Laboratories, Frederick, MD).
Autoradiography
Athymic nude mice (n = 1 per compound) were injected with either 47 MBq (1.27 mCi, 1940 Ci/mmol) of [125I] 4d for a 60 min conscious uptake or 9.1 MBq (246 μCi, 1000 Ci/ mmol) of [125I]11d for a 40 min conscious uptake. No anesthetics were used. Following the indicated uptake times, the mice were sacrificed by cervical dislocation and their brains were rapidly harvested and frozen over dry ice. The brains were then sectioned using a cryotome (HM Microm 550, Microm Intl. GmbH, Waldorf, Germany) to 20 μm thickness and adhered to charged glass slides (VWR, Radnor, PA) before exposing them to X-ray film (Kodak Biomax XR, Sigma, St. Louis, MO). Brain sections containing compound [125I]4d were exposed for 18 h, while sections containing compound [125I]11d were exposed for 24 h. Films were developed using a Kodak X-O-Mat film processor (Carestream, Rochester, NY). Exposures were digitized and displayed using an MCID Core system (MCID, Nottingham, UK) using the manufacturer's software.
Rodent Biodistribution
[124I]11d: Four male CD-1 mice were each injected intravenously with 1.85 MBq (50 μCi, 1000 Ci/mmol) of radiotracer in 100 μL of PBS. The mice underwent a 75 min anesthetic-free uptake period prior to sacrifice by cervical dislocation. Selected tissues (blood, muscle, cerebellum, olfactory bulb, hypothalamus, hippocampus, caudate putamen, frontal cortex, cortex, brain stem, thalamus, superior colliculus, and rest of brain) were rapidly dissected, weighed, and counted on a gamma counter (CS Compugamma 1282, LKB Wallac, Waverly, Vic) alongside diluted standard doses to obtain a percent injected dose per gram of wet tissue (% ID/g). Tissue uptake per region was averaged and graphed with standard deviation as indicated. [11C]11e: kinetic biodistribution was carried out at 15, 30, 45, and 60 min postinjection (n = 4 mice per time point) following the IV injection of 3.7 MBq (100 μCi, 1400 Ci/ mmol) of radiotracer in 100 μL of PBS. Similarly, for the autoblockade study, 3.7 MBq (11500 Ci/mmol) of radiotracer was injected alone or in combination with 4.5 mg/kg of unlabeled 11e (n = 4/group) in 100 μL of PBS. Time points for the blockade study were 60 min (tracer only and blocked) and 75 min (tracer only). Tissues were harvested and procedures were identical to those listed for [124I]11d.
In Vivo PET-CT Imaging
Two female athymic nude mice underwent dynamic positron emission tomography (PET) imaging. The mice were maintained under anesthesia with 2.5% isoflurane in oxygen (2L/min) and positioned side-by-side in a small animal SuperArgus PET/CT system (Sedecal, Madrid, Spain). One mouse was administered 8.14 MBq (220 μCi) of ([124I]11d) intravenously as a bolus. The other mouse received the same dose in addition to 150 mg/kg of ketamine. Dynamic PET imaging started simultaneously with dose administration and proceeded for 2 h, with the imaging field of view centered over the brain. A CT scan utilizing 360 projections at 60 keV was performed for anatomic coregistration purposes. Dynamic PET data were reconstructed using the manufacturer-provided 2D OSEM algorithm,21 and CT data were reconstructed with a manufacturer-provided standard filtered back-projection method. The PET reconstructed images were first coregistered with the CT using anatomic landmarks such as anterior eye socket to Harderian gland PET signal and displayed using AMIDE (http://amide.sourceforge.net/). Slices and 3D reconstructions were evaluated for brain uptake of radiotracer.
Supplementary Material
Acknowledgments
The technical assistance of Birgitte Nielsen is gratefully acknowledged. This work was supported by National Institutes of Health (P20GM103546 and R01NS097536 to K.B.H., R01NS065371 to S.F.T., and R01CA134675 to M.G.P.). A.P. acknowledges the financial support to the present research by the University of Milan (Piano di Sostegno alla Ricerca 2015/ 2017-Linea 2A).
Abbreviations Used
- NMDAR
N-methyl-d-aspartate receptor
- cLogD
calculated logarithm of distribution coefficient
- EtOAc
ethyl acetate
- AcOH
acetic acid
- TEA
trimethylamine
- Boc2O
di-tert-butyl dicarbonate
- TBTA
tris(benzyltriazolylmethyl)amine
- % ID/g
percentage injected dose per gram
- PET-CT imaging
positron emission tomography–computed tomography
- HEPES
4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid
- cRNA
complementary ribonucleic acid
- MBq
megabecquerel
Footnotes
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes: The authors declare the following competing financial interest(s): Stephen F. Traynelis is a co-founder of NeurOp Inc, is the PI on a research grant to Emory from Janssen, and is a paid consultant for NeurOp, Janssen, and Pfizer.
ORCID: Paola Conti: 0000-0003-2140-0567
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b01344.
Reporting the binding affinity for native NMDARs of compunds 4e and 11e; concentration–response data for 4a, 4d, 4e at NMDA receptor subtypes; ex vivo autoradiography using [125I]11d prodrug in brain sections (PDF)
Molecular formula strings (CSV)
References
- 1.Zhou Q, Sheng M. NMDA receptors in nervous system diseases. Neuropharmacology. 2013;74:69–75. doi: 10.1016/j.neuropharm.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 2.Buller AL, Larson HC, Schneider BE, Beaton JA, Morrisett RA, Monaghan DT. The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J Neurosci. 1994;14:5471–5484. doi: 10.1523/JNEUROSCI.14-09-05471.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogden KK, Traynelis SF. New advances in NMDA receptor pharmacology. Trends Pharmacol Sci. 2011;32:726–733. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassenbrock A, Vasdev N, Liang SH. Selected PET radioligands for ion channel linked neuroreceptor imaging: focus on GABA, NMDA and nACh receptors. Curr Top Med Chem. 2016;16:1830–1842. doi: 10.2174/1568026616666160315142457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naumiec GR, Jenko KJ, Zoghbi SS, Innis RB, Cai L, Pike VW. N′-3-(Trifluoromethyl)phenyl Derivatives of N-aryl-N′-methylguanidines as prospective PET radioligands for the open channel of the N-methyl-d-aspartate (NMDA) receptor: synthesis andstructure-affinity relationships. J Med Chem. 2015;58:9722–9730. doi: 10.1021/acs.jmedchem.5b01510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchigami T, Nakayama M, Yoshida S. Development of PET and SPECT probes for glutamate receptors. Sci World J. 2015;2015:716514. doi: 10.1155/2015/716514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Owens AA, Tebbutt AL, McGregor AL, Kodama K, Magar SS, Perlman ME, Robins DJ, Durant GJ, McCulloch J. Synthesis and binding characteristics of N-(1-naphthyl)-N′-(3-[125I]-iodophenyl)-N′-methylguanidine ([125I]-CNS 1261): a potential SPECT agent for imaging NMDA receptor activation. Nucl Med Biol. 2000;27:557–564. doi: 10.1016/s0969-8051(00)00102-5. [DOI] [PubMed] [Google Scholar]
- 8.Bressan RA, Erlandsson K, Stone JM, Mulligan RS, Krystal JH, Ell PJ, Pilowsky LS. Impact of schizophrenia and chronic antipsychotic treatment on [123I]CNS-1261 binding to N-methyl-d-aspartate receptors in vivo. Biol Psychiatry. 2005;58:41–46. doi: 10.1016/j.biopsych.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 9.(a) Conti P, Pinto A, Tamborini L, Grazioso G, De Sarro G, Bräuner-Osborne H, Szabo G, Gábor Hársing L, De Micheli C. Synthesis of conformationally constrained glutamic acid homologues and investigation of their pharmacological profiles. ChemMedChem. 2007;2:1639–1647. doi: 10.1002/cmdc.200700118. [DOI] [PubMed] [Google Scholar]; (b) Conti P, De Amici M, Grazioso G, Roda G, Pinto A, Hansen KB, Nielsen B, Madsen U, Bräuner-Osborne H, Egebjerg J, Vestri V, Pellegrini-Giampietro DE, Sibille P, Acher FC, De Micheli C. Synthesis, binding affinity at glutamic acid receptors, neuroprotective effects, and molecular modeling investigation of novel dihydroisoxazole amino acids. J Med Chem. 2005;48:6315–6325. doi: 10.1021/jm0504499. [DOI] [PubMed] [Google Scholar]; (c) Tamborini L, Pinto A, Mastronardi F, Iannuzzi MC, Cullia G, Nielsen B, De Micheli C. Conti, P. 3-Carboxy-pyrazolinalanine as a new scaffold for developing potent and selective NMDA receptor antagonists. Eur J Med Chem. 2013;68:33–37. doi: 10.1016/j.ejmech.2013.07.010. [DOI] [PubMed] [Google Scholar]; (d) Conti P, De Amici M, Grazioso G, Roda G, Barberis Negra FF, Nielsen B, Stensbøl TB, Madsen U, Bräuner-Osborne H, Frydenvang K, De Sarro G, Toma L, De Micheli C. Design, synthesis, and pharmacological characterization of novel, potent NMDA receptor antagonists. J Med Chem. 2004;47:6740–6748. doi: 10.1021/jm049409f. [DOI] [PubMed] [Google Scholar]
- 10.Conti P, Pinto A, Tamborini L, Madsen U, Nielsen B, Bräuner-Osborne H, Hansen KB, Landucci E, Pellegrini Giampietro DE, De Sarro G, Donato Di Paola E, De Micheli C. Novel 3-carboxy- and 3-phosphono-pyrazoline amino acids acting as potent and selective NMDA antagonists: design, synthesis and pharmacological characterization. ChemMedChem. 2010;5:1465–1475. doi: 10.1002/cmdc.201000184. [DOI] [PubMed] [Google Scholar]
- 11.Pfefferkorn JA, Choi C, Larsen SD, Auerbach B, Hutchings R, Park W, Askew V, Dillon L, Hanselman JC, Lin Z, Lu GH, Robertson A, Sekerke C, Harris MS, Pavlovsky A, Bainbridge G, Caspers N, Kowala M, Tait BD. Substituted pyrazoles as hepatoselective HMG-CoA reductase inhibitors: discovery of (3R,5R)-7-[2-(4-fluoro-phenyl)-4-isopropyl-5-(4-methyl-benzylcarbamoyl)-2H-pyrazol-3-yl]-3,5-dihydroxyheptanoic acid (PF-3052334) as a candidate for the treatment of hypercholesterolemia. J Med Chem. 2008;51:31–45. doi: 10.1021/jm070849r. [DOI] [PubMed] [Google Scholar]
- 12.Hansen KB, Bräuner-Osborne H, Egebjerg J. Pharmacological characterization of ligands at recombinant NMDA receptor subtypes by electrophysiological recordings and intracellular calcium measurements. Comb Chem High Throughput Screening. 2008;11:304–315. doi: 10.2174/138620708784246040. [DOI] [PubMed] [Google Scholar]
- 13.Cheng YC, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 14.Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- 15.Frizelle PA, Chen PE, Wyllie DJ. Equilibrium constants for (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) acting at recombinant NR1/NR2A and NR1/NR2B N-methyl-d-aspartate receptors: implications for studies of synaptic transmission. Mol Pharmacol. 2006;70:1022–1032. doi: 10.1124/mol.106.024042. [DOI] [PubMed] [Google Scholar]
- 16.Buller AL, Larson HC, Schneider BE, Beaton JA, Morrisett RA, Monaghan DT. The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J Neurosci. 1994;14:5471–5484. doi: 10.1523/JNEUROSCI.14-09-05471.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Akerman BL, Allen CD, Sylvain NP, North WG. Abstract 3631: NMDA receptors as possible therapeutic targets for cancer. Cancer Res. 2011;71:3631. [Google Scholar]; (b) North WG, Gao G, Jensen A, Memoli VA, Du J. NMDA receptors are expressed by small-cell lung cancer and are potential targets for effective treatment. Clin Pharmacol: Adv Appl. 2010;2:31–40. doi: 10.2147/CPAA.S6262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Song Z, He CD, Liu J, Sun C, Lu P, Li L, Gao L, Zhang Y, Xu Y, Shan L, Liu Y, Zou W, Zhang Y, Gao H, Gao W. Blocking glutamate-mediated signalling inhibits human melanoma growth and migration. Exp Dermatol. 2012;21:926–931. doi: 10.1111/exd.12048. [DOI] [PubMed] [Google Scholar]
- 18.Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of triheteromeric GluN1/ GluN2A/GluN2B NMDA receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jørgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF. Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-d-aspartate receptors. Mol Pharmacol. 2013;84:114–127. doi: 10.1124/mol.113.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clausen RP, Hansen KB, Cali P, Nielsen B, Greenwood JR, Begtrup M, Egebjerg J, Bräuner-Osborne H. The respective N-hydroxypyrazole analogues of the classical glutamate receptor ligands ibotenic acid and (RS)-2-amino-2-(3-hydroxy-5-methyl-4-isoxazolyl)-acetic acid. Eur J Pharmacol. 2004;499:35–44. doi: 10.1016/j.ejphar.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson M, Levkovitz R, Ben-Tal A, Thielemans K, Spinks T, Belluzzo D, Pagani E, Bettinardi V, Gilardi MC, Zverovich A, Mitra G. Enhanced 3D PETOSEM reconstruction using inter-update Metz filtering. Phys Med Biol. 2000;45:2417–2439. doi: 10.1088/0031-9155/45/8/325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.