

Abstract
Several group 11 metal complexes with chiral thiourea organocatalysts have been prepared and tested as organocatalysts. The promising results on the influence of metal‐assisted thiourea organocatalysts in the asymmetric Friedel–Crafts alkylation of indole with nitrostyrene are described. Better results with the metal complexes have been achieved because of the cooperative effects between the chiral thiourea and the metal. The synergic effect between both species is higher than the effect promoted by each one separately, especially for gold(I). These outcomes are attributed to a pioneering gold(I) activation of the thiourea catalysts, affording a more acidic and rigid catalytic complex than that provided by the thiourea alone. Furthermore, the use of the gold–thiourea organocatalyst allows reducing the catalyst loading to 1–3 mol %. This contribution could become an important starting point for further investigations opening a new line of research overlooked so far in the literature.
Keywords: alkylation, asymmetric catalysis, gold, organocatalysis, sulfur
Introduction
In the last decade, between the two main families of catalysts, namely, metal and enzymatic catalysis, a third complementary approach has emerged, organocatalysis,1 which has become an important tool to provide efficient and environmentally friendly access to enantiomerically pure compounds, including many drugs and bioactive natural products.2
Although organocatalysis has experimented great progress in the field of homogeneous catalysis, the organocatalysts still require more improvement to emulate and reach the achievements reported with metal or enzyme catalysts. With this aim, many efforts have been directed towards the synthesis of more efficient organocatalysts through the use of several strategies. One of them has been the development of bifunctional organocatalysts,3 following the multifunctional catalytic mode exhibited by enzymes. These complex systems have inspired the design of many catalytic systems such as chiral bifunctional thioureas/ureas,4 among others, which keep simultaneous activation of the nucleophile and the electrophile involved in the process (Figure 1).
Figure 1.

Bifunctional activation mode.
Another strategy to improve the reactivity of organocatalysts, which has received considerable attention in the last years, has been the use of two different catalysts in a cooperative way, a metal catalyst and an organocatalyst, which is called dual catalysis (Figure 2).5 This idea has emerged as a promising strategy for developing new and more valuable processes, and also takes advantage of simultaneous activation of the electrophile as well as the nucleophile by two different, but compatible and synergically acting catalysts.
Figure 2.

Activation by dual catalysis: combination of enamine nucleophiles and transition‐metal electrophiles.
The enormous success of chiral urea and thiourea compounds as hydrogen‐bond‐donor organocatalysts in asymmetric synthesis has led to a continuous improvement of these organocatalysts through the use of several modes of activation, trying to improve parameters such as catalysts loading, reaction time, and substrate scope for a given reaction. All these efforts made in the last years have materialized in several upgraded catalysts. Among them, Seidel and Ganesh have reported the use of an internal Brønsted acid forming a protonated thiourea catalyst (Figure 3 a),6 although it was not the most active catalyst in this work. Later, Smith's group and Probst et al. developed new conformationally well‐defined but flexible thiourea catalysts, stabilized by intramolecular hydrogen bonds (Figure 3 b).7, 8 More recently, Herrera and co‐workers have described the use of an external Brønsted acid (Figure 3 c)9 to improve the efficiency of the corresponding chiral thiourea catalysts.
Figure 3.

Activation of thiourea organocatalysts through an internal or an external Brønsted acid.
The use of an internal Brønsted acid produced significant rate acceleration and only a slight improvement in enantioselectivity for the Friedel–Crafts reaction.6 In contrast, the external Brønsted acid was able to assist thiourea catalysts as very effective catalytic species for promoting the enantioselective addition of indoles to nitroalkenes.9 The synergic effect between both species was demonstrated to be higher than the effect promoted by each one separately and better results in terms of enantioselectivity and reactivity were reached than with the corresponding thiourea alone.9 Mattson and co‐workers have also developed hybrid transition metals which act as hydrogen bond donor catalysts such as urea palladacycles10 inspired at the same time in boronates ureas developed by the same research group (Figure 3).11 However, in these examples the authors only use nonchiral ureas.12
In the search of new types of activation of thioureas as organocatalysts, and taking into account this novel concept together with our experience in the chemistry of group 11 metals,13 we were encouraged to use a Lewis acidic metal atom (Figure 4, III) instead of an external Brønsted acid. This will open up a novel and interesting line of research in which the metal center activates the thiourea catalyst, and this activation could be fine‐tuned by carefully choosing the Lewis acid character of the metal, its oxidation state and also its auxiliary ligands. Interestingly, the metal will not participate in the activation of none of the reagents. The coordination chemistry of gold to thiourea‐based ligands has been previously explored with other purposes, mainly because of the interesting biological properties displayed by the complexes. However, this work represents the first example in which coordination to chiral thioureas strongly activates them as organocatalysts.14
Figure 4.
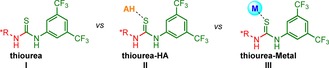
Activation of thiourea organocatalysts through a Lewis acidic metal atom (AH=Brønsted acid).
In the context of our research program focused on the design and synthesis of more active organocatalysts, we report here an unprecedented mode of activation of thiourea organocatalysts through the use of metallic Lewis acids. A wide range of group 11 metal‐thiourea complexes have been synthesized and used as single catalysts in asymmetric catalysis, taking advantages of the best part of both species in a synergic way.
Results and Discussion
To test our hypothesis, we started our study with the synthesis of a variety of group 11 metal complexes with thioureas as potential catalysts for the Friedel–Crafts alkylation reaction as a model process.
Thioureas T1‐3 were chosen as model catalytic structures because they exhibit different electronic and steric properties (Figure 5). Moreover, T3 was the best catalyst in our previously developed works.9, 15
Figure 5.

Model thioureas used in this work.
The exploration was started with T1 presenting less steric hindrance and without electronegative groups in the aromatic ring. The straightforward preparation of the metal complexes 1 a–g is shown in Scheme 1. There are neutral or cationic CuI, AgI, AuI, or AuIII species that have been perfectly identified and characterized by NMR spectroscopy (see Supporting Information).
Scheme 1.

Catalytic complex structures 1 a–g with Cu‐T1, Ag‐T1 and Au‐T1. i) Ag(OTf); ii) [Cu(NO3)(PPh3)2] or [Ag(OTf)(PPh3)] or [Au(OTf)(PPh3)]; iii) Ag(OTf) or [Au(tht)2]OTf; iv) [Au(C6F5)3(tht)].
Subsequently, the efficiency of these species in the benchmark reaction between indole 4 and nitrostyrene 5 was analyzed and the results of these tests are reported in Table 1.16 The results confirmed our hypothesis about the possible activation of a potential organocatalyst, because moderate to good yields and enantioselectivities were obtained in all the cases, whereas almost lack of reactivity was found with T1 (entry 12) or the metal precursor alone (entry 13), which proved the synergic effect between both species. Moreover, the new more hindered complexes likely make the transition state of this reaction more rigid and stable and consequently, able to induce enantioselectivity contrasting the results obtained with thiourea T1 (entry 12). Excellent reactivity values were found for the silver complexes [Ag(OTf)T1] 1 a (Tf=trifluoromethanesulfonyl, entry 2) and [Ag(T1)2]OTf 1 e (entry 8), and promising selectivities were achieved for the gold species [Au(PPh3)T1]OTf 1 d (entry 5) and [Au(T1)2]OTf 1 f (entry 10), with ee values of 42 and 30 %, respectively. We can conclude that coordination of a Lewis acid to the thiourea organocatalyst T1 greatly enhances reactivity and selectivity, compared with the values obtained with this thiourea species alone.
Table 1.
Screening of the reaction promoted by M–T1 (1 a–g) complexes.[a]

| Entry | M–T1 [mol %] | Solvent [mL] | T [°C] | t [d] | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1 | 1 a (10) | CH2Cl2 (0.5) | r.t. | 3 | 22 | 20 |
| 2 | 1 a (10) | CH2Cl2 (0.25) | r.t. | 3 | 83 | 14 |
| 3 | 1 b (10) | CH2Cl2 (0.5) | r.t. | 5 | 60 | 6 |
| 4 | 1 c (10) | CH2Cl2 (0.5) | r.t. | 4 | 38 | 18 |
| 5 | 1 d (10) | CH2Cl2 (0.5) | r.t. | 3 | 22 | 42 |
| 6 | 1 e (10) | CH2Cl2 (0.5) | r.t. | 3 | 60 | 20 |
| 7 | 1 e (10) | CH2Cl2 (0.25) | −17 | 6 | 60 | 32 |
| 8 | 1 e (10) | toluene (0.25) | r.t. | 3 | 75 | 8 |
| 9 | 1 e (10) | CHCl3 (0.25) | r.t. | 3 | 53 | 20 |
| 10 | 1 f (10) | CH2Cl2 (0.5) | r.t. | 3 | 19 | 30 |
| 11 | 1 g (10) | CH2Cl2 (0.5) | r.t. | 4 | 23 | 5 |
| 12 | T1 (10) | CH2Cl2 (0.5) | r.t. | 4 | 10 | Rac.[d] |
| 13 | [Au(tht)2]OTf (5) | CH2Cl2 (0.5) | r.t. | 3 | 10 | – |
[a] Amount of reagents: indole 4 (0.15 mmol) and nitrostyrene 5 (0.1 mmol). [b] Isolated yields after column chromatography. [c] Determined by chiral HPLC analysis (Chiralpak Daicel IA, 90:10 Hex/iPrOH, 1 mL min−1). [d] Racemic mixture.
These initial promising results encouraged us to examine other metal(M)–thiourea complexes varying the electronic properties of the thiourea catalyst. Thiourea T2 was the center of the subsequent study. As better values were obtained in terms of enantioselectivity with Au–T1 1 d and 1 f complexes and in terms of reactivity the best value was obtained with Ag–T1 1 a, in the ensuing screening, different Ag–T2 and Au–T2 catalysts were designed and tested (Scheme 2 and Table 2).
Scheme 2.

Catalytic complex structures 2 a–f with Ag‐T2 and Au‐T2. i) [Au(OTf)(PPh3)]; ii) [Ag(OTf)(PPh3)]; iii) Ag(OTf); iv) [Au(C6F5)(tht)]; v) [Au(C6F5)2(OEt2)2]ClO4; vi) [Au(tht)2]OTf.
Table 2.
Screening of the reaction promoted by M–T2 (2 a–f) complexes.[a]

| Entry | M‐T2 [%] | Solvent [mL] | T [°C] | t [d] | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1 | 2 a (10) | CH2Cl2 (0.5) | r.t. | 3 | 45 | 54 |
| 2 | 2 b (10) | CH2Cl2 (0.5) | r.t. | 3 | 37 | 16 |
| 3 | 2 c (10) | CH2Cl2 (0.5) | r.t. | 3 | 41 | 28 |
| 4 | 2 d (10) | CH2Cl2 (0.5) | r.t. | 3 | 19 | 8 |
| 5 | 2 e (10) | CH2Cl2 (0.5) | r.t. | 3 | 88 | 50 |
| 6 | 2 f (10) | CH2Cl2 (0.5) | r.t. | 4 | 60 | 40 |
| 7 | T2 (20) | CH2Cl2 (0.5) | r.t. | 4 | 22 | 20 |
[a] Amount of reagents: indole 4 (0.15 mmol) and nitrostyrene 5 (0.1 mmol). [b] Isolated yields after column chromatography. [c] Determined by chiral HPLC analysis (Chiralpak Daicel IA, 90:10 Hex/iPrOH, 1 mL min−1).
In this case, the better ee values were found with complexes 2 a and 2 e (Table 2, entries 1 and 5, respectively), although in terms of reactivity complex 2 e was the most active one (88 %, entry 5), followed by 2 f (60 %, entry 6). In summary, the silver(I) 2 c, the gold(I) 2 a and 2 f, and the gold(III) 2 e species bearing two thiourea ligands showed better results, as previously observed for T1 with complexes 1 e and 1 f (Table 1, entries 6–10). As previously found for T1, T2 activated with a metal Lewis acid affords better results than T2 alone (entry 7, Table 2), which again supports our main idea. Moreover, the reaction rate was increased if using 10 mol % of complex 2 e (entry 5) compared with that using 20 mol % of T2 alone (entry 7), probably because of the considerable decrease in the pKa of thiourea T2. With the aim of improving the reactivity and enantioselectivity of this process, we tested the efficiency of T3 synthesizing the same promising metal complexes 3 a–d containing Ag or Au (Scheme 3).
Scheme 3.

Synthesis of the M–T3 complexes 3 a–d. i) Ag(OTf); ii) [Ag(OTf)(PPh3)]; iii) [Au(OTf)(PPh3)]; iv) [Au(tht)2]OTf.
All these complexes were completely characterized and the data are collected in the Supporting Information. Subsequently, the efficiency of these species in the benchmark reaction between indole and nitrostyrene was analyzed and the results are reported in Table 3. As thiourea T3 reaches the best results in this reaction, and because we aimed to avoid high catalyst loading of thioureas as organocatalysts, we decided to test the reaction with only 5 mol % compared with the reported 20 mol % for T3.9, 15
Table 3.
Screening of the reaction promoted by M–T3 (3 a–d) complexes.[a]

| Entry | M–T3 [%] | Solvent [mL] | T [°C] | t [d] | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1 | 3 a (5) | CHCl3 (0.25) | r.t. | 2 | >95 | 32 |
| 2 | 3 b (5) | CHCl3 (0.25) | r.t. | 5 | 90 | 34 |
| 3 | 3 c (5) | CHCl3 (0.25) | r.t. | 2 | 79 | 48 |
| 4 | 3 d (5) | CHCl3 (0.25) | r.t. | 3 | >95 | 56 |
| 5 | T3 (20) | CH2Cl2 (0.5) | r.t. | 4 | 51 | 38 |
[a] Amount of reagents: indole 4 (0.15 mmol) and nitrostyrene 5 (0.1 mmol). [b] Isolated yields after column chromatography. [c] Determined by chiral HPLC analysis (Chiralpak Daicel IA, 90:10 Hex/iPrOH, 1 mL min−1).
The gold(I)–bis(thiourea) complex 3 d was the most active, leading to the best results in terms of reactivity and enantioselectivity (Table 3, entry 4). Remarkably, we used only 5 mol % of catalyst loading compared to 20 mol % in the former works with T3.9, 15 These data exceed those obtained with the catalyst thiourea T3 alone despite the lower catalyst loading (entry 5).
To finely tune the optimal reaction conditions with T3, we carefully checked the variation of other parameters such as solvent, temperature, and variation in the concentration of all species (Table 4), because we realized that small variations in all these parameters could play an important role in governing the enantioselectivity and reactivity of the process. Variations in the amount of indole afforded changes in the values of enantioselectivity and reactivity. However, at low temperature we discarded the use of only 1 equivalent of indole because the rate of the reaction would be too slow. An increase in the enantioselectivity was observed by lowering the reaction temperature to −15 from 25 °C (Table 5, entries 5–9) although the reaction rate was lower. Increasing the concentration of the reaction accelerates the rate of the process and better yields are obtained. Unfortunately, the large differences achieved at room temperature between the reaction performed with the thiourea T3 alone or with 3 d were not maintained at low temperature, probably because of a different mode of coordination between the thiourea catalyst and the metal at different temperatures. The best solvent was found to be CH2Cl2.
Table 4.
Screening of the reaction promoted by 3 d.[a]
| Entry | Cat. | Solvent | T | t | Yield[b] | ee [c] |
|---|---|---|---|---|---|---|
| [%] | [mL] | [°C] | [d] | [%] | [%] | |
| 1d | 3 d (5) | CH2Cl2 (0.25) | r.t. | 5 | 72 | 60 |
| 2d | 3 d (5) | CH2Cl2 (0.10) | r.t. | 5 | 84 | 56 |
| 3d | 3 d (5) | CHCl3 (0.25) | r.t. | 3 | 83 | 53 |
| 4e | 3 d (5) | CHCl3 (0.25) | r.t. | 3 | 82 | 48 |
| 5 | 3 d (5) | CH2Cl2 (0.50) | −15 | 3 | 15 | 63 |
| 6 | 3 d (5) | CH2Cl2 (0.25) | −15 | 5 | 40 | 76 |
| 7 | 3 d (5) | CH2Cl2 (0.10) | −15 | 5 | 66 | 72 |
| 8 | 3 d (5) | CHCl3 (0.25) | −15 | 4 | 38 | 53 |
| 9 | 3 d (5) | CHCl3 (0.10) | −15 | 4 | 60 | 58 |
| 10 | 3 d (5) | toluene (0.25) | −15 | 3 | 21 | 29 |
| 11 | 3 d (5) | CH3CN (0.25) | −15 | 3 | 38 | 33 |
| 12 | 3 d (5) | ClCH2CH2Cl (0.10) | −15 | 5 | 29 | 74 |
[a] Amount of reagents: indole 4 (0.15 mmol) and nitrostyrene 5 (0.1 mmol). [b] Isolated yields after column chromatography. [c] Determined by chiral HPLC analysis (Chiralpak Daicel IA, 90:10 Hex/iPrOH, 1 mL min−1). [d] Reaction performed with 1 equivalent of indole. [e] Reaction performed with 2 equivalents of indole.
Table 5.
Comparison of activity of thioureas T1‐3 versus [Au(thiourea)2]OTf (1 f, 2 f, 3 d).[a]
| Entry | Cat. [%] | Yield [%][b] | ee [%][c] |
|---|---|---|---|
| 1 | T1 (10) | 19 | Rac.[d] |
| 2 | 1 f (10) | 65 | 40 |
| 3 | 1 f (5) | 54 | 32 |
| 4 | 1 f (3) | 51 | 32 |
| 5 | 1 f (1) | 29 | 12 |
| 6 | T2 (10) | 37 | 26 |
| 7 | 2 f (10) | 64 | 48 |
| 8 | 2 f (5) | 60 | 44 |
| 9 | 2 f (3) | 63 | 40 |
| 10 | 2 f (10) | 56 | 25 |
| 11 | T3 (10) | 57 | 34 |
| 12 | T3 (5) | 25 | 30 |
| 13 | T3 (3) | 24 | 22 |
| 14 | T3 (1) | 21 | 8 |
| 15 | 3 d (10) | 94 | 60 |
| 16 | 3 d (5) | 93 | 56 |
| 17 | 3 d (3) | 95 | 54 |
| 18 | 3 d (1) | 83 | 50 |
[a] Experimental conditions: To a mixture of catalyst (mol %) and nitroalkene 5 (0.1 mmol) in CH2Cl2 (0.25 mL), indole 4 (0.15 mmol) was further added, in a test tube at room temperature. After the reaction time, product 6 was isolated by flash chromatography. [b] Isolated yields after column chromatography. [c] Determined by chiral HPLC analysis (Chiralpak Daicel IA, 90:10 Hex/iPrOH, 1 mL min−1). [d] Racemic.
With the optimal reaction conditions in hand, and since one of our concerns in organocatalysis is the high catalyst loading we decided to investigate the changes of reactivity and selectivity between the thioureas T1–T3 and the corresponding gold complexes [Au(thiourea)2]OTf, depending on the catalysts loading in the benchmark reaction (Table 5). T1 in a 10 mol % of catalyst loading gives very poor values (entry 1), with a yield of only 19 % and a racemic mixture. However, the gold complex [Au(T1)2]OTf 1 f in a 10 % produces a 65 % yield and an ee of 40 % (entry 2), which means a considerable increase in both reactivity and overall selectivity. The decrease in the catalyst loading to 3 mol % affords only slightly lower values of 51 % and 32 % ee (entry 3). The same tendency is observed with thiourea T2, with values increasing from 35 % yield and 26 % ee (entry 6) to 64 % and 48 % for the gold complex 2 f (entry 7). Again, these values are maintained to a lowering of the catalyst amount up to 3 %. For thiourea T3, which provided the best results in previous studies, the catalyst loading was lowered to 1 mol % without a significant decrease neither in the reactivity nor the enantioselectivity (entry 18). To illustrate this, in Figure 6 the comparative study between the thiourea T3 alone and the gold complex [Au(T3)2]OTf 3 d is shown for a variation in the catalyst loading.
Figure 6.

Comparative study of reactivity and selectivity between T3 and complex [Au(T3)2]OTf 3 d.
The effect of the metal over the electronic properties of the thiourea T3 is disclosed in the NMR spectra shown in Figure 7. At the same concentration (0.005 mmol catalyst and 500 μL CD2Cl2) in two NMR tubes both compounds, T3 and 3 d, were analyzed at room temperature.
Figure 7.

1H NMR experiments performed in CD2Cl2 at room temperature (400 MHz).
Some of the most relevant resonances in the thiourea catalysts T3 are downfield shifted because of coordination to the Au center in the new catalytic complex 3 d. A displacement of 0.9 ppm (signal from 8.01 ppm to 9.00 ppm) was observed for the thiourea N−H hydrogen atom, and 0.27 ppm (signal from 6.98 ppm to 7.25 ppm) was observed for the aminoindanol N−H hydrogen atom of T3. Additionally, a downfield shifting of the OH group in the aminoindanol scaffold was observed of 0.56 ppm (signal from 2.23 ppm to 2.79 ppm). These displacements would be in agreement with our initial hypothesis and the better values of yield found with complex 3 d with a more acidic thiourea skeleton. Both aliphatic CH−Het (Het=heteroatom) protons experience an upfield shifting (Figure 8).
Figure 8.

Chemical shifts experienced after coordination of T3 to gold(I).
Mechanistic study
Interestingly, the sense of the stereoselection in product 6 was in all cases the same as that expected if (R,R)‐aminoindanol thioureas T2 and T3 were the sole catalysts. This shows that chirality is preferentially governed by the thiourea catalyst, which prompted us to conclude that the metal is only activating the thiourea moiety rather than simply driving by itself some of the reagents into the transition state. Moreover, the metal does not activate this process as above mentioned. Based on our previous works (for TSI and TSII),9, 15 we proposed TSIII as the plausible mode of activation in this reaction (Figure 9).
Figure 9.

Proposed transition states for the Friedel–Crafts reaction.
Assumably, the M‐thiourea catalyst complex is the most reactive species, owing to the increased acidity of the NH in the thiourea after a synergic coordination with the metal atom, thereby increasing the reaction rate and favoring the activation of the substrates. Furthermore, improvement in the enantioselectivity could be attributed to the formation of a more rigid assembly in the transition state TSIII as combination of both structures. This mode of activation TSIII (Figure 9) would agree with the fact that the observed enantiomer is given by the enantioselectivity of the thiourea organocatalyst employed as obtained in the previous works.9, 15
As the gold atom is joined to two thiourea ligands in the best catalyst 3 d, we cannot discard that the same process occurs by both thiourea organocatalysts, which was supported by the better yields and enantioselectivities observed in this really congested catalyst, in comparison with the thiourea T3 alone.
Conclusions
The unprecedented activation of thiourea organocatalysts through the coordination of a metallic Lewis acid was described. Coordination of the metal produces the consequent acidification of the thiourea protons achieving a better activity in terms of conversion and selectivity in the benchmark reaction of addition of indole to nitrostyrene. Three thiourea organocatalysts T1–T3 with different electronic and steric requirements were used to prepare several group 11 metal complexes 1–3. All the tested M–thiourea complexes provided better results than the thioureas alone, and the gold complexes [Au(thiourea)2]OTf were the best in terms of reactivity and selectivity. After achieving the optimum experimental conditions a comparison of the activity of the thioureas versus the [Au(thiourea)2]OTf complexes in different catalysts loading were performed. As expected the higher values were achieved with thiourea T3 and [Au(T3)2]OTf 3 d showing a great improvement in the reactivity and selectivity values of the metal thiourea complex compared with the thiourea alone. It was possible to reduce the catalyst loading to 1 mol % without a significant decrease of the activity, although optimum values are afforded with 3 mol % catalyst loading. Thus, a new concept was proved demonstrating the cooperative effect between a metallic Lewis acid and thioureas providing metal complexes acting as organocatalysts.
Experimental Section
Instrumentation
Mass spectra were recorded on a BRUKER ESQUIRE 3000 PLUS, with the electrospray (ESI) technique. The attenuated total reflection (ATR–FTIR) spectra of solid samples were recorded on a PerkinElmer FT‐IR spectrometer equipped with a universal ATR sampling accessory. 1H and 13C{1H} APT NMR, including 2 D experiments, were recorded at room temperature on a BRUKER AVANCE 400 spectrometer (1H, 400 MHz; 13C, 100.6 MHz; 19F, 376.5 MHz) or on a BRUKER AVANCE II 300 spectrometer (1H, 300 MHz; 13C, 75.5 MHz; 19F, 282.3 MHz), with chemical shifts (ppm) reported relative to the solvent peaks of the deuterated solvent.17
Starting materials
All reactions were performed under air atmosphere and solvents were used as received without further purification or drying. The complexes [Ag(OTf)(PPh3)],18 [Au(tht)2]OTf19 (tht=tetrahydrothiophene), [Au(C6F5)3(tht)],20 [Au(C6F5)tht)],21 and [Au(C6F5)2(OEt)2]ClO4 22 were prepared following published procedures. [Au(OTf)(PPh3)] was obtained from [AuCl(PPh)3]20 with Ag(OTf) in dichloromethane. All other reagents were commercially available.
NMR spectra of all synthesized catalysts and the characterization of all new compounds are reported in the Supporting Information.
General procedure for the catalyzed Friedel–Crafts alkylation reaction
To a mixture of catalyst M–(T1–T3) (1–10 mol %), and nitroalkene 5 (0.1 mmol) at the indicated temperature (r.t. or −15 °C) in a test tube with CH2Cl2 (0.25 mL), indole 4 (0.15 mmol) was further added. After the appropriate reaction time (see Tables 1–5), the residue was purified by column chromatography (SiO2; hexane/EtOAc, 8:2) to afford final adduct 6. Yields and enantioselectivities are reported in Tables 1–5. Spectral and analytical data for compound 6 is in agreement with those previously reported in the literature 15a.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Authors thank the Ministerio de Economía y Competitividad (MINECO/FEDER CTQ2016‐75816‐C2‐1‐P), the High Council of Scientific Investigation (CSIC) (PIE‐201580I010) and Gobierno de Aragón‐Fondo Social Europeo (E77 and E104) for financial support of our research.
A. Izaga, R. P. Herrera, M. C. Gimeno, ChemCatChem 2017, 9, 1313.
Contributor Information
Dr. Raquel P. Herrera, Email: raquelph@unizar.es
Prof. M. Concepción Gimeno, Email: gimeno@unizar.es.
References
- 1.
- 1a. Berkessel A., Gröger H., Asymmetric Organocatalysis; Wiley-VCH, Weinheim, 2005; [Google Scholar]
- 1b. Enantioselective Organocatalysis (Ed.: P. I. Dalko), Wiley-VCH: New York, 2007; [Google Scholar]
- 1c. Science of Synthesis Asymmetric Organocatalysis (Eds.: B. List, K. Maruoka), Thieme Chemistry, 2012; [Google Scholar]
- 1d. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications (Ed.: P. I. Dalko), Wiley-VCH, Weinheim, 2013. [Google Scholar]
- 2.
- 2a. de Figueiredo R. M., Christmann M., Eur. J. Org. Chem. 2007, 2575–2600; [Google Scholar]
- 2b. Marqués-López E., Herrera R. P., Christmann M., Nat. Prod. Rep. 2010, 27, 1138–1167; [DOI] [PubMed] [Google Scholar]
- 2c. Marqués-López E., Herrera R. P. in Comprehensive Enantioselective Organocatalysis (Ed.: P. I. Dalko), Wiley-VCH, Weinheim, 2013, pp. 1359–1383; [Google Scholar]
- 2d. Alemán J., Cabrera S., Chem. Soc. Rev. 2013, 42, 774–793; [DOI] [PubMed] [Google Scholar]
- 2e. Sun B.-F., Tetrahedron Lett. 2015, 56, 2133–2140. [Google Scholar]
- 3.For selected reviews, see:
- 3a. Miyabe H., Takemoto Y., Bull. Chem. Soc. Jpn. 2008, 81, 785–795; [Google Scholar]
- 3b. Connon S. J., Chem. Commun. 2008, 2499–2510; [DOI] [PubMed] [Google Scholar]
- 3c. Chauhan P., Chimni S. S., RSC Adv. 2012, 2, 737–758; [Google Scholar]
- 3d. Narayanaperumal S., Rivera D. G., Silva R. C., Paixão M. W., ChemCatChem 2013, 5, 2756–2773; [Google Scholar]
- 3e. Xi Y., Shi X., Chem. Commun. 2013, 49, 8583–8585; [DOI] [PubMed] [Google Scholar]
- 3f. Serdyuk O. V., Heckel C. M., Tsogoeva S. B., Org. Biomol. Chem. 2013, 11, 7051–7071; [DOI] [PubMed] [Google Scholar]
- 3g. Fang X., Wang C.-J., Chem. Commun. 2015, 51, 1185–1197; [DOI] [PubMed] [Google Scholar]
- 3h. Sonsona I. G., Marqués-López E., Herrera R. P., Beilstein J. Org. Chem. 2016, 12, 505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.See for instance:
- 4a. Taylor M. S., Jacobsen E. N., Angew. Chem. Int. Ed. 2006, 45, 1520–1543; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1550–1573; [Google Scholar]
- 4b. Doyle A. G., Jacobsen E. N., Chem. Rev. 2007, 107, 5713–5743; [DOI] [PubMed] [Google Scholar]
- 4c. Zhang Z., Schreiner P. R., Chem. Soc. Rev. 2009, 38, 1187–1198; [DOI] [PubMed] [Google Scholar]
- 4d. Kotke M., Schreiner P. R. in Hydrogen Bonding in Organic Synthesis (Ed.: P. M. Pihko), Wiley-VCH, Weinheim, 2009, pp. 141–351; [Google Scholar]
- 4e. Marqués-López E., Herrera R. P., An. Quim. 2009, 105, 5; [Google Scholar]
- 4f. Marqués-López E., Herrera R. P. in New Strategies in Chemical Synthesis and Catalysis (Ed.: B. Pignataro), Wiley-VCH, Weinheim, 2012, pp. 175–199; [Google Scholar]
- 4g. Jakab G., Schreiner P. R. in Comprehensive Enantioselective Organocatalysis (Ed.: P. I. Dalko), Wiley-VCH, Weinheim, 2013, pp. 315–341, and references therein cited; [Google Scholar]
- 4h. Zhang Z., Bao Z., Xing H., Org. Biomol. Chem. 2014, 12, 3151–3162; [DOI] [PubMed] [Google Scholar]
- 4i. Auvil T. J., Schafer A. G., Mattson A. E., Eur. J. Org. Chem. 2014, 2633–2646. [Google Scholar]
- 5.For selected reviews about dual catalysis combining an organocatalyst and a metal catalyst, see:
- 5a. Shao Z., Zhang H., Chem. Soc. Rev. 2009, 38, 2745–2755; [DOI] [PubMed] [Google Scholar]
- 5b. Rueping M., Koenigs R. M., Atodiresei I., Chem. Eur. J. 2010, 16, 9350–9365; [DOI] [PubMed] [Google Scholar]
- 5c. Loh C. C. J., Enders D., Chem. Eur. J. 2012, 18, 10212–10225; [DOI] [PubMed] [Google Scholar]
- 5d. Allen A. E., MacMillan D. W. C., Chem. Sci. 2012, 3, 633–658; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Du Z., Shao Z., Chem. Soc. Rev. 2013, 42, 1337–1378; [DOI] [PubMed] [Google Scholar]
- 5f. Hopkinson M. N., Sahoo B., Li J.-L., Glorius F., Chem. Eur. J. 2014, 20, 3874–3886; [DOI] [PubMed] [Google Scholar]
- 5g. Dong X.-Q., Zhao Q., Li P., Chen C., Zhang X., Org. Chem. Front. 2015, 2, 1425–1431. [Google Scholar]
- 6. Ganesh M., Seidel D., J. Am. Chem. Soc. 2008, 130, 16464–16465. [DOI] [PubMed] [Google Scholar]
- 7. Jones C. R., Pantoş G. D., Morrison A. J., Smith M. D., Angew. Chem. Int. Ed. 2009, 48, 7391–7394; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7527–7530. [Google Scholar]
- 8.For a related and more recent example, see also: Probst N., Madarász A., Valkonen A., Pápai I., Rissanen K., Neuvonen A., Pihko P. M., Angew. Chem. Int. Ed. 2012, 51, 8495–8499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8623–8627. [Google Scholar]
- 9. Marqués-López E., Alcaine A., Tejero T., Herrera R. P., Eur. J. Org. Chem. 2011, 3700–3705. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Nickerson D. M., Angeles V. V., Auvil T. J., So S. S., Mattson A. E., Chem. Commun. 2013, 49, 4289–4291; [DOI] [PubMed] [Google Scholar]
- 10b. Nickerson D. M., Mattson A. E., Chem. Eur. J. 2012, 18, 8310–8314. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. So S. S., Burkett J. A., Mattson A. E., Org. Lett. 2011, 13, 716–719; [DOI] [PubMed] [Google Scholar]
- 11b. So S. S., Auvil T. J., Garza V. J., Mattson A. E., Org. Lett. 2012, 14, 444–447. [DOI] [PubMed] [Google Scholar]
- 12.For a more recent example of non-chiral ureas, see also: Hall E. A., Redfern L. R., Wang M. H., Scheidt K. A., ACS Catal. 2016, 6, 3248–3252. [Google Scholar]
- 13.See for example:
- 13a. Crespo O., Gimeno M. C., Jones P. G., Laguna A., López-de-Luzuriaga J. M., Monge M., Pérez J. L., Ramón M. A., Inorg. Chem. 2003, 42, 2061–2068; [DOI] [PubMed] [Google Scholar]
- 13b. Gimeno M. C., Laguna A. in Comprehensive Coordination Chemistry II, Silver and Gold, Vol. 6 (Eds.: J. A. McCleverty, T. J. Meyer), Elsevier, Oxford, 2004, pp. 911–1145; [Google Scholar]
- 13c. Gimeno M. C. in The Chemistry of Gold in Modern Supramolecular Gold Chemistry. Gold-Metal Interactions and Applications (Ed.: A. Laguna), Wiley-VCH, Weinheim, 2008, pp. 1–63; [Google Scholar]
- 13d. Gimeno M. C., Laguna A., Visbal R., Organometallics 2012, 31, 7146–7157; [Google Scholar]
- 13e. Goitia H., Nieto Y., Villacampa M. D., Kasper C., Laguna A., Gimeno M. C., Organometallics 2013, 32, 6069–6078. [Google Scholar]
- 14.The coordination chemistry of thioureas in metal complexes has been also explored for other purposes, such as for their biological properties:
- 14a. Yan K., Lok C.-N., Bierla K., Che C.-M., Chem. Commun. 2010, 46, 7691–7693; [DOI] [PubMed] [Google Scholar]
- 14b. Yang M., Pickard A. J., Qiao X., Gueble M. J., Day C. S., Kucera G. L., Bierbach U., Inorg. Chem. 2015, 54, 3316–3324; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Ali S., Yasin G., Zuhra Z., Wu Z., Butler I. S., Badshah A., ud Din I., Bioinorg. Chem. Appl. 2015, 386587. In material science: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. Choi Y. C., Yeom E. J., Ahn T. K., Seok S. I., Angew. Chem. Int. Ed. 2015, 54, 4005–4009; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4077–4081. Or as ligands in metal complexes: [Google Scholar]
- 14e. Koch K. R., Coord. Chem. Rev. 2001, 216, 473–488; [Google Scholar]
- 14f. Breuzard J. A. J., Christ-Tommasino M. L., Lemaire M., Top. Organomet. Chem. 2005, 15, 231–270; [Google Scholar]
- 14g. Pan J.-H., Yang M., Gao Q., Zhu N.-Y., Yang D., Synthesis 2007, 2539–2544. [Google Scholar]
- 15.For a selection of our works using these thioureas, see also:
- 15a. Herrera R. P., Sgarzani V., Bernardi L., Ricci A., Angew. Chem. Int. Ed. 2005, 44, 6576–6579; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6734–6737; [Google Scholar]
- 15b. Herrera R. P., Monge D., Martín-Zamora E., Fernández R., Lassaletta J. M., Org. Lett. 2007, 9, 3303–3306; [DOI] [PubMed] [Google Scholar]
- 15c. Roca-López D., Marqués-López E., Alcaine A., Merino P., Herrera R. P., Org. Biomol. Chem. 2014, 12, 4503–4510; [DOI] [PubMed] [Google Scholar]
- 15d. Schön E.-M., Marqués-López E., Herrera R. P., Alemán C., Díaz D. D., Chem. Eur. J. 2014, 20, 10720–10731; [DOI] [PubMed] [Google Scholar]
- 15e. Juste-Navarro V., Marqués-López E., Herrera R. P., Asian J. Org. Chem. 2015, 4, 884–889; [Google Scholar]
- 15f. Gimeno M. C., Herrera R. P., Cryst. Growth Des. 2016, 16, 5091–5099. [Google Scholar]
- 16.For the pioneering racemic version of this Friedel–Crafts alkylation reaction, see: Dessole G., Herrera R. P., Ricci A., Synlett 2004, 2374–2378. [Google Scholar]
- 17. Fulmer G. R., Miller A. J. M., Sherden N. H., Gottlieb H. E., Nudelman A., Stoltz B. M., Bercaw J. E., Goldberg K. I., Organometallics 2010, 29, 2176–2179. [Google Scholar]
- 18. Bardají M., Crespo O., Laguna A., Fischer A. K., Inorg. Chim. Acta 2000, 304, 7–16. [Google Scholar]
- 19. Usón R., Laguna A., Navarro A., Parisch R. V., Moore L. S., Inorg. Chim. Acta 1986, 112, 205–208. [Google Scholar]
- 20. Usón R., Laguna A., Organomet. Synth. 1986, 3, 322–342. [Google Scholar]
- 21. Usón R., Laguna A., Laguna M., Inorg. Synth. 1989, 26, 85–91. [Google Scholar]
- 22. Usón R., Laguna A., Arrese M. L., Synth. React. Inorg. Met.-Org. Chem. 1984, 14, 557–567. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary