

Abstract
The interaction of α‐synuclein (αS) with membranes is thought to be critical in the etiology of Parkinson's disease. Besides oligomeric αS aggregates that possibly form membrane pores, the aggregation of αS into amyloid fibrils has been reported to disrupt membranes. The mechanism by which aggregation affects the integrity of membranes is, however, unknown. Here, we show that whereas mature αS fibrils only weakly adhere to POPC/POPG giant unilamellar vesicles (GUVs), fibrillization of αS on the membrane results in large‐scale membrane remodeling. Fibrils that grow on the vesicle surface stiffen the membrane and make the initially spherical membrane become polyhedral. Additionally, membrane‐attached fibrils extract lipids. The lipid extraction and membrane remodeling of growing fibrils can consume the complete bilayer surface and results in loss of vesicle content. These observations suggest that there are several mechanisms by which growing fibrils can disrupt membrane function.
Keywords: alpha-synuclein, amyloids, lipid extraction, membrane remodelling, membranes
1. Introduction
The aggregation of proteins into cross β‐sheet structures is known as amyloid formation. This process is associated with several neurodegenerative diseases. Amyloid formation of the intrinsically disordered protein α‐synuclein (αS) is implicated as one of the causes of Parkinson's disease (PD). However, the role of αS amyloids in PD etiology is still poorly understood.
The physiological function of αS most likely involves a role in the regulation of the synaptic vesicle pool.1 In vitro studies have shown that αS binds membranes. This interaction results in a conformational change of the initially disordered random coil αS into an α‐helical structure.2 Depending on the membrane composition and therefore the αS binding affinity, partition depth, inter‐leaflet order and symmetry,3 the interaction of αS with membranes can lead to membrane remodeling. The binding of αS to the outer surface of negatively charged vesicles has been reported to generate curvature and to result in the tubulation of membranes. 2e Similar membrane tubules have been reported for vesicles composed of zwitterionic lipids after incubation with αS monomers.4
The aggregation of membrane‐bound αS has been hypothesized to cause membrane damage in PD. Protein fibrillization is known to play an important role in other protein aggregation diseases such as type II diabetes mellitus.5 In type II diabetes, fibril formation of the islet amyloid polypeptide (IAPP) at the vesicle surface leads to distortions that may be directly responsible for membrane leakage. Also, β2‐microglobulin (β2m) amyloid fibrils have been observed to bind membranes and cause vesicles to adopt teardrop‐like shapes.6 In this case, the membrane damage was hypothesized to result from lipid extraction at the points of distortion. The interaction with negatively charged lipids in membranes has been observed to trigger fiber formation of endostatin and lysozyme.7 This interaction has been directly linked to the observed cytotoxicity of the process which has been attributed to the uptake of plasma membrane by the fibrils.7 Aggregation of αS on supported lipid bilayers has been reported to result in the extraction of lipids8 and co‐aggregation of αS with small unilamellar vesicles (SUVs) results in lipid‐protein aggregates with distinct structures.[9] Fibrillization of αS also facilitates the leakage of dye molecules encapsulated in LUVs and finally results in the formation of mixed supra‐fibrillar lipid‐protein aggregates.10 Although it is becoming increasingly clear that αS fibril formation on the membrane causes membrane damage, the nature of this damage has not yet been visualized. Here, we used giant unilamellar vesicles (GUVs) as a model system to visualize the membrane deformations and remodeling resulting from the interaction with αS amyloid fibrils.
2. Results and Discussion
Vesicles composed of negatively charged phospholipids are reported to bind both monomeric and oligomeric αS with a higher affinity than vesicles composed of zwitterionic phospholipids,[11] this may also be the case for αS amyloid fibrils. In a control experiment we therefore tested if αS fibrils bind GUVs composed of the zwitterionic phospholipid POPC and if this binding results in vesicle deformation. A few minutes after 10 μm ThT stained pre‐formed fibrils were added to the solution containing the GUVs, we did not observe an accumulation of fibrils or fibrillar aggregates at the vesicle surface (Figure S1A). In the presence of αS fibrils, immobilized POPC vesicles remained spherical and intact for at least 26 hours. Vesicles in the presence of fibrils were indistinguishable from control vesicles in buffer solution (Figure S1B). Subsequently, the effect of fibril growth on the morphology of POPC GUVs was investigated. The aggregation of αS was performed in quiescent conditions at 37 °C. To speed up aggregation, αS monomers were incubated with preformed fibril seeds. As observed for preformed fibrils, growing fibrils did not bind to POPC GUVs and the vesicles remained intact and spherical for up to at least 26 hours (Figure S1C and D).
Our results suggest that the electrostatic interactions, between the negatively charged membrane and the positively charged lysines at the N‐terminal part of the protein, that are important for monomer membrane interactions12 may also play a critical role in the binding of amyloid fibrils to GUV membranes. The mature pre‐formed αS amyloid fibrils and the fibrils grown in the presence of POPC vesicles may therefore not have a high enough affinity for net neutral membranes to bind POPC GUVs.
In contrast to the zwitterionic POPC membranes, the negatively charged membranes have a high affinity for monomeric αS.11 When GUVs composed of a 1:1 mixture of the zwitterionic phospholipid POPC and the negatively charged phospholipid POPG were incubated with 5 μm αS monomers of which a fraction was labelled with AlexaFluor488 the fluorescently labelled protein was, as expected, observed to colocalize with the membrane label (Figure S2). In these conditions differences in αS concentrations between the vesicle interior and exterior are difficult to observe. After incubation of POPC/POPG with 100 μm αS monomers doped with 1 mol % αS‐AlexaFluor647, the AlexaFluor647 labelled αS could be observed in the solution surrounding the GUVs, the vesicle interior stayed devoid of labelled protein. The fluorescently labelled protein could not translocate over the membrane suggesting that the membrane remains intact upon αS binding (Figure S3). A sphericity analysis indicates that membranes do not undergo large‐scale deformations in the presence of αS monomers (Figure 3).
Figure 3.
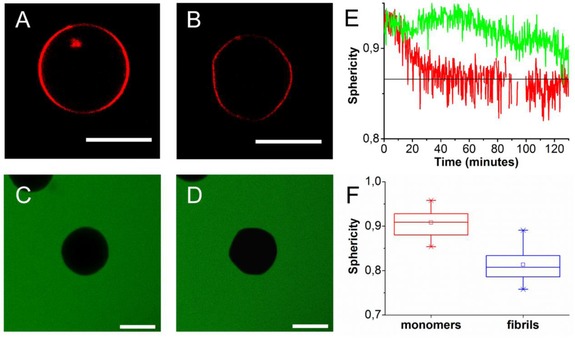
Fibrillization‐induced deformation of POPC/POPG GUVs: A) An initially spherical vesicle is incubated with 95 μm αS monomers and 5 μm fibril seeds B) as a result of fibril growth it deforms and becomes a polyhedron (Δt=4 hours). When POPC/POPG vesicles were incubated with 20 μm αS seeds and 80 μm αS monomers no influx of calcein dye (green) could be observed. C) Immobilized vesicle at t=0 and D) t=160 minutes after the start of the experiment. E) To quantify the shape changes the sphericity of the vesicles was followed in time. The black line indicates the sphericity of an icosahedron, the green and red line follows the shape changes of the GUVs shown in A, B and C, D respectively. E) Sphericity of POPC:POPG 1:1 GUVs in the presence of αS monomers (red) and after incubation with growing αS fibrils. The average sphericity (open squares), the mean sphericity (middle line) and the 25 and 75 percentile (box) are shown. All scale bars: 10 μm.
To visualize if the preformed fibrils bind to POPC/POPG 1:1 membranes, fibrils stained with ThT were incubated with immobilized POPC/POPG GUVs. Images show that fibrils can be found in close proximity of the immobilized vesicles (Figure 1 A). Some fibrils seem to stick to the GUVs. Figure 1 A shows a fibril that co‐localizes with the vesicle surface at one end. To investigate if fibrils bind the vesicle kymographs were obtained. Kymographs in which the vesicle position was fixed show that whereas the position of the fibril close to the membrane is relatively stable, the fluctuations in fibril position become larger further away from the vesicle (Figure 1 B). This is best observed when the width of the fluctuations is plotted as a function of the position along the fibril as visualized in Figure 1 C. This clearly indicates that fibrils can bind to the vesicle surface.
Figure 1.
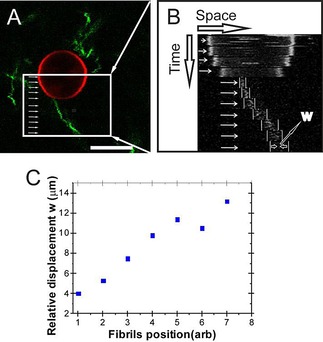
Binding of αS fibrils to POPC/POPG GUVs: A) Pre‐stained ThT fibrils (50 μm equivalent monomer concentration) were incubated with the immobilized GUVs. Kymographs were obtained at different positions along the fibril contour in the region indicated by the white box. The white arrows indicate the positions that were followed in time. Scale bar: 10 μm. B) Kymographs of the different positions indicate that whereas the position of the vesicle is fixed, larger fluctuations in position are observed at one end of the fibril compared to the other. C) Quantification of the maximal thermal displacement along the length of the fibril.
In contrast to what has been observed for β2m fibrils, these mature membrane‐bound αS fibrils were, however, not able to deform the membrane.6 The difference in the observation for β2m fibrils and our results may be a consequence of the different length scales involved. Deformation of highly curved membranes, such as the LUVs used in the experiments with β2m fibrils, may be more prominent because the relative deformation is larger than in less curved membranes. Additionally, the adhesion strength of the β2m fibrils may be higher than that of the αS fibrils. Besides the vesicle radius and adhesion strength, the contour and persistence length of the fibril and bending rigidity of the membrane will determine if deformations can occur. The adhesion strength of the long and stiff mature αS fibrils was apparently not high enough to zipper and bend the fibril onto the membrane or to deform the membrane itself.
In contrast to the addition of preformed fibrils, fibrillization of αS on the membrane has been reported to damage POPC/POPG LUVs.10 To directly visualize fibrillization induced changes in the membrane integrity or the GUV morphology with confocal microscopy, the immobilized GUVs were incubated with fibrillar seeds and monomers. Figure 2 shows the resulting vesicles three hours after the start of the experiments. In this experiment, the fibril seeds were labelled with ThT while a fraction of the monomers was labelled with AlexaFluor647. At the near physiological salt conditions used, fibrils are known to cluster.13 Fibril seed clusters are visible as bright ThT positive regions in the image. Labelled monomers add to fibrils in these clusters and cause fibril elongation into the solution. The Rh‐DOPE signal that co‐localizes with some of the fibrils that grow into the solution suggests that growing fibrils extract lipids from the bilayer. The co‐localization of labelled monomers, ThT positive fibrils and the GUV surface indicates that fibrils also grow at the membrane of both unilamellar and multilamellar GUVs (Figure 2, Figure S4). Besides extracting lipids, the fibrillization at the membrane causes the initially nearly spherical vesicles develop facets (Figure 2, Figure S4).
Figure 2.
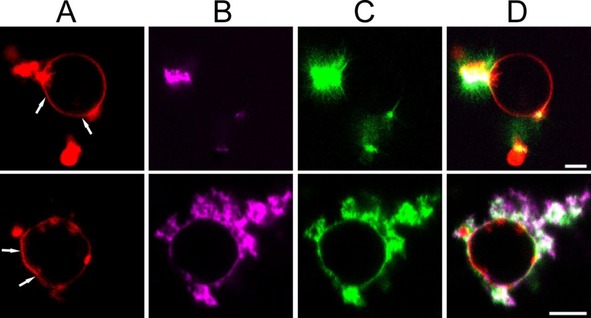
Fibrillization‐induced membrane deformations: POPC/POPG GUVs doped with Rh‐DOPE were incubated with 20 μm (equivalent monomer concentration) ThT labelled fibril seeds and 80 μm αS monomers (1 mol % αS‐AlexaFluor647) at 37°C in quiescent conditions. Images were taken 3 hours after the start of the experiment. A) Rh‐DOPE fluorescence (red). B) αS‐Alexa647 fluorescence (magenta). C) ThT fluorescence (green). D) Overlay of A, B and C. The addition of αS to fibril seeds results in the growth of ThT positive amyloid fibrils from the seeds at both the membrane and in solution. The white arrows indicate some of the places where fibril growth has resulted in deviations from the normally nearly spherical vesicle shape. Scale bar: 5 μm.
The differences in the ability to deform the membrane between mature αS fibrils and fibrils grown in the presence of αs binding vesicles may be caused by small structural differences. Several polymorphs of amyloid fibrils have been observed for αS.14 The prevalent fibril polymorph or fold of amyloid fibrils grown on membranes may be slightly different from those grown in solution. This may result in a different propensity of fibrils to remodel the GUV membrane.
To further characterize the GUV remodeling—from almost spherical to faceted—by growing fibrils, we followed single GUVs in time for up to four hours. Initially, thermal fluctuations cause shape fluctuations of the immobilized vesicles that are also typically observed for vesicles in solution.15 But in time, the shape fluctuations decrease and membranes become rigid (Figure 3). A sphericity analysis of the vesicle shape as a function of time shows that the process differs from vesicle to vesicle. Figure 3 E shows two examples; for one GUV a continuous decrease in sphericity S is observed until, after approximately 40 minutes, it reaches a constant value which agrees with the sphericity of an icosahedron (S=0.87), while for the other GUV the sphericity fluctuates to a lower S in time. The distribution of observed sphericity values is shown Figure 2 F. The transition from a circular cross‐section to a polygonal cross‐section suggests that the initially approximately spherical vesicles become polyhedra.
There are several factors that may contribute to the observed vesicle‐shape change in the presence of aggregating αS. The aggregation of αS in the solution surrounding the vesicle coincides with a change in the osmotic pressure; the concentration of particles in solution changes when monomers aggregate into fibrils. The decrease in the osmotic pressure of the bulk solution compared to the vesicle interior is expected to result in diffusion of water into the vesicle. However, even if we assume that all the αS monomers aggregate into a single fibril, the membrane tension stays an order of magnitude lower than the typical lysis tension of vesicles.16 The GUV is therefore not expected to lyse. The GUV shape changes in the presence of aggregating αS does also not follow the expected change in osmotic pressure. In our experiments, vesicles do not round up as expected but become faceted instead. The faceting must therefore be a direct result of the formation of amyloid fibrils at the membrane.
The observed faceting of the vesicles requires water efflux. To check if this efflux of water is associated with the formation of permanent pores or edge defects, which allow for volume loss, calcein was added to the immobilized vesicles. In the absence of seeds and monomers, the calcein was not able to cross the membrane. As a result, no calcein fluorescence could be observed in the interior of the vesicle. When subsequently αS fibril seeds and monomers were added to the vesicle solution this again resulted in the shape changes described above. However, these shape changes of vesicles did not result in the influx of calcein (Figure 3 C, D). We therefore conclude that the shape change and concomitant loss of volume from the vesicles do not coincide with the formation of permanent pores or defects. The sharp edges of the polyhedron apparently do not result in membrane defects that allow calcein to equilibrate over the vesicle membrane. Volume loss therefore probably results from diffusion of water over the membrane.
As for many other proteins that interact via an amphipathic α‐helix, the membrane remodeling capacity of αS is thought to result from an increase in the area of the lipid layer into which the protein inserts. Part of the N‐terminal region involved in αS membrane interaction is solvent exposed in the fibril. Supposing that stiff αS fibrils bind membranes by inserting N‐terminal α‐helices, the thus created area excess may support elongated, highly curved structures, such as the vertices of polyhedra. The results presented in Figure 2 and S4, however, suggest that the fibrils are not solely present at the vertices, the labelled proteins and ThT signal are more homogenously distributed over the vesicles surface.
Considering that most of the surface of the faceted vesicles is finally covered with amyloid fibrils, the observed shape changes suggest that the attachment of fibrils has either made the initially fluid membrane elastic, resulting in a buckling transition or that more rigid domains appear in the membrane. SUVs composed of phospholipids have been reported to become faceted when incubated below the lipid phase transition temperature. The higher bending rigidity of the gel phase membrane cannot support the high curvature of the SUVs which results in buckling.17 It is known that particle, cation or protein binding can result in a transition from liquid‐crystalline to a crystalline phase.18 The appearance of crystalline or more ordered domains should be visible in the distribution of Rh‐DOPE fluorescence over the membrane. During the transition from the spherical to faceted vesicles, we do not observe any domain formation in the Rh‐DOPE distribution. This suggests that the membrane stays liquid. The coupling of the membrane to a shell of membrane binding and strongly interacting fibrils13 is therefore probably responsible for the formation of an elastic shell or stiffer domains. The shape of the faceted vesicle does not look like the perfect icosahedra observed for elastically homogeneous materials. Moreover, the angles between the edges are not very sharp and vesicles are not leaking from the edges. This suggests that instead of a buckled shell, large rigid domains appeared in the membrane. These rigid domains are separated by soft (fluid) interfaces that bend to release stress. A first estimate of the fibril binding energy required to support this deformation confirms the feasibility of this mechanism (SI).
Besides the shape change into polyhedra, the data presented in Figure 2 and Figure S4 suggest that lipids are possibly extracted from the GUV membrane by the growing fibrils. The effect of the lipid extraction even becomes clearer in Figure 4. While some vesicles, such as the GUVs marked with an asterisk that appears in the field of view after the start of the experiment, develop facets, other vesicles additionally appeared to shrink and finally completely disappeared from the field of view. When the focal plane was moved close to the surface, we observed small vesicles and fibrillar, Rh‐DOPE‐containing structures (Figure 4 M‐N). After 32 hours the previously reported mixed lipid‐protein aggregates were found.10
Figure 4.
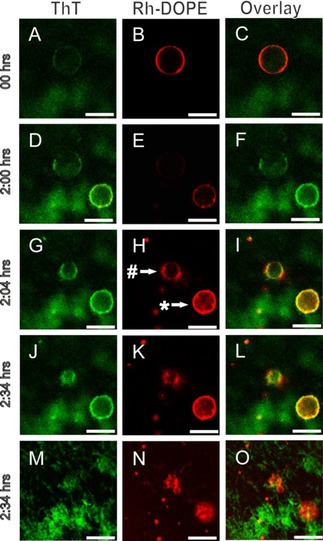
Membrane faceting and tubulation induced by fibril formation of αS at the membrane surface. POPC/POPG vesicles were incubated with 50 μm seeds and 50 μm αS monomers at 37 °C in quiescent conditions. The images show time‐dependent changes in vesicle morphology induced by αS fibrillization on the membrane surface (A‐O). Vesicles were doped with Rh‐DOPE (red) and fibrils stained with ThT (green). The # and * indicate a tubulated and faceted vesicle, respectively. Images M‐O were taken on the glass surface. Scale bar: 10 μm.
Fibril adsorption and the growth of fibrillar aggregates on the membrane surface were visualized using the amyloid‐binding dye ThT (Figure 4). The ThT intensity on polyhedral vesicles is high compared to the ThT intensity on the shrinking vesicles indicating that the initial vesicle deformation mode depends on the fibril density on the membrane surface (Figure 4 F, I). To support the ThT data, AFM images of ThT positive, Rh‐DOPE‐containing structures on a mica surface were obtained. These images confirm the fibrillary nature of the surface attached structures (Figure S5).
For some proteins such as dynamin, the polymerization of a helical coat around the membrane is thought to create membrane tubules. The interaction of αS, in different stages or states of aggregation, has also been reported to result in membrane remodeling. In the presence of monomeric αS, POPG GUVs and POPC LUVs develop into membrane tubules.4, 19 It is, however, unlikely the observed fibrilar structures represent such membrane tubules. The fibril‐like structures observed after seeded aggregation in the presence of GUVs on mica surfaces typically have a height of <10 nm in AFM images (Figure S5). Considering that this height is very close to the diameter of solution grown fibrils,14b it unlikely that these structures represent membrane tubules wrapped with amyloid fibrils. It is more likely that they represent amyloids that have extracted lipids from the GUV membrane. Additionally, the extraction of lipids by amyloid fibrils has been reported in the literature. Amyloids fibrils of endostatin, lysozyme, IAPP and αS have been shown to take up lipids during aggregation.7, 8, 10, 20 In the long term, this extraction of lipids from the membrane is thought to cause a disruption of the membrane barrier.7, 8, 10, 20 On longer timescales, this lipid extraction is probably responsible for formation of the previously reported Lewy body like lipid containing supra‐fibrillar aggregates.10
3. Conclusions
In conclusion, we have shown that in contrast to membrane binding of mature amyloid fibrils, the growth of αS amyloid fibrils on the membrane results in membrane deformations. These deformations depend on membrane coverage and include both lipid extraction and the formation of rigid membrane facets. If these deformation modes play a role, the loss of membrane function and the formation of lipid‐containing protein deposits like Lewy bodies in αS aggregation diseases remains to be established.
Experimental Section
Expression and Purification of αS
The expression and purification of human wild type αS and the cysteine mutant 140C αS were performed as previously described. The expression and purification of human wild type α‐synuclein (αS) and the cysteine mutant 140C αS were performed as previously described.10, 14b The protein concentrations were estimated by measuring the absorbance at 276 nm on a Shimadzu spectrophotometer, using a molar extinction coefficient of 5600 m −1 cm−1 for αS and 5745 m −1 cm−1 for 140C αS.
Fibril Formation
αS amyloid fibrils were prepared using a 100 μm αS solution in 10 mm tris‐Cl, 100 mm NaCl, pH 7.4. To induce aggregation, this solution was incubated at 37 °C and was agitated at 500 rpm for seven days. The fibrils were subsequently dialyzed against an equiosmolar buffer containing 10 mm tris‐Cl, 100 mm NaCl and 100 mm glucose pH 7.4. Small fibril fragments, which we call fibril seeds, were prepared by fragmenting mature fibrils on ice using a tip sonicator (Branson Ultrasonics). ThT assays indicate that fibril seeds produced with this method efficiently speed up αS aggregation by directly enhancing fibril growth.10
Formation of Giant Unilamellar Vesicles
1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho (1’‐rac‐glycerol) (sodium salt) (POPG) and 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl) (ammonium salt) (Liss Rhod PE) lipids were purchased from Avanti Polar Lipids and were used without any further purification. Lipid mixtures were prepared from 10 mg mL−1 stock solutions of POPC and POPG in chloroform. To allow visualization of the vesicles using fluorescence microscopy 0.05 % of Lissamine Rhodamine PE was included in the mixtures. Biotinylated lipids were incorporated to immobilize vesicles on streptavidin‐coated glass slides.
GUVs were prepared by electro‐formation using a custom‐built platinum (Pt) wire electrode with a diameter of 0.762 mm in a 1 mL cuvette. The lipids suspended in chloroform were deposited on the wire drop by drop, and the solvent was evaporated using a nitrogen stream. After deposition of the lipids on the electrodes, they were transferred to the cuvette filled with 700 μl of electro‐swelling buffer containing 100 mm KCl, 100 mm sucrose and 10 mm tris‐Cl and connected to a frequency generator (PCGUI 1000 Velleman). The electro‐swelling was done at 500 Hz, and during detachment of vesicles the frequency was gradually decreased from 500 to 50 Hz.21 For all experimental conditions, the electro‐swelling was done at room temperature using the same frequency generator settings.
Interaction of αS Monomers and Fibrils with GUVs
To study the interaction of αS monomers with GUVs, POPC/POPG and POPC GUVs doped with Rh‐DOPE were incubated with a mixture of 1 mol % Alexa‐fluorophore‐labelled αS 140C and 99 mol % wt αS monomers at a total protein concentration of 100 μm. The experiments with αS monomers were performed at room temperature in quiescent conditions. To study the binding of αS fibrils to membranes, GUVs composed of POPC/POPG and POPC phospholipids were immobilized on streptavidin‐coated glass slides and incubated with 10 μm pre‐formed αS amyloid fibrils stained with 10 μm thioflavinT (ThT). The streptavidin‐coated slides were prepared by incubating standard glass slides in a 100 μg mL−1 streptavidin in PBS buffer for 15 minutes. After this incubation step, the slides were rinsed with the appropriate buffer used in the experiments. The samples were imaged using a 63× Plan‐Apochromat 1.4 NA oil immersion objective on a Zeiss LSM510 microscope.
Kymographs
To follow the thermal fluctuations of fibrils attached to GUVs in time, a kymograph was prepared. For this purpose, the red and green channels of the images were converted to 8‐bit grey level. Using the Image J analysis module Template Matching all frames were aligned so that, in time, the center of the vesicle remained at the same location. The fluorescence intensity was traced in time along the horizontal lines shown in Figure 3 (A and B). In this way kymographs were constructed at 11 different locations, starting from the center of the vesicles, passing through the contact point of fibrils with vesicles, up to the end of the fibril. The width of the thermal fluctuations w was determined by measuring the maximum distance travelled by the fluorescently labelled filament on the timescale of the experiment.
Deformation of Giant Unilamellar Vesicles
To investigate the effect of fibril growth on the GUVs, we incubated 5–50 μm αS seeds (equivalent monomer concentration) with 95–50 μm αS monomers in the presence of immobilized POPC/POPG (1:1) GUVs and followed the sample for up to four hours. The vesicles were incubated at room temperature in quiescent conditions and the fibrils were visualized by the addition of 10 μm ThT. For end‐point measurement, the monomeric αS 140C was labelled with AlexaFluor647. In another set of experiments, 500 μm of calcein was incubated with the GUVs to study the integrity of the vesicle membrane in the presence of 20 μm αS seeds and 80 μm αS monomers. The change in morphology of vesicles was visualized in time using a 63× Plan‐Apochromat 1.4 NA oil immersion objective on a Zeiss LSM510 microscope. To characterize the shape change, the sphericity (S) of the deformed vesicles was analysed using the Image J plugin Morphology.22 Here the sphericity is given by the ratio between the radii of the inscribed and enclosed circles of the cross‐section of the nearly spherical and deformed vesicles.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Kirsten A. van Leijenhorst‐Groener for the production of the recombinant αS protein. This project is financially supported by a VIDI grant (700.59.423 H.C. and M.M.A.E.C.) from the Netherlands Organization for Scientific Research (NWO)
H. Chaudhary, V. Subramaniam, M. M. A. E. Claessens, ChemPhysChem 2017, 18, 1620.
References
- 1.
- 1a. Auluck P. K., Caraveo G., Lindquist S., Ann. Rev. Cell Dev. Biol. 2010, 26, 211–233; [DOI] [PubMed] [Google Scholar]
- 1b. Snead D., Eliezer D., Exp. neurobiol. 2014, 23, 292–313; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Fakhree M. A., Zijlstra N., Raiss C. C., Siero C. J., Grabmayr H., Bausch A. R., Blum C., Claessens M. M., Sci. Rep. 2016, 6, 30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Ferreon A. C., Gambin Y., Lemke E. A., Deniz A. A., Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Georgieva E. R., Ramlall T. F., Borbat P. P., Freed J. H., Eliezer D., J. Am. Chem. Soc. 2008, 130, 12856–12857; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Jao C. C., Hegde B. G., Chen J., Haworth I. S., Langen R., Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Trexler A. J., Rhoades E., Biochemistry 2009, 48, 2304–2306; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Mizuno N., Varkey J., Kegulian N. C., Hegde B. G., Cheng N., Langen R., Steven A. C., J. Biol. Chem. 2012, 287, 29301–29311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braun A. R., Lacy M. M., Ducas V. C., Rhoades E., Sachs J. N., J. Am. Chem. Soc. 2014, 136, 9962–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang Z., de Messieres M., Lee J. C., J. Am. Chem. Soc. 2013, 135, 15970–15973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engel M. F., Khemtemourian L., Kleijer C. C., Meeldijk H. J., Jacobs J., Verkleij A. J., de Kruijff B., Killian J. A., Hoppener J. W., Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milanesi L., Sheynis T., Xue W. F., Orlova E. V., Hellewell A. L., Jelinek R., Hewitt E. W., Radford S. E., Saibil H. R., Proc. Natl. Acad. Sci. USA 2012, 109, 20455–20460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Zhao H., Jutila A., Nurminen T., Wickstrom S. A., Keski-Oja J., Kinnunen P. K., Biochemistry 2005, 44, 2857–2863; [DOI] [PubMed] [Google Scholar]
- 7b. Gorbenko G. P., Ioffe V. M., Kinnunen P. K., Biophys. J. 2007, 93, 140–153; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Zhao H., Tuominen E. K., Kinnunen P. K., Biochemistry 2004, 43, 10302–10307. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Reynolds N. P., Soragni A., Rabe M., Verdes D., Liverani E., Handschin S., Riek R., Seeger S., J. Am. Chem. Soc. 2011, 133, 19366–19375; [DOI] [PubMed] [Google Scholar]
- 8b. Iyer A., Petersen N. O., Claessens M. M., Subramaniam V., Biophys. J. 2014, 106, 2585–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hellstrand E., Nowacka A., Topgaard D., Linse S., Sparr E., PloS one 2013, 8, e77235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chaudhary H., Stefanovic A. N., Subramaniam V., Claessens M. M., FEBS Lett. 2014, 588, 4457–4463. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Zhu M., Li J., Fink A. L., J. Biol. Chem. 2003, 278, 40186–40197; [DOI] [PubMed] [Google Scholar]
- 11b. Lorenzen N., Lemminger L., Pedersen J. N., Nielsen S. B., Otzen D. E., FEBS Lett. 2014, 588, 497–502. [DOI] [PubMed] [Google Scholar]
- 12. Zarbiv Y., Simhi-Haham D., Israeli E., Elhadi S. A., Grigoletto J., Sharon R., Neurobiol. Dis. 2014, 70, 90–98. [DOI] [PubMed] [Google Scholar]
- 13. Semerdzhiev S. A., Dekker D. R., Subramaniam V., Claessens M. M., ACS Nano 2014, 8, 5543–5551. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Sidhu A., Segers-Nolten I., Subramaniam V., Biochim. Biophys. Acta 2014, 1844, 2127–2134; [DOI] [PubMed] [Google Scholar]
- 14b. van Raaij M. E., Segers-Nolten I. M., Subramaniam V., Biophys. J. 2006, 91, L96–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Evans E., Needham D., J Phys Chem. 1987, 91, 4219–4228. [Google Scholar]
- 16.
- 16a. Olbrich K., Rawicz W., Needham D., Evans E., Biophys. J. 2000, 79, 321–327; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Rawicz W., Smith B. A., McIntosh T. J., Simon S. A., Evans E., Biophys. J. 2008, 94, 4725–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andersson M., Hammarstrom L., Edwards K., J Phys Chem. 1995, 99, 14531–14538. [Google Scholar]
- 18. Dubois M., Lizunov V., Meister A., Gulik-Krzywicki T., Verbavatz J. M., Perez E., Zimmerberg J., Zemb T., Proc. Natl. Acad. Sci. USA 2004, 101, 15082–15087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Varkey J., Isas J. M., Mizuno N., Jensen M. B., Bhatia V. K., Jao C. C., Petrlova J., Voss J. C., Stamou D. G., Steven A. C., Langen R., J. Biol. Chem. 2010, 285, 32486–32493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sparr E., Engel M. F., Sakharov D. V., Sprong M., Jacobs J., de Kruijff B., Hoppener J. W., Killian J. A., FEBS Lett. 2004, 577, 117–120. [DOI] [PubMed] [Google Scholar]
- 21. Pott T., Bouvrais H., Meleard P., Chem. Phys. Lipids 2008, 154, 115–119. [DOI] [PubMed] [Google Scholar]
- 22.G. Landini, Proceedings of the Second ImageJ User and Developer Conference, Luxembourg 2008, p116–121.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary