

Abstract
Adult bone mass is maintained through a balance of the activities of osteoblasts and osteoclasts. Although Notch signaling has been shown to maintain bone homeostasis by controlling the commitment, differentiation, and function of cells in both the osteoblast and osteoclast lineages, the precise mechanisms by which Notch performs such diverse and complex roles in bone physiology remain unclear. By using a transgenic approach that modified the expression of delta‐like 1 (DLL1) or Jagged1 (JAG1) in an osteoblast‐specific manner, we investigated the ligand‐specific effects of Notch signaling in bone homeostasis. This study demonstrated for the first time that the proper regulation of DLL1 expression, but not JAG1 expression, in osteoblasts is essential for the maintenance of bone remodeling. DLL1‐induced Notch signaling was responsible for the expansion of the bone‐forming cell pool by promoting the proliferation of committed but immature osteoblasts. However, DLL1‐Notch signaling inhibited further differentiation of the expanded osteoblasts to become fully matured functional osteoblasts, thereby substantially decreasing bone formation. Osteoblast‐specific expression of DLL1 did not alter the intrinsic differentiation ability of cells of the osteoclast lineage. However, maturational arrest of osteoblasts caused by the DLL1 transgene impaired the maturation and function of osteoclasts due to a failed osteoblast‐osteoclast coupling, resulting in severe suppression of bone metabolic turnover. Taken together, DLL1‐mediated Notch signaling is critical for proper bone remodeling as it regulates the differentiation and function of both osteoblasts and osteoclasts. Our study elucidates the importance of ligand‐specific activation of Notch signaling in the maintenance of bone homeostasis. J. Cell. Physiol. 232: 2569–2580, 2017. © 2016 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals Inc.
Integrity of the mammalian skeleton is maintained through two distinct mechanisms. Bone modeling during fetal and early postnatal life, in which osteoblasts and osteoclasts act independently in response to physiological stress, determines the shape and size of bone. In bone remodeling that occurs throughout life, old bone is removed by osteoclasts and then replaced with bone newly formed by osteoblasts, thereby maintaining the proper bone mass. An imbalance in this coupled activity of osteoclasts and osteoblasts leads to pathologic conditions, such as osteosclerosis, osteopetrosis, and osteoporosis (Feng and McDonald, 2011).
Notch signaling is an evolutionarily conserved signaling pathway and plays a central role in cell fate decisions and maintenance of tissue homeostasis in various organs during embryonic development as well as in postnatal life. Although the importance of Notch signaling in bone homeostasis has long been suggested through a number of in vitro studies (Tezuka et al., 2002; Sciaudone et al., 2003; Yamada et al., 2003; Deregowski et al., 2006; Vujovic et al., 2007; Sekine et al., 2012), Notch signal modifications in target cells have often produced conflicting results regarding the differentiation and/or function of the respective cell types. Recently, a series of in vivo murine genetic studies, in which Notch signaling was specifically activated or abolished at the various and specific stages of osteoblast lineage development, confirmed that Notch signaling regulates skeletal development as well as adult bone mass (Engin and Lee, 2010; Tao et al., 2010; Chen et al., 2014). These studies established the idea that the effects of Notch signaling in bone physiology are cell‐context dependent, meaning that Notch exerts different effects depending on the differentiation status of target cells. However, how Notch carries out such diverse roles in bone cells remains elusive. Notch can be activated only through cell–cell contact, and in theory, binding of Notch to any one of the at least four Notch ligands, namely Jagged1, Jagged2, delta‐like ligand 1 (DLL1), and DLL4, can trigger the Notch signaling cascade. Nevertheless, whether any or all of the Notch ligands can elicit identical cellular events in bone cells has not yet been investigated. Since Jagged1 and DLL1 had been shown to be upregulated at the site of bone regeneration (Nobta et al., 2005), we decided to investigate how these two Notch ligands participate in bone homeostasis. To assess the roles of Jagged1 and DLL1 in bone metabolism in vivo, we utilized two transgenic mouse strains expressing either human Jagged1 (JAG1) or DLL1 under the control of the osteoblast‐specific col1a1 promoter. It has been shown that while JAG1–expression in osteoblasts resulted in slightly decreased trabecular bone mass without altering total bone volume (Negishi et al., 2014), DLL1‐expressing transgenic mice demonstrated abnormally dense bone (Ito et al., 2012), indicating that JAG1 and DLL1 induce distinct cellular responses in bone cells. Since DLL1‐expressing mice recapitulated the phenotypes of osteoblast‐specific Notch gain‐of‐function mice (Engin et al., 2008), we sought to determine the roles of DLL1‐mediated Notch signaling activation in the maintenance of bone homeostasis.
Here, we report that DLL1‐mediated Notch signaling regulates the differentiation and function of both osteoblast and osteoclast lineage cells during bone remodeling. A primary role of DLL1‐mediated Notch signaling is to induce proliferation of osteoblast progenitors to expand the pool of bone‐forming cells. In addition, DLL1‐Notch signaling regulates osteoblast–osteoclast coupling by controlling the differentiation of osteoblast progenitors into mature osteoblast/osteocytes, which are responsible for local production of RANK ligand (RANKL) and osteoprotegerin (OPG), thus maintaining the integrity of bone.
Materials and Methods
Mice
Transgenic mice that express human DLL1 or JAG1 under the control of a 2.3‐kb osteoblast‐specific promoter region of mouse Col1a1 promoter were generated initially on a non‐obese diabetic/severe combined immunodeficient/IL2Rγnull (NOG) background. More than three founder mice were obtained in each transgenic line. Each founder mouse within the same transgenic line demonstrated a similar bone phenotype (Ito et al., 2012; Negishi et al., 2014). The NOG‐DLL1‐Tg and NOG‐JAG1‐Tg mice were backcrossed with C57BL/6 mice more than ten times before being used for this study. Conditional knockout mice for the DLL1 gene were created by crossing the DLL1‐floxed mice with the RosaCreERT2 mice (Seibler et al., 2003; Hozumi et al., 2004). The DLL1 gene was removed by treating mice with tamoxifen (2 mg/20 g) for 4 consecutive days. Mice, between E15.5 and 8 weeks old, were used at the time points specified in individual experiments. Both male and female mice were used. All experiments were performed using age‐ and sex‐matched littermates. Mice were maintained in the animal facility of the Tokai University School of Medicine under specific‐pathogen‐free conditions with ad libitum access to sterilized food and water. All animal experiments were approved by the Animal Care Committee of Tokai University.
Bone histomorphometric analysis and microcomputed tomography analysis
Both static and dynamic histomorphometry and CT scanning were conducted independently at the Kureha Special Laboratory (Fukushima, Japan). Briefly, femurs and tibiae were fixed and dehydrated in ethanol. Femurs were subjected to CT scanning using Scan Xmate‐A09S (Comscantechno, Kanagawa, Japan). Three‐dimensional microstructural image data were reconstructed, and structural indices were calculated using 3D‐BON software (Ratoc Systems Inc, Tokyo, Japan). Dehydrated and degreased tibiae were embedded undecalcified in glycol methacrylate acrylic resin, sliced to 3 μm in thickness, and stained with Toluidine blue. Bone formation and resorption parameters were measured in a defined area of secondary spongiosa between 600 and 2100 μm from the growth plate using an OsteoPlan II morphometry system (Carl Zeiss, Thornwood, NY). Double labeling was performed via intraperitoneal calcein (Nacalai Tesque, Kyoto, Japan) injection twice at an interval of 3 days. Mice were sacrificed one day after the last injection. The terminology and units recommended by the American Society for Bone and Mineral research were used in this study (Parfitt et al., 1987).
Skeletal preparation, histology, immunohistochemistry, and image analysis
Whole‐mount skeletal preparations were established using a standard protocol described elsewhere. For histology, bones were fixed overnight in 4% paraformaldehyde at 4°C and decalcified in 0.2 M EDTA for 1–4 weeks depending on the age of the animals. Bones were embedded in paraffin, and 3 μm longitudinal sections were obtained. For immunohistochemical staining, sections were dewaxed, rehydrated, and subjected to an antigen‐retrieval procedure. Endogenous peroxidase and nonspecific binding of antibody were blocked by treating sections with 0.3% H2O2 in methanol and 5% normal sera, respectively. Sections were incubated overnight at 4°C with the antibodies listed below. The specific binding of primary antibodies was detected using a standard avidin‐biotin peroxidase method (Vector Laboratories, Burlingame, CA), the universal immunoenzyme polymer method (Nichirei, Tokyo, Japan), or a tyramid signal amplification system (DakoCytomation, Denmark), followed by visualization with DAB. In some experiments, sections were stained for tartrate‐resistant acid phosphatase (TRAP) to identify osteoclasts. The number of osterix‐positive osteoblasts or TRAP‐positive osteoclasts in the secondary spongiosa was obtained by using Image J software (http://rsbweb.nih.gov/ij/).
Antibodies
For immunostaining, the following antibodies were used at the indicated concentrations: anti‐DLL1 (1:50, goat polyclonal, Santa Cruz Biotechnology, Dallas, TX), anti‐Jagged1 (1:100, goat polyclonal, Santa Cruz Biotechnology), anti‐DLL4 (1:50, rabbit polyclonal, BioRad, Hercules, CA; Novus Biologicals, Littleton, CO), anti‐Hes1 (1:250, rabbit polyclonal, Abcam, Cambridge, UK), anti‐osterix (1:5000, rabbit polyclonal, Abcam), anti‐RANK ligand (RANKL) (1:100, goat polyclonal, Santa Cruz), anti‐Runx2 (1:100, mouse monoclonal, clone 8G5, MBL, Nagoya, Japan), anti‐CD31 (1:10, rat monoclonal, clone MEC13.3, BD Biosciences, San Jose, CA).
Preparation of BM cells
For in vitro culture of osteoblasts and osteoclasts, bone marrow (BM) cell fractions were prepared as described previously with slight modifications (Morikawa et al., 2009). Briefly, femurs, tibiae, and humeri were dissected and ground in 1 mL phosphate‐buffered saline (PBS) using a mortar and pestle. PBS that contained BM cells, including both hematopoietic and non‐hematopoietic cells, was transferred to a small tube. After centrifugation, cell‐free PBS, hereafter called BM liquid, was collected and saved for protein quantification. Following treatment with 0.15 M NH4Cl cell lysis buffer, cells, hereafter called BM cells, were resuspended in α‐MEM containing 10% FBS (10%MEM) and used for osteoclast culture. Bone fragments were digested with 0.2% collagenase (Wako Chemicals, Osaka, Japan) for 1 h at 37°C. Liquid cell suspension was passed through a 0.45 μm filter, centrifuged, treated with 1 ml of sterile distilled water to remove hematopoietic cells, and washed with PBS containing 2% FBS. The remaining cells, hereafter called mesenchymal stem cell (MSC)‐enriched BM cells, were used for osteoblast culture.
In vitro osteoblast culture
MSC‐enriched BM cells were plated at a density of 5 × 104 cells/cm2 in 10%MEM. Cells were harvested at several time points for gene expression analyses, alkaline phosphatase (ALP) staining, and ALP activity analyses. ALP enzyme activity was measured using the LabAssay ALP kit (Wako chemical).
In vitro osteoclast culture
BM cells plated at a density of 5 × 104 cells/cm2 were cultured with 20 ng/ml each of macrophage‐colony stimulating factor (M‐CSF) and RANKL (both from R&D systems, Minneapolis, MN) for 6–7 days. For mixed culture, BM cells were plated at a density of 3 × 106 cells/cm2 and stimulated with 10−7 M dexamethasone (Dex) and 10−8 M 1α,25‐dihydroxhy vitamin D3 (VD3) for 11–13 days. Culture supernatant of this mixed culture was saved for cytokine quantification analyses. Co‐culture experiments were conducted as described previously (Xing et al., 2002) with slight modifications. Briefly, primary osteoblasts were isolated from calvariae of 4‐ to 6‐day‐old DLL1 and non‐transgenic littermates using a sequential collagenase/dispase digestion, propagated, and plated on to 24‐well tissue culture plates (2.5 × 104 cells/well). BM cells obtained as described above were cultured overnight in 10%MEM containing 10 ng/ml M‐CSF. Non‐adherent BM cells (2 × 105 cells/well) were seeded onto a monolayer of primary osteoblasts and cultured for 9–13 days in the presence of 10−7 M Dex and 10−8 M VD3. At the end of the culture, wells were fixed with 3.7% formaldehyde/PBS and stained for TRAP. TRAP‐positive cells containing 3 or more nuclei were designated as osteoclasts and counted under the microscope.
RNA purification and gene expression analysis
Total RNA was purified from cultured cells and calvariae using Isogen II solution (Nippon Gene Co, Toyama, Japan) according to the manufacturer's instructions and was reverse transcribed into cDNA. Expression levels of osteoblast differentiation markers, Notch signaling molecules, and osteoclast differentiation markers were quantified using appropriate Taqman® probes, listed below (Applied Biosystems, Foster City, CA). Jagged2: Mm01325629_m1, Hes1: Mm01342805_m1, Hey1: Mm00468865_m1, Col1a1: mm0080166_g1, ALP: Mm00475834_m1, osteocalcin (bglap3): Mm03413826_m1, Runx2: Mm00501580_m1, osterix (sp7): Mm04209856_m1, RANKL (tnfsf11): Mm00441906_m1, Sema3b: Mm00436477_m1.
Enzyme‐linked immunosorbent assay (ELISA)
The concentration of RANKL, osteoprotegerin (OPG), M‐CSF in plasma, BM liquid, and culture supernatant was determined using Quantikine kits (R&D systems). The concentrations of carboxylated osteocalcin (Gla‐osteocalcin), an indicator of bone formation, and undercarboxylated osteocalcin (Glu‐osteocalcin), an indicator of bone resorption, were determined using High Sensitive EIA kits (Takara, Shiga, Japan).
Statistics
Data were analyzed using GraphPad Prism, version 5.0 (GraphPad Software). Student's two‐tailed unpaired (or paired, when applicable) t‐test was used to determine the significance of the difference between the means of two groups. A normal distribution of the data was confirmed using the Kolmogorov‐Smirnov test. The mean ± SD is presented in each graph. P values <0.05 were considered significant. NS stands for not significant.
Results
Phenotypes of Notch ligand transgenic mice
In our attempt to determine the ligand‐specific effects of Notch signaling in the physiology of bone in normal immune environment, previously described NOG‐DLL1‐Tg and NOG‐JAG1‐Tg mice, in which human DLL1 or JAG1 was expressed under the control of the osteoblast‐specific 2.3 kb col1a1 promoter on NOG background, were backcrossed with C57BL/6 mice. In the bone marrow (BM) of both non‐transgenic mice and DLL1‐C57BL/6 transgenic mice, hereafter called DLL1 mice, endogenous murine Jagged1 and DLL4 were prominently expressed both the bone lining and the matrix embedded osteoblasts. Meanwhile, endogenous expression of DLL1 was detected only weakly in osteoblasts of non‐transgenic mice. The col1a1‐driven upregulation of DLL1 augmented the expression of DLL1 in the BM stroma as well as in osteoblasts without affecting DLL4 or Jagged1 expression levels (Supplementary Fig. S1a). It did not affect the expression of Jagged2 in calvaria either (Supplementary Fig. S1b). The results indicate the specificity of the transgene expression in our experimental system. DLL1 mice, both male and female, were smaller than their non‐transgenic littermates and exhibited a general feature of osteosclerosis (Fig. 1a and Supplementary Fig. S1c). In analyses of skeletal preparations, both the proximal and distal portions of limbs appeared short and thick, and the calvaria was also short (Fig. 1b and Supplementary Fig. S1d). These osteosclerotic phenotypes were confirmed by histological analyses in 3‐ and 6‐week‐old mice and μCT analysis of 6‐week‐old mice (Fig. 1c and Supplementary Fig. S1e). Unexpectedly, despite their abnormally high bone mass, bone metabolic turnover in DLL1 mice was severely compromised. Quantitative histomorphometry revealed substantial decreases in the mature osteoblast surface area and the bone formation rate as well as in the osteoclast surface area and eroded surface area in both 3‐ and 6‐week‐old DLL1 mice (Fig. 1d and Supplementary Fig. S1f). Consistent with these histological measurements, plasma levels of both carboxylated (Gla‐Ocn) and undercarboxylated (Glu‐Ocn) osteocalcin were markedly decreased (Fig. 1e). Gla‐Ocn has a high affinity for bone minerals and is often considered a biochemical marker for bone formation. On the other hand, the acidic pH in resorption lacunae decarboxylates Gla‐Ocn bound to calcium in the bone; hence, the circulating level of Glu‐Ocn reflects bone‐resorption activity (Karsenty and Ferron, 2012). The significant decreases in these biomarkers confirmed that the osteosclerotic phenotype of DLL1 mice was caused by functional suppression of both osteoblasts and osteoclasts. In contrast to this pathological bone phenotype in DLL1 mice, osteoblast‐specific expression of JAG1 on a C57BL/6 background had a mild effect on bone. The cortical bone of JAG1‐expressing mice was thick but porous, resembling cancellous bone, suggesting an enhancement in bone‐resorption activity (Supplementary Fig. S1g). These results are consistent with our hypothesis that particular Notch ligands are responsible for distinct biological events in bone physiology.
Figure 1.
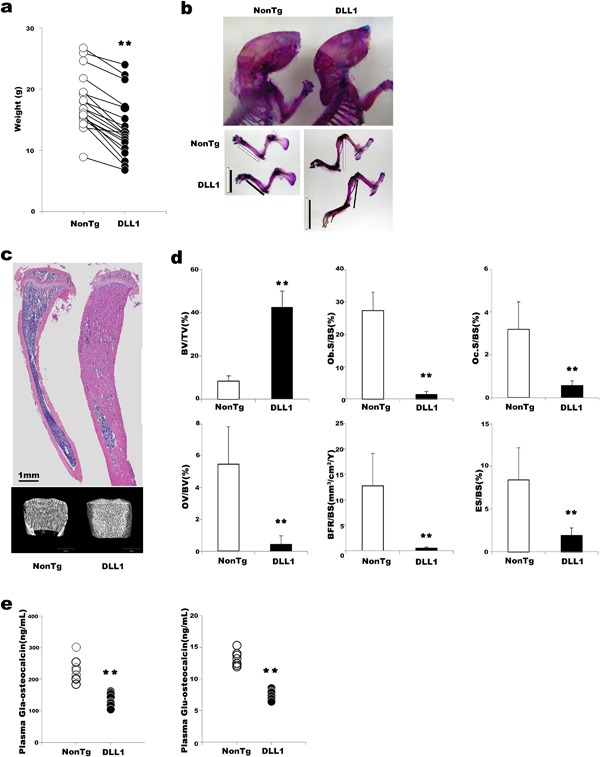
Phenotypes of DLL1 mice (a) Body weights of non‐transgenic and DLL1 littermates at 3–8 weeks of age. Horizontal lines indicate analytical pairs. A total of 19 non‐transgenic and DLL1 littermate pairs were analyzed. (b) Representative pictures of skeletal preparations at 4 weeks of age. Eight non‐transgenic and three DLL1 mice were analyzed. Bars indicate the lengths of the distal portions of limbs. (c) Representative H&E staining of bone section and μCT analysis at 6 weeks of age. Six non‐transgenic and DLL1 littermate pairs were analyzed. At least two slides were stained in each littermate. (d) Static bone morphometric analysis at 6 weeks of age (n = 8). Error bars, mean ± SD; **P < 0.01. (e) ELISA analyses of carboxylated and decarboxylated osteocalcin in plasma of 4‐week‐old mice. n = 8.
Effects of DLL1‐mediated Notch signaling on osteoblast differentiation in vivo
Because DLL1 overexpression produced a stronger effect on the postnatal bone phenotype than did JAG1 overexpression, we evaluated the roles of DLL1‐mediated Notch signal activation in bone physiology. We asked whether DLL1‐mediated Notch signaling functioned in perinatal bone formation. Using routine histological observation, no distinct morphological differences were noted in the endochondral ossification of DLL1 and non‐transgenic littermates at E15.5 (Fig. 2a and Supplementary Fig. S2a). The difference between the two groups first became noticeable at E18.5. Non‐hematopoietic fibroblast‐like cells progressively dominated in the BM of DLL1 mice, and at 1 week after birth, fibroblastic cells with the morphology of early osteoblasts outnumbered hematopoietic cells. This was in stark contrast to the BM of non‐transgenic mice, in which hematopoietic cells migrating through the vascular network proliferated vigorously, making BM a primary site of adult hematopoiesis. Since immunohistochemical staining using a CD31 antibody revealed a similar level of vascular invasion into the BM cavity at the beginning of BM hematopoiesis, the abnormally high content of fibroblast‐like cells in the BM of DLL1 mice was due not to poor migration of hematopoietic cells from other embryonic hematopoietic sites but rather to in situ proliferation of immature osteoblast‐like cells, as confirmed by positive staining with a Ki67 antibody (Fig. 2b). In addition, activation of Notch signaling was detected by Hes1 immunostaining of BM sections (Fig. 2b) as well as by quantitative analyses of Hes1 and Hey1 expression in calvaria and adherent BM cells obtained from DLL1 mice (Fig. 2c). These results indicate that expression of the DLL1 transgene in osteoblasts induces marked proliferation of Notch signal‐activated non‐hematopoietic cells.
Figure 2.
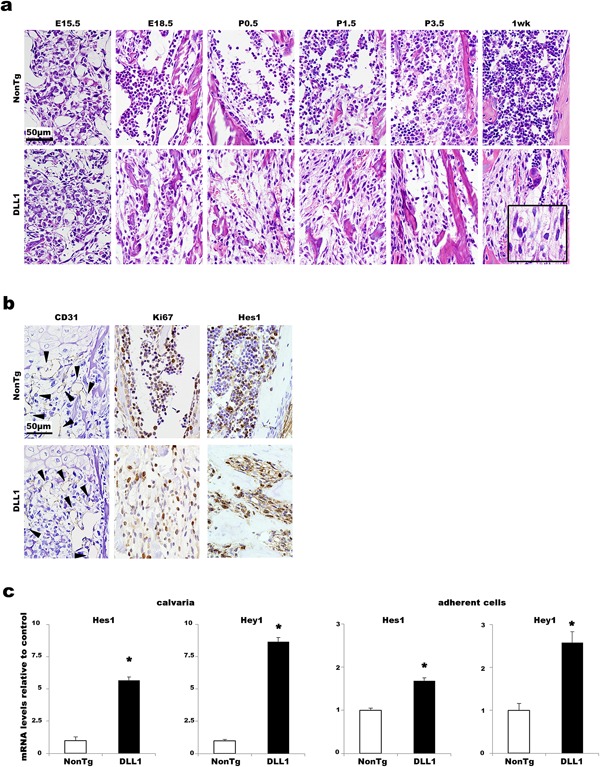
Effects of osteoblast‐specific DLL1 expression in BM histomorphology. (a) Representative H&E staining of hind limbs of non‐transgenic and DLL1 littermates between E15.5 and 1 week. Beginning at E18.5, fibroblast‐like cells are evident in DLL1 specimens. An insert at 1 week of DLL1 image shows higher magnification. Two to six littermate pairs were analyzed at each time point. At least two slides were stained for each sample. (b) Representative images of immunostaining for CD31 at E15.5 and for Ki67 and Hes1 at P5.5. Note that numerous Ki67‐ and Hes1‐positive fibroblastic cells are evident in DLL1 BM, whereas in the non‐transgenic littermate, the majority of Ki67‐ and Hes1‐positive cells appear to be round hematopoietic cells. Arrowheads indicate CD31 positive endothelial cells of vasculature. Two littermate pairs were analyzed at each time point. At least two independent staining procedures were performed for each antibody. (c) Representative quantitative‐PCR analyses for Hes1 and Hey1 expression in calvaria and adherent BM cells. RNA purified from calvaria (n = 7) and adherent BM cells (n = 3) obtained from non‐transgenic and DLL1 littermates was analyzed. All analyses were performed in quadruplicate wells. Gene expression levels are shown relative to non‐transgenic controls. Error bars, mean ± SD; *P < 0.05.
To unequivocally characterize the fibroblast‐like cells proliferating in the BM of DLL1 mice, hind limbs of embryos and neonates were stained for osteoblast differentiation markers. In non‐transgenic mice, Runx2‐ and osterix‐positive osteoblasts were easily detected around primary spongiosa of developing bone at E15.5 and E18.5, but their expressions became restricted in cells on the surface or embedded in the bone at later time points (Fig. 3a). In contrast, those osteoblast marker‐positive cells were ubiquitously distributed in the BM of DLL1 mice throughout the time points analyzed (Fig. 3b). Even at 6 weeks of age, the number of Runx2‐ and osterix‐positive immature osteoblasts per bone surface was significantly higher in DLL1 mice (Fig. 3c and Supplementary Fig. S2b) than in their non‐transgenic littermates. This result did not contradict our earlier histomorphometric analyses, i.e., fewer mature osteoblasts on bone surface and a concomitant decrease in bone formation, but rather indicated perturbations in osteoblast maturation in DLL1 mice. Consistently, osteoid thickness and cortical bone mineral density, key parameters for matrix production and mineralization, respectively, were both markedly decreased in DLL1 mice at 3 and 6 weeks (Fig. 3d and Supplementary Fig. S2c), another indication of reductions in functional osteoblasts. These results indicate that DLL1‐Notch signaling plays diverse roles in osteoblast development.
Figure 3.
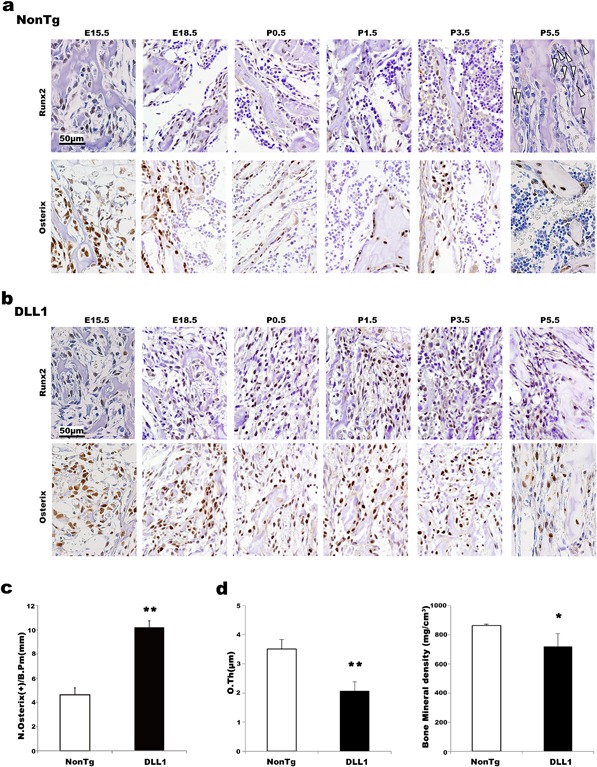
Effects of osteoblast‐specific DLL1 expression in osteoblast differentiation in vivo. Immunostaining for Runx2 and osterix. Representative images of BM samples from non‐transgenic (a) and DLL1 mice (b) are shown. Arrowheads at P5.5 of non‐transgenic specimens indicate Runx2‐positive cells. Two to three littermate pairs were analyzed at each time point. At least two independent staining procedures were performed for each sample. (c) Quantification of osterix‐positive cells in bone surface of the 6‐week‐old BM specimen. Osterix‐positive cells in 20 random fields were counted for each mouse. Pooled data obtained from three independent littermate pairs were used for analyses. Error bars, mean ± SD; **P < 0.01. (d) Osteoid thickness and mineral density of non‐transgenic and DLL1 mice at 6 weeks of age (n = 8). Error bars, mean ± SD; *P < 0.05, **P < 0.01.
Roles of DLL1 mediated Notch signal activation in osteoblast development in vitro
To determine the endogenous function of DLL1‐mediated Notch signaling in the physiology of osteoblast development, in vitro osteoblast differentiation experiments were conducted without adding any cytokines or osteogenic agents using MSC‐enriched BM cells obtained from DLL1 mice, mice with conditional deletion of the DLL1 gene (Dll1‐floxed), and their respective littermates. In a classical colony forming unit (CFU) assay that identified both fibroblast‐like (CFU‐F) and alkaline phosphatase (ALP)‐expressing osteoblast colonies (CFU‐ALP), DLL1 mice showed approximately 4 times as many osteoblast progenitors as their littermates (Fig. 4a). Consistent with this, mesenchymal stem cell (MSC)‐enriched BM cells obtained from DLL1 mice demonstrated significantly higher expression levels of Collagen 1, a marker for committed osteoblast progenitors, at days 3, 7, and 10 and of Alp, an early osteoblast marker, at days 3 and 7 (Fig. 4b). In contrast, the expression levels of Collagen 1 and Alp, as well as the number of colonies, were all reduced in Dll1‐floxed cultures (Fig. 4c and Supplementary Fig. S3a), indicating the physiological importance of DLL1 during the early phase of osteoblast differentiation. ALP histochemistry and enzyme activity analyses confirmed the gene expression analyses (Supplementary Fig. S3a and b). Conversely, the expression of Osteocalcin, a marker for mature functional osteoblasts, was severely reduced in the DLL1 culture while mildly elevated in Dll1‐floxed culture at days 7 and 10 (Fig. 4b and c). These results indicate that DLL1 expression in osteoblasts must be regulated properly to ensure both the early and late phases of the physiological development of osteoblast
Figure 4.
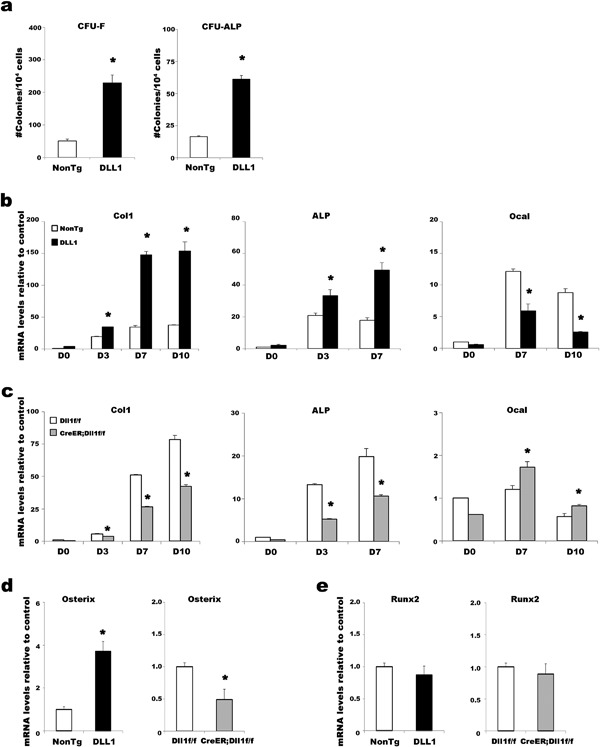
Roles of DLL1 in osteoblast differentiation in vitro. (a) CFU assays for quantifying fibroblast and osteoblast colonies formed from the BM cells of non‐transgenic and DLL1 mice. Representative data from seven independent experiments are shown. All experiments were performed in triplicate wells. Error bars, mean ± SD; *P < 0.05. (b and c) Time course analyses of the expression levels of osteoblast markers during cultures of DLL1 and Dll1‐floxed mice, as indicated. Col1: Collagen 1, ALP: Alkaline phosphatase, Ocal: Osteocalcin. Osterix (d) and Runx2 (e) expression in MSC‐enriched BM cells. In (b–e), representative data from three independent experiments are shown. All analyses were performed in triplicate wells. Bars in each graph represent relative levels of mRNA compared to non‐transgenic controls. Error bars, mean ± SD; *P < 0.05.
To further delineate the mechanisms by which DLL1‐mediated Notch signaling regulates osteoblast differentiation and maturation, expression levels of transcriptional regulators were analyzed. The transcription factors Runx2 and osterix govern the commitment to osteoblast‐lineage cells from MSCs, subsequent differentiation, and further maturation into functional osteoblasts. At the beginning of culture, the expression of Osterix was more than 3‐fold higher in DLL1 culture than in the controls, whereas Osterix expression was decreased by half in Dll1‐floxed culture compared to the controls (Fig 4d), implicating the involvement of DLL‐Notch signaling in osterix‐dependent proliferation of osteoblast progenitors. On the contrary, no significant changes were detected in Runx2 transcription in either DLL1 or Dll1‐floxed mice compared with their respective littermates, implying that DLL1‐mediated Notch signaling was not involved in a Runx2 transcription‐dependent process of osteoblast commitment from MSCs. These results delineated the mechanisms underlying the pathological bone phenotype seen in DLL1 mice at the cellular and molecular level: transgenic expression of DLL1 drove the proliferation of early osteoblast cells but caused a maturational arrest of those cells.
Effects of DLL1‐mediated Notch signal activation on osteoclast differentiation in vivo and in vitro
We have thus far determined the cause of the suppression of bone formation as a maturational arrest in osteoblast differentiation. In addition, severely compromised bone turnover was a distinct feature of our DLL1 mice. To gain further insight into the processes suppressing bone‐resorption activity in DLL1 mice, we examined the differentiation of cells in the osteoclast lineage. Until late stages of embryonic development (E15.5 and E18.5), there were no noticeable changes in the number or morphology of osteoclasts: Relatively small tartrate‐resistant acid phosphatase (TRAP)‐positive osteoclasts were observed in similar numbers in the BM of DLL1 mice and non‐transgenic littermates. However, the difference between the two groups became apparent immediately after birth (Fig. 5a). As early as P0.5, multinucleated large osteoclasts covering the bone surface formed resorption lacunae, an indicator of active osteoclast function, in the BM of non‐transgenic littermates. In contrast, osteoclasts in DLL1 mice were smaller and were not particularly associated with the bone surface. In addition, consistent with the earlier histomorphometric analyses of adult mice, microscopic quantification of TRAP‐stained BM sections showed a marked reduction in the number of osteoclasts per bone surface in DLL1 mice even at 1 week after birth (Fig. 5b) without substantial changes in the total number of TRAP‐positive cells in the observed fields. These results revealed that down‐regulation of bone‐resorption activity began at the beginning of bone remodeling.
Figure 5.
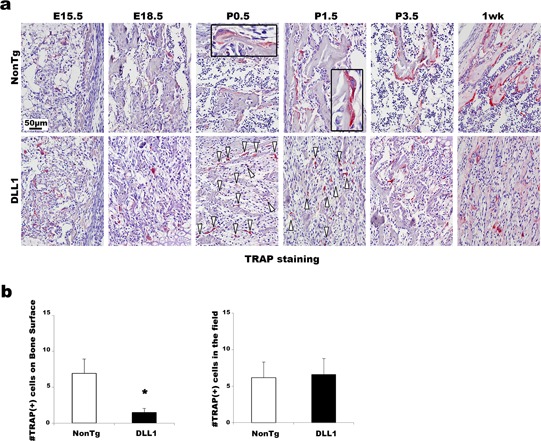
Effects of osteoblast‐specific DLL1 expression in osteoclast differentiation in vivo. (a) Representative TRAP staining pictures of BM specimens from E15.5 to 1 week. Arrowheads in DLL1 images indicate osteoclasts. Inserts in non‐transgenic images are higher magnification. Two littermate pairs were analyzed at each time point. At least two slides were stained for each sample. (b) Microscopic quantification of the number of TRAP(+) cells on the bone surface and in the entire field at 1‐week‐old samples. Seven random fields were counted for each sample. Pooled data from two independent littermate pairs were used for analyses. Error bars, mean ± SD; *P < 0.05.
To identify the cause of defective bone‐resorption activity at the cellular level, we first examined the ability of hematopoietic cells to form osteoclasts in vitro. Stimulation of hematopoietic cells with macrophage colony‐stimulating factor (M‐CSF) and RANK ligand (RANKL) can produce osteoclasts. When BM hematopoietic cells of DLL1 mice and non‐transgenic littermates were cultured in the presence of M‐CSF and RANKL, osteoclasts were formed in similar numbers (Fig. 6a), confirming that the number and the functionality of osteoclast precursors within the hematopoietic population were not altered by osteoblast‐specific activation of DLL1‐mediated Notch signaling. Since osteoblasts are the major source of cytokines regulating osteoclast differentiation during bone remodeling in vivo, we examined osteoclast differentiation in the context of osteoblast‐osteoclast coupling. When whole BM cells were stimulated with 1,25‐dihydroxyvitamin D3 (VD3) and dexamethasone, a condition known to promote osteoclast formation via osteoblast‐derived factors (Muguruma and Lee, 1998; Takahashi et al., 1988a,1988b), considerably fewer osteoclasts were detected in DLL1 cultures (Fig. 6b), indicating alterations in osteoblast–osteoclast coupling. To identify the cause of this defective osteoblast–osteoclast coupling, we conducted co‐culture experiments using calvarial primary osteoblasts and BM hematopoietic cells obtained from DLL1 and non‐transgenic littermates. Consistent with our results described above, there were no differences between DLL1 and non‐transgenic mice in the numbers of osteoclasts formed from BM hematopoietic cells. On the contrary, significantly fewer numbers of osteoclasts were formed when BM hematopoietic cells were cultured with osteoblasts obtained from DLL1 mice (Fig. 6c), confirming that the defective osteoclast differentiation and function observed in DLL1 mice were indeed due to the defect in osteoblasts.
Figure 6.
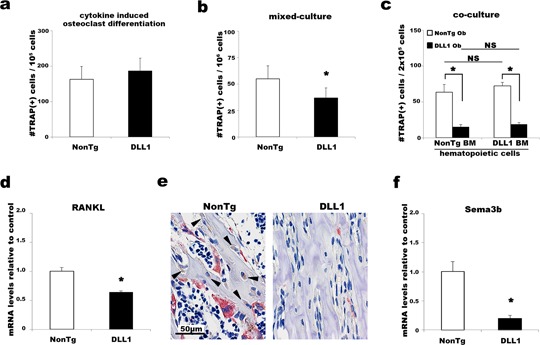
Effects of osteoblast‐specific DLL1 expression in osteoclast differentiation in vitro. (a) The numbers of TRAP(+) osteoclasts formed in cytokine‐induced culture, (b) mixed culture, and (c) osteoblast‐osteoclast co‐culture in vitro. (a and b) Representative data from six independent experiments are shown. (c) Representative data from three independent experiments using calvarial osteoblasts obtained from a total of three non‐transgenic and six DLL1 littermates. Ob, osteoblasts. Experiments were performed at least in triplicate wells. Error bars, mean ± SD; *P < 0.05. Quantitative PCR analyses of Rankl (d) and Semaphorin 3B (f) expression in non‐transgenic and DLL1 osteoclast culture. Representative data from five independent experiments are shown. Analyses were performed at least in triplicate wells. Error bars, mean ± SD; *P < 0.05. (e) Immunostaining of 4‐week‐old mice. BM specimens were stained for RANKL and then for TRAP. Arrowheads indicate RANKL‐positive, bone‐lining, and matrix‐embedded osteoblasts. Four independent littermate pairs at the ages of 2–4 weeks were stained.
To further clarify the roles of DLL1‐mediated Notch signaling in osteoblast–osteoclast coupling, we measured the concentrations of cytokines important for osteoclastogenesis in the plasma, BM liquid, and culture supernatant of DLL1 and non‐transgenic littermates. Although the concentration of M‐CSF in the supernatant did not differ between the DLL1 and non‐transgenic cultures, the DLL1 cultures showed a significant reduction in RANKL production and a slight elevation in osteoprotegerin (OPG), a decoy receptor for RANKL that acts as an inhibitor of osteoclast differentiation, which resulted in a considerably smaller RANKL/OPG ratio, an important determinant of skeletal integrity (Table 1). Reductions in RANKL production were confirmed by quantitative gene expression analyses as well as immunohistochemical staining of BM specimens with RANKL antibody followed by TRAP staining, revealing a lack of RANKL production in osteoblasts/osteocytes and concomitant poor osteoclast differentiation in DLL1 mice (Fig. 6d and e). In contrast, the systemic levels of RANKL, OPG, and M‐CSF did not differ between DLL1 and non‐transgenic littermates (supplementary Fig. S4), highlighting the importance of local, i.e., osteoblast/osteocyte‐specific, production of RANKL in osteoclast differentiation during bone remodeling. Also interesting was a significant decrease in Semaphorin 3B (Sema3B) expression in DLL1 cultures (Fig. 6f). Sema3B is expressed in osteoblasts and induces osteoclastogenesis in response to VD3 stimulation (Sutton et al., 2008). Although, a previous study indicated that transgenic expression of Sema3B did not alter the expression levels of any cytokines critical for osteoclastogenesis, we observed an association of the transgenic expression of DLL1 in osteoblasts with marked down‐regulation of both Sema3B and Rankl transcriptional activity. These results demonstrated that the abnormal osteoclast differentiation and function observed in DLL1 mice were not due to a cell‐autonomous defect in osteoclast lineage but instead to a defective environment that could not properly supply osoteclastogenic cytokines. In other words, a direct effect of transgenic expression of DLL1 in osteoblast was a blockage in osteoblast maturation, which in turn inhibited the differentiation and maturation of osteoclasts, thereby resulting in severe suppression of bone metabolism. Altogether, our results demonstrate the significance of DLL1‐mediated Notch signaling in osteoblast–osteoclast coupling during bone remodeling.
Table 1.
Quantification of cytokines produced in osteoclastogenic culture
| M‐CSF (pg/ml) | RANKL (pg/ml) | OPG (pg/ml) | RANKL/OPG | |
|---|---|---|---|---|
| Non‐transgenic | 45.0 ± 10.5 | 298.3 ± 46.7 | 83.9 ± 6.4 | 3.6 ± 0.5 |
| DLL1 | 42.9 ± 6.4 | 165.2 ± 8.2* | 87.1 ± 3.7 | 1.9 ± 0.01* |
BM cells of non‐transgenic and DLL1 mice were stimulated with Dex and VD3. ELISA analyses were performed on culture supernatant collected at Day 8 of culture. Representative data from three independent experiments are shown. All experiments were performed in at least triplicate wells. Mean ± SD is shown.
*P < 0.05.
Discussion
Although the importance of Notch signaling in bone physiology has been shown by a number of in vivo and in vitro studies, the mechanisms by which Notch signaling regulates bone homeostasis remain elusive. Since Notch regulates many types of cellular events, we hypothesized that the effects of Notch signal activation are determined by the ligands that bind to Notch at particular times and places. In this study, we investigated the effects of DLL1‐induced Notch signaling in the differentiation and function of bone cells.
Mice expressing human DLL1 under the control of a Col1a1 promoter on C57BL/6 background exhibited severe osteosclerosis that was caused by the proliferation of Notch‐activated osterix‐positive immature osteoblasts. Importantly, postnatal deletion of DLL1 resulted in a decrease in the expression of Osterix and the number of osteoblast progenitors, leading to a significant inhibition of osteoblast differentiation, indicating that DLL1‐Notch signaling is indispensable in the osterix‐dependent proliferation of osteoblasts, an event necessary for the expansion of bone‐forming cells. On the contrary, transgenic expression or deletion of DLL1 in osteoblasts did not affect Runx2 transcription in our experiments. Notch has been shown to negatively control two important Runx2‐dependent events in osteoblast development (Rossert et al., 1995; Kalajzic et al., 2002; Hilton et al., 2008; Zanotti et al., 2008). One is the inhibition of osteoblast commitment from MSCs through the downregulation of Runx2 transcriptional activity, which did not appear to involve DLL1‐Notch signaling, suggesting that other Notch ligands are responsible for this process. The other is the negative effect on the differentiation and maturation of immature osteoblasts into functional osteoblasts, plausibly through physical interaction with Runx2. In this study, DLL1 expression in osteoblasts caused substantial decreases in the numbers of mature osteoblasts both in vitro and in vivo, while deletion of DLL1 slightly upregulated the maturation of cells already committed to the osteoblast lineage in vitro. Interestingly, histological analyses detected markedly higher numbers of Runx2‐positive osteoblasts in the BM of DLL1 mice. Since Runx2 expression was unaffected by the DLL1 transgene, the persistence of Runx2‐positive osteoblasts was not due to the increase in transcription but may be due in part to the suppression of Runx2 degradation, as seen in mice lacking the zinc finger adapter protein Schnurri‐3, a protein important for the maintenance of adult bone mass (Jones et al., 2006; Glimcher et al., 2007). As overexpression of Runx2 in osteoblasts has been shown to inhibit osteoblast differentiation at a later stage (Liu et al., 2001), the continuous expression of Runx2 protein is likely to be a cause of the maturational arrest of osteoblasts in our DLL1 mice. Taken together, DLL1‐mediated Notch signaling appeared to act as a regulatory switch for at least two critical points in the osteoblast differentiation: a positive regulator for the expansion of bone‐forming cells and a negative regulator for the functional maturation of osteoblasts.
A profound loss of bone‐resorbing osteoclasts is another pathologic feature of DLL1 bone. DLL1 mice demonstrated severely suppressed bone metabolic turnover, a phenotype quite similar to a dentin matrix protein 1 promoter‐specific gain‐of‐Notch‐function mouse (Dmp‐1 mouse), in which Notch signaling was preferentially activated in osteocytes (Canalis et al., 2013). Unlike the Dmp‐1 mouse, in which a systemic upregulation of OPG production is a main cause of the osteosclerosis, the circulating levels of all of the cytokines important for maintaining bone homeostasis, including OPG, were unchanged in the DLL1 mice. Even in the in vitro experiment that demonstrated a substantial decrease in osteoclast formation, no meaningful change was noted in the OPG level compared to non‐transgenic culture. In contrast, a significant reduction in RANKL production was confirmed at both the gene expression and protein levels in DLL1 osteoblasts/osteocytes. Furthermore, a substantial decrease in Sema3B expression was detected in DLL1 culture. Semaphorins are secreted and membrane‐bound proteins that regulate developmental processes in many organs. As proteins expressed by both osteoblasts and osteoclasts, semaphorins have recently been identified as critical regulators of the coupling process in bone remodeling (Jongbloets and Pasterkamp, 2014; Sims and Martin, 2014). Sema3B is a soluble semaphorin produced by osteoblasts and induces osteoclastogenesis upon VD3 treatment. Transgenic expression of Sema3B was shown to promote osteoclast formation by enhancing the action of RANKL without changing the levels of Rankl transcript (Sutton et al., 2008). Although Sema3B knockout mice have yet to be described, we demonstrated that consistent activation of DLL1‐Notch signaling simultaneously reduced the expression levels of Sema3B and Rankl. Based upon these observations, it is evident that DLL1‐Notch signaling regulates the production of RANKL from matrix‐embedded osteoblasts, a critical determinant of bone remodeling (Nakashima et al., 2011; Xiong et al., 2011), conceivably by controlling the maturation of osteoblasts into RANKL‐producing osteoblasts/osteocytes, or possibly by directly modulating Rankl expression through a Notch‐semaphorin cascade, thus maintaining the proper bone mass.
Our study revealed unique roles of DLL1‐mediated Notch signaling in the process of bone remodeling. DLL1‐Notch signaling directly controls the physiological expansion and differentiation of osteoblasts and indirectly affects the maturation and function of osteoclasts, thereby ensuring the homeostasis of bone mass. Deregulations in Notch signaling have been implicated in the development of bone cell neoplasia (Zhang et al., 2008). Given the importance of DLL1‐Notch signaling in the proliferation and differentiation of osteoblast cells, we plan to investigate the relationship between DLL1 expression and bone neoplasia.
Authors’ Contribution
YM and KH designed the experiments and wrote the paper. YM and TY performed the experiments. HW and TU analyzed the gene expression data. KH and MI created transgenic mice. KA approved data.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Figure S1.
Supporting Figure S2.
Supporting Figure S3.
Supporting Figure S4.
Acknowledgments
We would like to thank Dr. Y. Nakano, Mr. K. Hirano, members of the Support Center for Medical Research and Education, and the Center for Regenerative Medicine at Tokai University for technical assistance.
Conflict of Interest: The authors declare no conflicts of interest regarding this article.
Contributor Information
Yukari Muguruma, Email: mugu@is.icc.u-tokai.ac.jp.
Kiyoshi Ando, Email: andok@keyaki.cc.u-tokai.ac.jp.
Literature Cited
- Canalis E, Parker K, Feng JQ, Zanotti S. 2013. Osteoblast lineage‐specific effects of notch activation in the skeleton. Endocrinology 154:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lee BH, Bae Y. 2014. Notch signaling in skeletal stem cells. Calcif Tissue Int 94:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. 2006. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta‐catenin but not bone morphogenetic protein signaling. J Biol Chem 281:6203–6210. [DOI] [PubMed] [Google Scholar]
- Engin F, Lee B. 2010. NOTCHing the bone: Insights into multi‐functionality. Bone 46:274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang MM, Chen Y, Wang L, Zheng H, Sutton RE, Boyce BF, Lee B. 2008. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med 14:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, McDonald JM. 2011. Disorders of bone remodeling. Annu Rev Pathol 6:121–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH, Jones DC, Wein MN. 2007. Control of postnatal bone mass by the zinc finger adapter protein Schnurri‐3. Ann N Y Acad Sci 1116:174–181. [DOI] [PubMed] [Google Scholar]
- Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, Long F. 2008. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med 14:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozumi K, Negishi N, Suzuki D, Abe N, Sotomaru Y, Tamaoki N, Mailhos C, Ish‐Horowicz D, Habu S, Owen MJ. 2004. Delta‐like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat Immunol 5:638–644. [DOI] [PubMed] [Google Scholar]
- Ito R, Negishi N, Irie N, Matsuo K, Suzuki D, Katano I, Hayakawa E, Kawai K, Kamisako T, Eto T, Ogura T, Hozumi K, Ando K, Aiso S, Tamaoki N, Habu S, Ito M. 2012. Osteosclerosis and inhibition of human hematopoiesis in NOG mice expressing human delta‐like 1 in osteoblasts. Exp Hematol 40:953–963. [DOI] [PubMed] [Google Scholar]
- Jones DC, Wein MN, Oukka M, Hofstaetter JG, Glimcher MJ, Glimcher LH. 2006. Regulation of adult bone mass by the zinc finger adapter protein Schnurri‐3. Science 312:1223–1227. [DOI] [PubMed] [Google Scholar]
- Jongbloets BC, Pasterkamp RJ. 2014. Semaphorin signalling during development. Development 141:3292–3297. [DOI] [PubMed] [Google Scholar]
- Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, Rowe D. 2002. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res 17:15–25. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Ferron M. 2012. The contribution of bone to whole‐organism physiology. Nature 481:314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. 2001. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol 155:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi‐Hara C, Nagoshi N, Sunabori T, Shimmura S, Miyawaki A, Nakagawa T, Suda T, Okano H, Matsuzaki Y. 2009. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 206:2483–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muguruma Y, Lee MY. 1998. Isolation and characterization of murine clonogenic osteoclast progenitors by cell surface phenotype analysis. Blood 91:1272–1279. [PubMed] [Google Scholar]
- Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh‐Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H. 2011. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234. [DOI] [PubMed] [Google Scholar]
- Negishi N, Suzuki D, Ito R, Irie N, Matsuo K, Yahata T, Nagano K, Aoki K, Ohya K, Hozumi K, Ando K, Tamaoki N, Ito M, Habu S. 2014. Effective expansion of engrafted human hematopoietic stem cells in bone marrow of mice expressing human Jagged1. Exp Hematol 42:487–494. [DOI] [PubMed] [Google Scholar]
- Nobta M, Tsukazaki T, Shibata Y, Xin C, Moriishi T, Sakano S, Shindo H, Yamaguchi A. 2005. Critical regulation of bone morphogenetic protein‐induced osteoblastic differentiation by Delta1/Jagged1‐activated Notch1 signaling. J Biol Chem 280:15842–15848. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. 1987. Bone histomorphometry: Standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2:595–610. [DOI] [PubMed] [Google Scholar]
- Rossert J, Eberspaecher H, de Crombrugghe B. 1995. Separate cis‐acting DNA elements of the mouse pro‐alpha 1(I) collagen promoter direct expression of reporter genes to different type I collagen‐producing cells in transgenic mice. J Cell Biol 129:1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaudone M, Gazzerro E, Priest L, Delany AM, Canalis E. 2003. Notch 1 impairs osteoblastic cell differentiation. Endocrinology 144:5631–5639. [DOI] [PubMed] [Google Scholar]
- Seibler J, Zevnik B, Kuter‐Luks B, Andreas S, Kern H, Hennek T, Rode A, Heimann C, Faust N, Kauselmann G, Schoor M, Jaenisch R, Rajewsky K, Kuhn R, Schwenk F. 2003. Rapid generation of inducible mouse mutants. Nucleic Acids Res 31:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine C, Koyanagi A, Koyama N, Hozumi K, Chiba S, Yagita H. 2012. Differential regulation of osteoclastogenesis by Notch2/Delta‐like 1 and Notch1/Jagged1 axes. Arthritis Res Ther 14:R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NA, Martin TJ. 2014. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep 3:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton AL, Zhang X, Dowd DR, Kharode YP, Komm BS, Macdonald PN. 2008. Semaphorin 3B is a 1,25‐dihydroxyvitamin D3‐induced gene in osteoblasts that promotes osteoclastogenesis and induces osteopenia in mice. Mol Endocrinol 22:1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ, Suda T. 1988a. Osteoblastic cells are involved in osteoclast formation. Endocrinology 123:2600–2602. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Yamana H, Yoshiki S, Roodman GD, Mundy GR, Jones SJ, Boyde A, Suda T. 1988b. Osteoclast‐like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology 122:1373–1382. [DOI] [PubMed] [Google Scholar]
- Tao J, Chen S, Lee B. 2010. Alteration of Notch signaling in skeletal development and disease. Ann N Y Acad Sci 1192:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezuka K, Yasuda M, Watanabe N, Morimura N, Kuroda K, Miyatani S, Hozumi N. 2002. Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res 17:231–239. [DOI] [PubMed] [Google Scholar]
- Vujovic S, Henderson SR, Flanagan AM, Clements MO. 2007. Inhibition of gamma‐secretases alters both proliferation and differentiation of mesenchymal stem cells. Cell Prolif 40:185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Bushnell TP, Carlson L, Tai Z, Tondravi M, Siebenlist U, Young F, Boyce BF. 2002. NF‐kappaB p50 and p52 expression is not required for RANK‐expressing osteoclast progenitor formation but is essential for RANK‐ and cytokine‐mediated osteoclastogenesis. J Bone Miner Res 17:1200–1210. [DOI] [PubMed] [Google Scholar]
- Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. 2011. Matrix‐embedded cells control osteoclast formation. Nat Med 17:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yamazaki H, Yamane T, Yoshino M, Okuyama H, Tsuneto M, Kurino T, Hayashi S, Sakano S. 2003. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood 101:2227–2234. [DOI] [PubMed] [Google Scholar]
- Zanotti S, Smerdel‐Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. 2008. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology 149:3890–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Yang Y, Zweidler‐McKay PA, Hughes DP. 2008. Critical role of notch signaling in osteosarcoma invasion and metastasis. Clin Cancer Res 14:2962–2969. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Figure S1.
Supporting Figure S2.
Supporting Figure S3.
Supporting Figure S4.