

Abstract
When pediatric, adolescent, and young adult patients present with a bone sarcoma, treatment decisions, especially after relapse, are complex and require a multidisciplinary approach. This review presents scenarios commonly encountered in the therapy of bone sarcomas with the goal of objectively presenting a consensus, multidisciplinary management approach. Little variation was found in the authors' group with respect to local control or systemic therapy. Clinical trials were universally prioritized in all settings. Decisions regarding relapse therapies in the absence of a clinical trial had very minor variations initially, but a consensus was reached after a literature review and discussion. This review presents a concise document and figures as a starting point for evidence‐based care for patients with these rare diseases. This framework allows prospective decision making and prioritization of clinical trials. It is hoped that this framework will inspire and focus future clinical research and thus lead to new trials to improve efficacy and reduce toxicity. Cancer 2017;123:2206–2218. © 2017 American Cancer Society.
Keywords: adolescent and young adult (AYA), chemotherapy, Ewing sarcoma, osteosarcoma, pathways, pediatric
Short abstract
This review presents a pathway for the management of common clinical scenarios that arise in the treatment of bone sarcomas in children, adolescents, and young adults. Clinical trials should be prioritized when they are available, and for those times when trials are unavailable, a consensus, multidisciplinary management approach to bone sarcomas is presented.
INTRODUCTION
The most common malignancies of bone in the first 3 decades of life are osteosarcoma, Ewing sarcoma, and chondrosarcoma. The treatment of these tumors is complex, and management decisions are best made in a multidisciplinary environment with input from experienced orthopedists, radiotherapists, radiologists, pathologists, and oncologists.
Several factors, including the variability in primary tumor locations, sites and extent of metastatic disease, feasibility of surgical resection, molecular features, growth rate, and histologic variation, complicate care for these relatively rare cancers. Although there are many case reports, retrospective studies, and review articles, there are few prospective, randomized trials beyond first‐line therapy to guide clinical decisions for individual patients with bone sarcoma, and this increases the relative value of expert opinion. There also may be a reluctance from experts to comment on the care of more complicated patients with whom they are not directly involved, and sometimes not all of the relevant patient‐specific details are considered when treatment options are offered. Available therapy guidelines often simply list options and include all reasonable therapy possibilities rather than suggesting a specific course.1 In the relapsed setting, data to inform decisions are even more limited and often come from small, single‐arm clinical trials without a perspective on how treatments should be prioritized or even if they should be considered at all. Finally, although these tumors are often considered pediatric neoplasms, the peak incidence is in teenagers and young adults, who may be managed by physicians less familiar with these rare tumors.
Here we address select scenarios to highlight consensus treatment approaches. We also emphasize the importance of enrolling patients into clinical trials. Because the purpose of this article is to propose a treatment pathway rather than provide a comprehensive review of the field, we apologize in advance to the many authors whose important contributions could not be cited because of word limitations. In addition, we recognize that our recommendations include off‐label uses of many agents, and for that reason, they should be applied with the understanding that they are often based on small published trials and not on the same rigorous level of evidence required for Food and Drug Administration approval. Although variations in recommendations for care certainly exist, we found surprisingly little disagreement among the members of our group. We expect that our pathway will lead to clearer decisions on treatment options for patients and physicians throughout the greater oncology community. We anticipate disagreements with some recommendations and view the resulting discussions as opportunities for clinical investigation. We hope that this approach will help to focus future research on important unsolved management questions, maximize clinical trial enrollment, and minimize off‐label use of ineffective agents.
METHODS
This project began at the February 2016 National Pediatric Cancer Foundation's Sunshine Project retreat when we were discussing appropriate prioritization of phase 1 and 2 clinical trials for relapsed sarcoma patients. We recognized that no comprehensive list of agents and clinical outcome data existed to help drive decision making, and we thus set out to develop a consensus pathway for treating common pediatric, adolescent, and young adult bone sarcomas. We diagramed our opinions into a pathway covering the initial workup, the importance of multidisciplinary therapy, the role of clinical trials, the communication of risk and long‐term toxicities, the approaches to surveillance, and the best established therapies for relapsed disease. Teleconferences were convened biweekly over the course of 6 months.
Question 1: What Is the Appropriate Initial Workup of a Bone Sarcoma?
Most patients with a bone sarcoma present with swelling or pain that waxes and wanes; it classically wakes the patient from sleep at night and occasionally culminates in a pathologic fracture. The initial evaluation (Fig. 1) would typically include plain films revealing an aggressive lesion and prompt a referral to an orthopedic surgeon with malignant bone tumor expertise. Additional imaging with magnetic resonance imaging of the entire bone of concern is necessary to demonstrate the extent and characteristics of the lesion and to evaluate the patient for skip lesions. Next, a core‐needle biopsy or an open biopsy should be considered. For challenging anatomic locations, interventional radiographic techniques may be incorporated. Our consensus is that either technique is acceptable. We typically pursue a core biopsy to minimize patient morbidity, although because of the more limited sampling, this technique occasionally may not provide sufficient material for a diagnosis or research studies. Once the diagnosis is confirmed, the proper staging and treatment can be determined. In addition, consideration at this point should be given to genetic testing or counseling and also to supportive care consultations (eg, for fertility preservation). We acknowledge that the workup is similar to that described in the National Comprehensive Cancer Network guidelines. It differs by providing more detail, omitting lactate dehydrogenase, specifically mentioning the biopsy method, and also not including consideration of a computed tomography scan of the primary tumor site.
Figure 1.
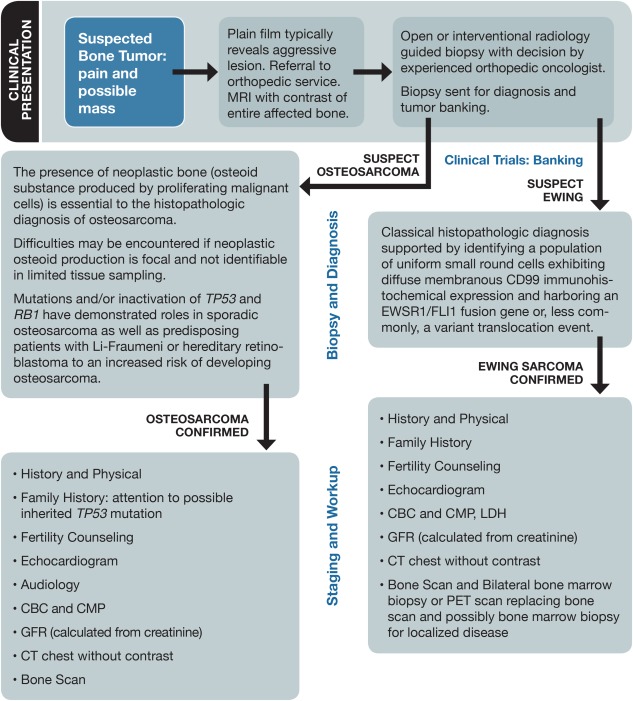
Workup for newly suspected bone sarcoma. CBC indicates complete blood count; CMP, complete metabolic panel; CT, computed tomography; GFR, glomerular filtration rate; LDH, lactate dehydrogenase; MRI, magnetic resonance imaging; PET, positron emission tomography.
Question 2: What Is Needed to Accurately Diagnose and Stage a Newly Diagnosed Osteosarcoma Patient?
Once osteosarcoma is diagnosed, the extent of disease must be determined (Fig. 1). In preparation for potential chemotherapy toxicity, standard baseline organ assessments should be obtained (Fig. 1). Effective communication of the long‐term risks of therapy, including the risks of infertility, cardiotoxicity, and/or second cancers, is needed before the initiation of therapy. Chemotherapy and surgical options, including clinical trial options, should be discussed as part of the informed consent process.
Question 3: What Is the Best Initial Treatment Strategy for a Newly Diagnosed Osteosarcoma Patient?
Osteosarcoma is the most common primary malignant bone tumor in children, adolescents, and young adults.
The current standard of care for localized osteosarcoma is neoadjuvant chemotherapy followed by complete surgical resection of the primary disease and then adjuvant chemotherapy, ideally in a clinical trial when one is available (Fig. 2). We recommend methotrexate, doxorubicin, and cisplatin (MAP) chemotherapy for the first‐line treatment, as reinforced by previous clinical trials.2, 3 Some studies questioned the addition of methotrexate; however, high‐dose methotrexate is now included as standard therapy since subsequent studies have demonstrated a benefit.4 In addition, standard MAP therapy includes a cumulative doxorubicin dose of 450 mg/m2, which is higher than the 300 mg/m2 threshold commonly considered to place patients at risk for long‐term cardiotoxicity. Our consensus regarding cardioprotection is to use dexrazoxane with bolus‐dose doxorubicin on days 1 and 2 when the dose of doxorubicin is in excess of 300 mg/m2. An alternative and acceptable plan is to use dexrazoxane with all doses of doxorubicin, although we recognized in discussion that universal acceptance of this has not yet been achieved among all authors. A continuous infusion of doxorubicin has been advocated by some when dexrazoxane is not being used, although conflicting data regarding the efficacy in preventing cardiotoxicity have led some to not use this approach.5, 6 Although there are no data regarding the optimal timing of local control, most teams recommend two 5‐week cycles of neoadjuvant MAP therapy, which can facilitate limb salvage.7 For curative intent, regardless of location, complete surgical resection of all disease must be the goal. In the appendicular skeleton, limb‐preserving resection and reconstruction with endoprostheses and, in some sites, an allograft or rotationplasty are the standard of care. An amputation may be performed when the limb cannot be preserved or in cases in which the functional outcome may be improved by this technique. The combination of improved imaging modalities, the standard use of neoadjuvant chemotherapy, and the advancement of complex surgical techniques has led to higher limb‐salvage rates.8 Surgical local control for an axial skeleton osteosarcoma is complex, and when the disease is not amenable to surgical resection or the margins are positive, radiation therapy has been shown to improve local control rates.9 Higher total radiation doses or hypofractionation (larger daily doses) may improve local control. Stereotactic radiosurgery may allow shorter courses of treatment while still providing durable local control.10
Figure 2.
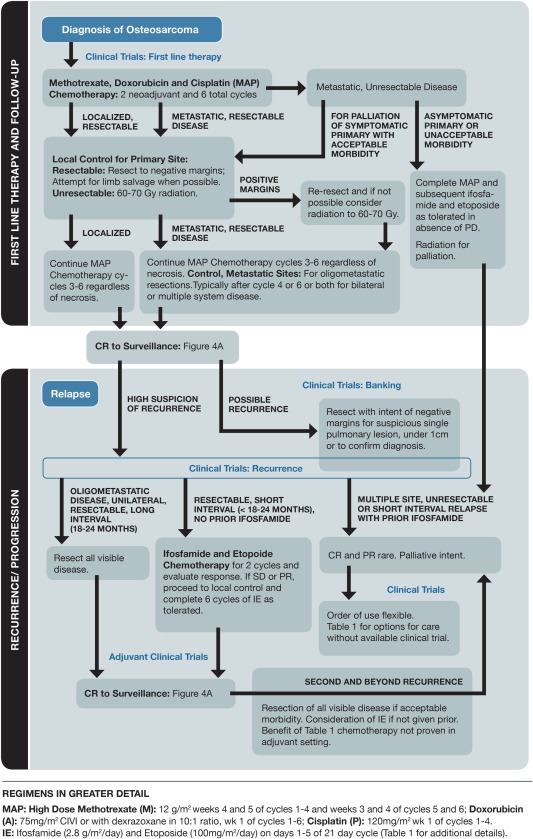
Pathway for treating newly diagnosed and relapsed osteosarcoma. CIVI indicates continuous intravenous infusion; CR, complete response; IE, ifosfamide and etoposide; MAP, methotrexate, doxorubicin, and cisplatin; PD, progressive disease; PR, partial response; SD, stable disease.
Tumor necrosis after neoadjuvant chemotherapy has prognostic significance.11 Although different histopathologic subtypes of high‐grade, intraosseous conventional osteosarcoma have been identified (eg, osteoblastic, chondroblastic, and fibroblastic), no relation between these subtypes and the treatment or prognosis appears to exist.12 Recently, the European and American Osteosarcoma Study (EURAMOS) group completed a large phase 3, randomized study that failed to demonstrate improved event‐free survival by modifying adjuvant chemotherapy for either patients with poor necrosis by the addition of ifosfamide and etoposide (IE) or patients with good necrosis by the addition of pegylated interferon‐α‐2b.13 Therefore, we recommend continuing MAP adjuvant chemotherapy regardless of tumor necrosis. Posttherapy follow‐up is outlined in Figure 4A.
Figure 4.

Recommendations for tumor surveillance after treatment. CBC indicates complete blood count; CMP, complete metabolic panel; CT, computed tomography; CXR PA, chest X‐ray, posterior anterior; PET, positron emission tomography.
Question 4: What Is the Standard Primary Therapy for Osteosarcoma Patients With Equivocal Pulmonary Nodules?
Although most patients present with localized osteosarcoma, up to 30% present with metastatic disease, mostly in the lungs. Equivocal pulmonary lesions are commonly encountered, especially with high‐resolution computed tomography scanning. Following guidelines proposed by EURAMOS, we would consider 1 or more pulmonary/pleural nodules ≥ 1 cm or 3 or more pulmonary nodules ≥ 0.5 cm to be consistent with a diagnosis of metastatic osteosarcoma, whereas smaller lesions are indeterminate without pathologic confirmation. Patients with equivocal lung nodules have 2 treatment options. One option is to recommend wedge resection (with negative margins) at the time of diagnosis. This strategy is preferred to needle biopsy because it will confirm the pathology of the nodule and also is the optimal therapy. The alternative is to observe the nodules and assess the response to chemotherapy. If a pulmonary nodule does not change size after chemotherapy and there is evidence of a treatment effect on the primary tumor, then we believe that the nodule is less likely to be a tumor. Continued close surveillance after adjuvant chemotherapy is critical in these situations. Nodules that decrease in size or mineralize with chemotherapy or that have no alternative etiology (eg, endemic mycosis) should prompt the clinician to recommend surgical resection. The optimal timing for metastatectomy remains unclear. Our recommendation is to proceed with pulmonary metastatectomy after 4 cycles of MAP chemotherapy (followed by 2 more cycles after surgery) or at the end of treatment (Fig. 2).14
Question 5: How Do You Treat Patients With Recurrent Osteosarcoma of the Lung?
For patients who relapse, the first step is to confirm the diagnosis of recurrent disease (Fig. 1). Patients with definitive evidence of a pulmonary recurrence (nodules ≥ 1 cm, multiple nodules ≥ 5 mm) likely do not require a biopsy. However, clinician judgment should dictate whether the identity of a pulmonary nodule is unclear. In cases of smaller nodules, management is typically based on the feasibility of biopsy, and often, further observation is warranted. Although we recognize that additional nodules can develop, allowing time to clarify the etiology is not felt to compromise the quality and goals of therapy. Once recurrence has been confirmed, enrollment in a therapeutic clinical trial is encouraged. Current clinical trials typically include patients with either unresectable disease or adjuvant studies attempting to prevent recurrence after the complete resection of pulmonary disease. However, for those with an early relapse, although we would consider a clinical trial, we would typically recommend IE. We would prioritize a clinical trial for oligometastatic disease in the neoadjuvant setting using a new agent with ifosfamide or IE. Preoperative chemotherapy is typically recommended when there is more than 1 lesion at recurrence, the lesion or lesions are not surgically resectable, or there has been a short time to recurrence. Although data are limited and different time points have been used as the cutoff, significantly worse outcomes have consistently been described for patients who relapse <24 months after their diagnosis.15, 16 The Children's Oncology Group has also assessed survival after the relapse of osteosarcoma and found similarly worse outcomes for those who relapse before 24 months (M. Isakoff, MD, oral communication, October, 2016). For those with relapsed disease within the first 24 months after the diagnosis, our first‐line chemotherapy recommendation is IE. Although an overall survival benefit has not been fully assessed with this regimen, trials using IE have demonstrated response rates between 10% and 33%.17, 18 We usually give 2 neoadjuvant cycles followed by 4 to 6 adjuvant cycles according to the response and tolerability, and we are more likely to recommend adjuvant cycles with favorable radiographic changes and/or histologic demonstration of pathologic necrosis (Fig. 2). However, there are not sufficient data to drive these recommendations, and the adverse effects of this regimen are significant. Thus, the process of informed consent in this scenario is critically important, and patients should be made aware of the risks and potential benefits. After 2 cycles of chemotherapy, surgical resection of all pulmonary nodules is strongly encouraged because it is the only curative option and the main determinant of the outcome.19 For those patients with a resectable relapse more than 24 months after the diagnosis, we typically do not add chemotherapy and proceed with observation alone (Fig. 2). Comparing the goal efficacy threshold for a given clinical trial with an individual patient's recurrence risk along with the expected toxicity of a study regimen would be an important consideration when one is deciding whether or not to recommend adjuvant chemotherapy trials for patients with long‐interval recurrences that have been resected.
Question 6: What Is the Best Management Strategy for Patients With Multiply Recurrent or Unresectable Osteosarcoma?
The treatment of relapsed/refractory bone metastatic osteosarcoma is particularly challenging, and the prognosis is dismal. Multivariate analyses have revealed factors that affect the risk of recurrence, including the size and number of lesions, the time to recurrence, and the site, and these may influence decision making for providers and families. Although there may not be an absolute right answer, there are options based on the balance of toxicity, quality of life, and prognosis. For interested patients with an adequate performance status, we strongly advocate clinical trial enrollment in these situations and even prioritize phase 1 studies over nontrial options (Table 1). The combination of gemcitabine and docetaxel has been used for patients with refractory sarcoma, including osteosarcoma, with partial responses and stable disease seen in approximately 20% of patients with refractory disease with a tolerable adverse‐effect profile.23 Data on sorafenib support its use as well, with progression‐free survival in a heavily pretreated, single‐arm patient cohort that exceeded 40% at 3 months.22 In addition to chemotherapy, palliative radiotherapy may be effective in the treatment of painful lesions. In our discussions, anecdotal variations in the consensus, such as trying therapies with low response rates for patients with an excellent performance status when trials were not available, did emerge.
Table 1.
Relapse Regimens for Osteosarcoma and Ewing Sarcoma
| Agents and Schedule | Primary Toxicities | Cumulative No. | Estimated or Communicated PFS at 4 mo | Median Time to Progression | Combined Response Rate | Partial Response | Complete Response | Toxicitya | |
|---|---|---|---|---|---|---|---|---|---|
| Osteosarcoma | Ifosfamide + etoposide: iv, inpatient, d 1‐5 of 21‐d cycle17, 18, 20, 21 | Myelosuppression (requires myeloid growth factor), neurotoxicity, alopecia | 83 | 80% | N/A | 27% | 25% | <5% without surgery | Very high |
| Sorafenib: po, bid, continuously22 | Rash (hand‐foot skin reaction), hypertension, mucositis | 35 | 46% (mean, 4 mo) | 4 mo | 9% | 9%b | 0 | Low | |
| Gemcitabine + docetaxel: iv, outpatient, d 1 and 8 of 21‐d cycle23, 24, 25, 26, 27 | Myelosuppression (requires myeloid growth factor), cumulative neurotoxicity, allergic reactions, alopecia | 68 | 40% | 4 mo | 16% | 16% | <5% without surgery | High | |
| Ewing sarcoma | Cyclophosphamide + topotecan: iv, outpatient, d 1‐5 of 21‐d cycle28, 29 | Myelosuppression (requires myeloid growth factor), alopecia | 71 | Early progression: < 25% Late progression: > 75% | N/A | 35% | 10% | 25% | Medium |
| Temozolomide + irinotecan ± vincristine: outpatient, po or iv and d 1‐5 for irinotecan, po and d 1‐5 for temozolomide (vincristine on d 1)30, 31 | Diarrhea (treated with loperamide and antibiotic pretreatment) | 102 | 55%‐70% | N/A | 53% | 27% | 26% | Medium | |
| High‐dose ifosfamide: iv, inpatient, d 1‐5 of 21‐d cycle32 | Myelosuppression (requires myeloid growth factor), neurotoxicity, alopecia | 35 | N/A | N/A | 34% | 29% | 5% | Very high | |
| Gemcitabine + docetaxel: iv, outpatient, d 1 and 8 of 21‐d cycle23, 24, 33 | Myelosuppression (requires myeloid growth factor), cumulative neurotoxicity, allergic reaction, alopecia | 24 | >25% | 5 mo | 29% | 25% | <5% without radiation | High | |
| Oral etoposide: po, daily for several weeks on, 1‐ to 2‐wk break as tolerated34 | Myelosuppression, secondary malignancy | 58 | 33% | N/A | 19% | 19% | <5% | Low |
Abbreviations: bid, twice daily; iv, intravenous; N/A, not available; PFS, progression‐free survival; po, by mouth; d, day.
Very high, high, medium, or low.
Response Evaluation Criteria in Solid Tumors.
Question 7: What Is the Role of Molecular Testing and In Vivo Assays and When Is Systemic Therapy No Longer Recommended for a Patient With Bone Sarcoma?
Recent advances in molecular testing have led to the exciting possibility that treatment choices might be directed on the basis of the identification of so‐called actionable mutations in a patient's individual tumor. Pediatric data publicly released by Foundation Medicine35 have revealed a limited number of genomic alterations found in any 1 tumor, and there are no prospective data supporting the idea that choosing a therapy on the basis of this type of analysis improves the response rate or survival. Thus, we would encourage obtaining these data through participation in a clinical trial so that the value of these analyses can be rigorously tested and their effectiveness can be established or refuted conclusively.
Deciding when further disease‐directed therapy does more harm rather than providing a chance for a benefit remains a significant challenge, especially for patients with a good performance status. In these circumstances, molecular testing or testing with a xenograft‐based drug sensitivity assay is especially tempting because of the belief that so‐called targeted therapies are better tolerated than traditional cytotoxic agents. Pursuing these approaches would preferably be done in the context of a clinical trial, but in the absence of a trial option, careful consideration should be given to the paucity of prospective data and the very real potential toxicity of even commonly prescribed tyrosine kinase inhibitors. Deciding whether to recommend further therapy on the basis of tests such as these, especially when the alternative is to discontinue any additional cancer‐directed treatment, should be made after extensive discussion with the patient and/or family and preferably with the input of a molecular tumor board or the equivalent. A full discussion of the ethics of deciding when it is appropriate to recommend discontinuation of further attempts at systemic therapy is beyond the scope of this review.
Question 8: What Is Needed to Accurately Diagnose and Stage a Newly Diagnosed Ewing Sarcoma Patient?
The incidence of Ewing sarcoma, like that of osteosarcoma, peaks in the teenage years. However, Ewing sarcoma is much more varied in primary tumor sites, with one‐half arising from the axial skeleton (eg, the pelvis or ribs) and up to 20% originating in soft tissues.12 Our staging strategy is described in Figure 1. Although bone scans are less expensive and involve less radiation, positron emission tomography/computed tomography scans have greater sensitivity and specificity for detecting metastatic disease and are, therefore, recommended.36 Because bone marrow metastases may occur, bilateral bone marrow aspirates and biopsies have been routinely performed for newly diagnosed patients. However, the yield of finding marrow disease in patients without metastases identified by positron emission tomography or other imaging studies is extremely low, and outside a clinical trial, a marrow evaluation is unnecessary for these patients.37, 38
Question 9: What Are the Key Components of Therapy for Ewing Sarcoma and Should Therapy Differ for Young Adults Versus Children?
Like the treatment for osteosarcoma, the treatment for Ewing sarcoma combines local management of the primary tumor with chemotherapy to eradicate micrometastatic disease. Most patients start treatment with neoadjuvant chemotherapy for approximately 12 weeks, and this is followed by local control with radiation, surgery, or both. Further chemotherapy is then administered to reach a cumulative total of at least 14 cycles. An early landmark study showed the benefit of adding IE to the backbone of vincristine, doxorubicin, and cyclophosphamide (VDC) for younger patients with localized tumors.39 Outcomes were further improved by compression of the schedule; cycles were initiated every 2 weeks as tolerated instead of every 3 weeks.40 This interval compression of VDC‐IE is now the standard of care in North America. In Europe, drugs are grouped differently, and multiple cycles of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) constitute the standard chemotherapy regimen.41 A direct comparison of VIDE and VDC‐IE is being performed in the ongoing Euro‐Ewing 12 study (EudraCT number 2012‐002107‐17). In the largest retrospective studies, age does not necessarily appear to be an independent prognostic factor, and in general, young adults tolerate aggressive chemotherapy similarly to children.42 For these reasons, cooperative group trials have included patients up to the age of 50 years. Therefore, we advocate the enrollment of young adults into these trials or treatment according to a pediatric regimen when therapy is outside a clinical study.
Question 10: What Factors Influence Local Control of Ewing Sarcoma?
The choice of local control depends on the location of the tumor and the ability to obtain negative surgical margins. Resection of the tumor is usually performed if possible with acceptable morbidity. Radiation can provide equally durable local control of the tumor but does carry the associated long‐term risk of a secondary malignancy.43, 44 However, if surgery would cause excess morbidity, radiotherapy can provide similar treatment outcomes.45 In addition, radiotherapy is indicated for patients with positive surgical margins. In Europe, radiotherapy is also administered to patients with a poor histologic response (≤90% necrosis) after neoadjuvant chemotherapy, although this approach is not used by the authors. Posttherapy follow‐up is outlined in Figure 4B.
Question 11: What Is the Approach for Metastatic Ewing Sarcoma?
Metastases are identified at diagnosis in approximately one‐fourth of Ewing sarcoma patients when the previously mentioned osteosarcoma lung nodule size criteria are used. The decision to biopsy lung nodules is based on the number and size of nodules as well as the overall staging results. Because we recommend whole‐lung radiotherapy after the completion of chemotherapy for any patient with pulmonary metastasis and we recognize that small nodules often are quickly resolved with chemotherapy, biopsy of equivocal lung nodules before treatment is started should be considered if feasible.
The outcome of patients with disseminated Ewing sarcoma is correlated with the extent of metastases. Those with metastases limited to the lungs have an expected 5‐year overall survival rate of 40%, whereas fewer than 20% of patients with bone and/or bone marrow metastases are long‐term survivors.46 Enrollment in a clinical trial is especially attractive for these patients because the standard treatment has not improved patient survival in decades, and the intensification of conventional therapy or even myeloablative chemotherapy with stem cell transplantation has not been reported to be advantageous. Combining conventional cytotoxic chemotherapy with targeted drugs that have demonstrated single‐agent activity is a reasonable strategy for investigation. Although the use of extended low‐dose maintenance therapy for patients in remission after conventional treatment is intriguing, the benefits of this approach have not been established, and this is not routinely used by the authors.47
Question 12: What Is the Approach to Relapsed Ewing Sarcoma?
Most Ewing sarcoma relapses occur within 2 years of the initial diagnosis, with metastases typically to the lungs and/or bone. The patients at highest risk for recurrence are those who had metastatic disease at the initial presentation, pelvic tumors, or a poor histologic response to neoadjuvant chemotherapy. The decision to biopsy suspicious lesions depends on the context of risk factors, the extent of new findings, and the possibility of a reasonable alternative explanation for the findings. As a group, patients with recurrent Ewing sarcoma have a dismal overall prognosis, with an estimated 5‐year overall survival rate of <15%.48 However, certain prognostic factors, such as the timing of the recurrence, may help to identify a subset of patients most likely to benefit from salvage therapy. For example, although patients with an early relapse (within 2 years of the initial diagnosis) have a < 10% chance of long‐term survival, up to one‐fourth of those with a later relapse may potentially be cured. Patients with local recurrences also tend to fare better than those with systemic or combined recurrences, particularly if further local therapy can be administered in addition to chemotherapy. In patients with metastatic or recurrent disease with better long‐term prognoses, stereotactic body radiotherapy may offer a quicker treatment course that provides durable local control.10
Treatment decisions should take into account these prognostic factors, prior therapy, and individual patient characteristics, including organ function and the desire for further treatment. In contrast to recurrent osteosarcoma, for which surgical metastatectomy may be the only therapy used for some patients, chemotherapy is generally recommended for all recurrent Ewing sarcoma patients (Fig. 3). Enrollment in a clinical trial should be strongly considered, even at the first relapse. Early‐phase clinical trials are generally well tolerated in adolescents and young adults and offer the chance to identify promising new therapies.
Figure 3.
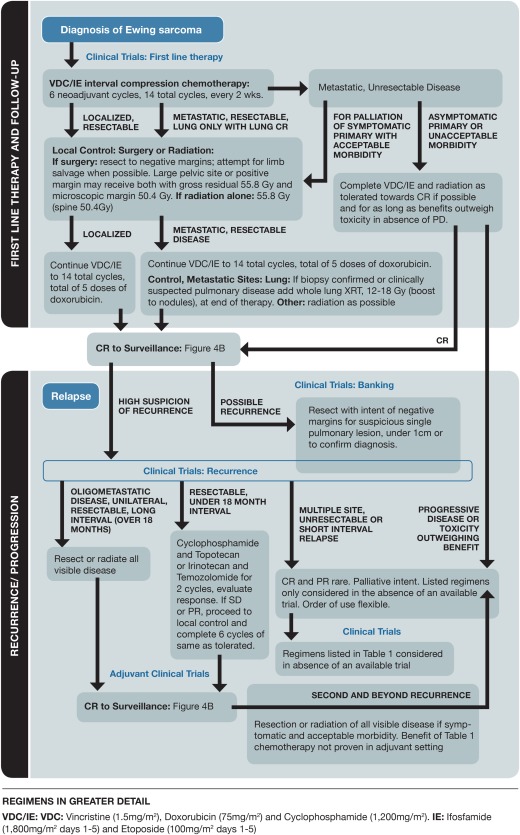
Pathway for treating newly diagnosed and relapsed Ewing sarcoma. CR, complete response; IE, ifosfamide and etoposide; PD, progressive disease; PR, partial response; SD, stable disease; VDC, vincristine, doxorubicin, and cyclophosphamide; XRT, X‐ray therapy.
If the patient is not enrolled in a clinical trial, several different treatment regimens may be considered (Table 1). Overall, the consensus among authors is to use a combination of irinotecan and temozolomide as the first option, primarily because of the favorable toxicity profile and the option of giving both agents orally for the patient's convenience. When they are given orally, drug exposures similar to those from intravenous administration can be seen as long as the dose of irinotecan is adjusted to account for the limited bioavailability and prophylactic cephalosporins are used to reduce irinotecan‐associated diarrhea.30 Interestingly, the patterns of resistance to topotecan seem different than the patterns of resistance to irinotecan, such that one regimen may still have activity despite the failure of the other. Responses to cyclophosphamide and topotecan therapy were correlated with the relapse interval, with no responses when the disease was progressing on prior therapy and with a rate greater than 50% response rate when 2 years had passed between the prior therapy and the initiation of therapy.28 Once camptothecins have failed, possible adverse effects, the patient's performance status and marrow reserve, and the schedule of administration often affect decisions about salvage therapy, especially with young adults. For example, the frequency of intravenous chemotherapy administration, the need for growth factor and transfusion support, and even the likelihood of certain toxicities such as nausea or alopecia may all factor into treatment planning.
The median postrecurrence survival ranges from 9 to 17 months and depends on the extent of disease. Although a cure is elusive, many patients experience some temporary response or disease stabilization with the aforementioned therapies. Patients with painful lesions typically respond well to palliative doses of radiation.49 It is not unusual for patients to receive more than 1 salvage regimen, and each change in treatment allows additional opportunities for clinical trial enrollment to be considered. Question 7 addresses our opinion on the role of emerging molecular technologies and when additional care may not be in the best interest of the patient. In addition, for patients who have a declining performance status or insist on the convenience of less complicated treatment regimens, both oral etoposide and pazopanib have been reported to produce occasional responses.34, 50 Our current impression is that exomic testing of tumor tissue with institutional or commercially available platforms only rarely identifies actionable mutations for this translocation‐driven malignancy.
Finally, the use of high‐dose chemotherapy with autologous stem cell support has gained some interest for treating patients with recurrent Ewing sarcoma, although published studies have been limited by the lack of an adequate control group.51, 52 For example, patients undergoing high‐dose therapy must have previously responded to initial conventional salvage treatments, have organ function that will allow high‐dose therapy, and have adequate stem cells available for collection. Although encouraging results may be seen in these select patients, there is not sufficient evidence at present to support this intensive approach as standard care for all relapsed patients.
Question 13: What Is the Multimodal Approach to Therapy for Chondrosarcomas?
There are several subtypes of chondroblastic malignancies seen in the young adult years. These often present with pain in the axial skeleton and have particular radiographic features, including scalloping, to suggest the diagnosis. Surgical management is critical for any chance of a cure, and for resectable pulmonary metastases, we also advocate metastatectomy. Conventional chondrosarcoma is chemotherapy‐resistant, possibly because of the cartilaginous matrix and difficulty with effective delivery of systemic agents to malignant cells, and adjuvant systemic therapy would not be suggested.53 Radiation therapy is sometimes recommended for positive margins. If these patients develop metastases or have an unresectable local recurrence, molecular testing, particularly for isocitrate dehydrogenase mutations, and clinical trials would be the main consideration.54 Other types of chondrosarcoma may be more sensitive to chemotherapy, with dedifferentiated chondrosarcoma being treated similarly to osteosarcoma in the young population and mesenchymal chondrosarcoma being treated similarly to Ewing sarcoma.55
In conclusion, we have described a consensus approach to the most common bone sarcomas of childhood, adolescence, and young adulthood. Despite the many options for any given situation, we report a consensus from multiple centers and have outlined a general approach that can be taken for a variety of first‐line and relapse clinical scenarios.
Although there may not be one correct treatment regimen, especially in the setting of relapse, we hope that emphasizing clinical trials and presenting the modest or limited activity of available regimens will lead to more consistent treatment and allow future improvements in patient care.
FUNDING SUPPORT
Damon R. Reed and Michael S. Isakoff received funding from the National Pediatric Cancer Foundation (www.nationalpcf.org) for this work. Michael S. Isakoff also received funding from the Reid R. Sacco Adolescent and Young Adult Cancer Alliance. Masanori Hayashi was supported from the SARC Sarcoma SPORE Career Development Award (5U54CA168512‐02).
CONFLICT OF INTEREST DISCLOSURES
Damon R. Reed reports personal fees from EMD Serrano and Janssen Oncology outside the submitted work. Julia Bridge reports a grant from Cepheid and personal fees from the National Comprehensive Cancer Network and Bristol‐Myers Squibb outside the submitted work.
REFERENCES
- 1. National Comprehensive Cancer Network . NCCN guidelines for treatment of cancer by site: bone cancer (version 2.2017). https://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed November 14, 2016.
- 2. Link MP, Goorin AM, Horowitz M, et al. Adjuvant chemotherapy of high‐grade osteosarcoma of the extremity. Updated results of the Multi‐Institutional Osteosarcoma Study. Clin Orthop Relat Res. 1991;270:8–14. [PubMed] [Google Scholar]
- 3. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33:3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaffe N, Gorlick R. High‐dose methotrexate in osteosarcoma: let the questions surcease—time for final acceptance. J Clin Oncol. 2008;26:4365–4366. [DOI] [PubMed] [Google Scholar]
- 5. Schwartz CL, Wexler LH, Krailo MD, et al. Intensified chemotherapy with dexrazoxane cardioprotection in newly diagnosed nonmetastatic osteosarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2016;63:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Dalen EC, van der Pal HJ, Kremer LC. Different dosage schedules for reducing cardiotoxicity in people with cancer receiving anthracycline chemotherapy. Cochrane Database Syst Rev. 2016;3:CD005008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goorin AM, Schwartzentruber DJ, Devidas M, et al. Presurgical chemotherapy compared with immediate surgery and adjuvant chemotherapy for nonmetastatic osteosarcoma: Pediatric Oncology Group Study POG‐8651. J Clin Oncol. 2003;21:1574–1580. [DOI] [PubMed] [Google Scholar]
- 8. Rougraff BT, Simon MA, Kneisl JS, Greenberg DB, Mankin HJ. Limb salvage compared with amputation for osteosarcoma of the distal end of the femur. A long‐term oncological, functional, and quality‐of‐life study. J Bone Joint Surg Am. 1994;76:649–656. [DOI] [PubMed] [Google Scholar]
- 9. Ozaki T, Flege S, Kevric M, et al. Osteosarcoma of the pelvis: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol. 2003;21:334–341. [DOI] [PubMed] [Google Scholar]
- 10. Brown LC, Lester RA, Grams MP, et al. Stereotactic body radiotherapy for metastatic and recurrent Ewing sarcoma and osteosarcoma. Sarcoma. 2014;2014:418270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bishop MW, Chang YC, Krailo MD, et al. Assessing the prognostic significance of histologic response in osteosarcoma: a comparison of outcomes on CCG‐782 and INT0133—a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer. 2016;63:1737–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fletcher CDM, Unni KK, Mertens F, eds. WHO Classification of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2013. [Google Scholar]
- 13. Bielack SS, Smeland S, Whelan JS, et al. Methotrexate, doxorubicin, and cisplatin (MAP) plus maintenance pegylated interferon alfa‐2b versus MAP alone in patients with resectable high‐grade osteosarcoma and good histologic response to preoperative MAP: first results of the EURAMOS‐1 good response randomized controlled trial. J Clin Oncol. 2015;33:2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhattasali O, Vo AT, Roth M, et al. Variability in the reported management of pulmonary metastases in osteosarcoma. Cancer Med. 2015;4:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrari S, Briccoli A, Mercuri M, et al. Postrelapse survival in osteosarcoma of the extremities: prognostic factors for long‐term survival. J Clin Oncol. 2003;21:710–715. [DOI] [PubMed] [Google Scholar]
- 16. Leary SE, Wozniak AW, Billups CA, et al. Survival of pediatric patients after relapsed osteosarcoma: the St. Jude Children's Research Hospital experience. Cancer. 2013;119:2645–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marti C, Kroner T, Remagen W, Berchtold W, Cserhati M, Varini M. High‐dose ifosfamide in advanced osteosarcoma. Cancer Treat Rep. 1985;69:115–117. [PubMed] [Google Scholar]
- 18. Harris MB, Cantor AB, Goorin AM, et al. Treatment of osteosarcoma with ifosfamide: comparison of response in pediatric patients with recurrent disease versus patients previously untreated: a Pediatric Oncology Group study. Med Pediatr Oncol. 1995;24:87–92. [DOI] [PubMed] [Google Scholar]
- 19. Kempf‐Bielack B, Bielack SS, Jurgens H, et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol. 2005;23:559–568. [DOI] [PubMed] [Google Scholar]
- 20. Miser JS, Kinsella TJ, Triche TJ, et al. Ifosfamide with mesna uroprotection and etoposide: an effective regimen in the treatment of recurrent sarcomas and other tumors of children and young adults. J Clin Oncol. 1987;5:1191–1198. [DOI] [PubMed] [Google Scholar]
- 21. Gentet JC, Brunat‐Mentigny M, Demaille MC, et al. Ifosfamide and etoposide in childhood osteosarcoma. A phase II study of the French Society of Paediatric Oncology. Eur J Cancer. 1997;33:232–237. [DOI] [PubMed] [Google Scholar]
- 22. Grignani G, Palmerini E, Dileo P, et al. A phase II trial of sorafenib in relapsed and unresectable high‐grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol. 2012;23:508–516. [DOI] [PubMed] [Google Scholar]
- 23. Navid F, Willert JR, McCarville MB, et al. Combination of gemcitabine and docetaxel in the treatment of children and young adults with refractory bone sarcoma. Cancer. 2008;113:419–425. [DOI] [PubMed] [Google Scholar]
- 24. Rapkin L, Qayed M, Brill P, et al. Gemcitabine and docetaxel (GEMDOX) for the treatment of relapsed and refractory pediatric sarcomas. Pediatr Blood Cancer. 2012;59:854–858. [DOI] [PubMed] [Google Scholar]
- 25. Qi WX, He AN, Tang LN, Shen Z, Lin F, Yao Y. Efficacy and safety of gemcitabine‐docetaxel combination therapy for recurrent or refractory high‐grade osteosarcoma in China: a retrospective study of 18 patients. Jpn J Clin Oncol. 2012;42:427–431. [DOI] [PubMed] [Google Scholar]
- 26. Song BS, Seo J, Kim DH, Lim JS, Yoo JY, Lee JA. Gemcitabine and docetaxel for the treatment of children and adolescents with recurrent or refractory osteosarcoma: Korea Cancer Center Hospital experience. Pediatr Blood Cancer. 2014;61:1376–1381. [DOI] [PubMed] [Google Scholar]
- 27. Palmerini E, Jones RL, Marchesi E, et al. Gemcitabine and docetaxel in relapsed and unresectable high‐grade osteosarcoma and spindle cell sarcoma of bone. BMC Cancer. 2016;16:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jurgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47:795–800. [DOI] [PubMed] [Google Scholar]
- 29. Saylors RL III, Stine KC, Sullivan J, et al. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol. 2001;19:3463–3469. [DOI] [PubMed] [Google Scholar]
- 30. Wagner LM. Fifteen years of irinotecan therapy for pediatric sarcoma: where to next? Clin Sarcoma Res. 2015;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson P, Kopp L, Anderson N, et al. Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing's sarcoma and osteosarcoma). Expert Opin Investig Drugs. 2008;17:1703–1715. [DOI] [PubMed] [Google Scholar]
- 32. Ferrari S, del Prever AB, Palmerini E, et al. Response to high‐dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer. 2009;52:581–584. [DOI] [PubMed] [Google Scholar]
- 33. Mora J, Cruz CO, Parareda A, de Torres C. Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol. 2009;31:723–729. [DOI] [PubMed] [Google Scholar]
- 34. Podda MG, Luksch R, Puma N, et al. Oral etoposide in relapsed or refractory Ewing sarcoma: a monoinstitutional experience in children and adolescents. Tumori. 2016;102:84–88. [DOI] [PubMed] [Google Scholar]
- 35. Foundation Medicine . https://pediatric-data.foundationmedicine.com/dashboard. Accessed September, 2016.
- 36. Treglia G, Taralli S, Bertagna F, et al. Usefulness of whole‐body fluorine‐18‐fluorodeoxyglucose positron emission tomography in patients with neurofibromatosis type 1: a systematic review. Radiol Res Pract. 2012;2012:431029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kopp LM, Hu C, Rozo B, et al. Utility of bone marrow aspiration and biopsy in initial staging of Ewing sarcoma. Pediatr Blood Cancer. 2015;62:12–15. [DOI] [PubMed] [Google Scholar]
- 38. Anderson PM. Futility versus utility of marrow assessment in initial Ewing sarcoma staging workup. Pediatr Blood Cancer. 2015;62:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. [DOI] [PubMed] [Google Scholar]
- 40. Womer RB, West DC, Krailo MD, et al. Randomized controlled trial of interval‐compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2012;30:4148–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Juergens C, Weston C, Lewis I, et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO‐E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer. 2006;47:22–29. [DOI] [PubMed] [Google Scholar]
- 42. Scurr M, Judson I. How to treat the Ewing's family of sarcomas in adult patients. Oncologist. 2006;11:65–72. [DOI] [PubMed] [Google Scholar]
- 43. Paulino AC, Fowler BZ. Secondary neoplasms after radiotherapy for a childhood solid tumor. Pediatr Hematol Oncol. 2005;22:89–101. [DOI] [PubMed] [Google Scholar]
- 44. Kuttesch JF Jr, Wexler LH, Marcus RB, et al. Second malignancies after Ewing's sarcoma: radiation dose‐dependency of secondary sarcomas. J Clin Oncol. 1996;14:2818–2825. [DOI] [PubMed] [Google Scholar]
- 45. DuBois SG, Krailo MD, Gebhardt MC, et al. Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group. Cancer. 2015;121:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18:3108–3114. [DOI] [PubMed] [Google Scholar]
- 47. Ray‐Coquard I, Le Cesne A. A role for maintenance therapy in managing sarcoma. Cancer Treat Rev. 2012;38:368–378. [DOI] [PubMed] [Google Scholar]
- 48. Leavey PJ, Mascarenhas L, Marina N, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi‐modality therapy: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2008;51:334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koontz BF, Clough RW, Halperin EC. Palliative radiation therapy for metastatic Ewing sarcoma. Cancer. 2006;106:1790–1793. [DOI] [PubMed] [Google Scholar]
- 50. Alcindor T. Response of refractory Ewing sarcoma to pazopanib. Acta Oncol. 2015;54:1063–1064. [DOI] [PubMed] [Google Scholar]
- 51. Rasper M, Jabar S, Ranft A, Jurgens H, Amler S, Dirksen U. The value of high‐dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer. 2014;61:1382–1386. [DOI] [PubMed] [Google Scholar]
- 52. Barker LM, Pendergrass TW, Sanders JE, Hawkins DS. Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol. 2005;23:4354–4362. [DOI] [PubMed] [Google Scholar]
- 53. Italiano A, Mir O, Cioffi A, et al. Advanced chondrosarcomas: role of chemotherapy and survival. Ann Oncol. 2013;24:2916–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. [DOI] [PubMed] [Google Scholar]
- 55. Frezza AM, Cesari M, Baumhoer D, et al. Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur J Cancer. 2015;51:374–381. [DOI] [PubMed] [Google Scholar]