

Abstract
Secukinumab is a human monoclonal antibody with demonstrated efficacy for moderate to severe psoriasis; it binds to and neutralizes interleukin (IL)‐17A. The pharmacokinetic (PK) parameters of secukinumab were best described by a 2‐compartment model. Only weight was included in the final model, as other covariates did not affect clinical relevance. The estimated serum clearance of secukinumab was 0.19 L/day, with interindividual variability (IIV) of 32% coefficient of variation (CV), and low total volume of distribution (central compartment volume, 3.61 L with IIV of 30% CV; peripheral compartment volume, 2.87 L with IIV of 18% CV). The bioavailability of secukinumab after subcutaneous dosing was approximately 73%, with an absorption rate of 0.18/day with IIV of 35% CV. The PK profile of secukinumab was linear, with no evidence of a dose dependence of clearance. Clearance and volume of secukinumab varied with body weight in an allometric relationship. The time to maximum serum concentration at steady state occurred approximately 6 days after dosing for both secukinumab 300 mg and secukinumab 150 mg. Overall, the PK properties of secukinumab were typical of a 150‐kDa human IgG1 antibody interacting with a soluble target.
Keywords: IL‐17A, monoclonal antibody, pharmacodynamics, population pharmacokinetics, psoriasis, secukinumab
The immunopathogenesis of psoriatic skin inflammation is largely driven by cytokines regulated by T‐helper 17 (Th17) cells.1, 2, 3 In genetically susceptible individuals, environmental triggers and cutaneous pathogens stimulate dendritic cells to produce interleukin (IL)‐23, which promotes Th17 differentiation. In turn, this leads to elevated levels of serum and cutaneous Th17 cells in patients with psoriasis.3, 4, 5, 6, 7 Continued exposure of Th17 cells to IL‐23 leads to enhanced production of IL‐17A, which has been identified as a central cytokine effector of skin changes associated with psoriasis.1, 2, 8 Increased IL‐17A production promotes keratinocyte activation and production of inflammatory mediators, creating a cycle of inflammation that perpetuates lesion formation. Thus, IL‐17A has been identified as a key therapeutic target for the treatment of psoriasis,2, 6, 8 and biologic agents that target the IL‐17 pathway have shown promising efficacy and safety in clinical trials.9, 10, 11, 12, 13
One such biologic agent is secukinumab, an immunoglobulin G1κ (IgG1κ) monoclonal antibody that potently and selectively binds to and neutralizes IL‐17A. In pivotal phase 3 trials of patients with moderate to severe plaque psoriasis, at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75) was achieved by 77% to 82% of patients after 12 weeks of treatment with subcutaneous secukinumab 300 mg, and PASI 75 response rates ranged from 67% to 72% for patients treated with secukinumab 150 mg.9 Investigator's Global Assessment modified 2011 responses of 0 (clear) or 1 (almost clear) at week 12 were reported by 63% to 65% of patients who received secukinumab 300 mg and by 51% who received secukinumab 150 mg.9 In these trials, secukinumab was generally well tolerated, with the most commonly reported adverse events including nasopharyngitis, upper respiratory tract infection, and headache.9, 12, 13, 14
Findings from a dose‐ranging study in patients with psoriasis15 showed that, at therapeutic doses, secukinumab concentration increased in a dose‐proportional manner. Overall, the pharmacokinetic (PK) properties of secukinumab are typical of an IgG1 antibody because it exhibits good stability, long persistence in the body, and high selectivity and specificity.16, 17 Furthermore, with a novel technique, open‐flow microperfusion, the concentration of secukinumab was reported as 6.8 μg/mL in lesional skin 7 days after a single 300‐mg subcutaneous injection of secukinumab.18 In patients with psoriasis, interstitial fluid concentrations of secukinumab in lesional and nonlesional skin were in the range of 28% to 39% relative to serum concentrations.18 Relevant cytokines and markers were also analyzed directly in the skin through this method. At baseline, levels of IL‐17A and human β‐defensin 2 (hBD‐2), a downstream marker of IL‐17A signaling and psoriasis disease severity,19, 20, 21 were significantly higher in psoriatic skin lesions, and levels of hBD‐2 significantly and rapidly decreased following administration of secukinumab.22 The objective of this study was to further characterize the PK properties of secukinumab using pooled results from 6 clinical studies with either intravenous or subcutaneous administration in patients with psoriasis. In addition, pharmacodynamic (PD) biomarkers of secukinumab activity are explored based on the measurement of total IL‐17A concentrations in the phase 3 JUNCTURE trial.13
Methods
Some studies were multicenter, and each site complied with the Declaration of Helsinki. All protocols were approved by each site's institutional review board or ethics committee. Informed consent was provided by all patients.
Population PK Modeling
As mentioned, the relationship between the dose, regimen, and serum concentration of secukinumab was characterized based on pooled data from 6 clinical studies in patients with psoriasis. Included studies (described in Table 1) comprised 5 phase 1 or 2 studies15, 23, 24 and 1 phase 3 study (ERASURE)9 that assessed a range of subcutaneous (25, 75, 150, and 300 mg) and intravenous (1, 3, and 10 mg/kg) secukinumab dosing regimens.
Table 1.
Secukinumab clinical studies constituting the PK analysis data set
| Study | Primary Objective | Design | Treatment Regimensa | PK Sampling | Mean Age of Patients Receiving Secukinumab |
|---|---|---|---|---|---|
| Proof of concept, Hueber et al23 | Efficacy | Randomized, double‐blind, parallel‐group, placebo‐controlled | Single dose (n = 36)
|
Predose, end of infusion, 1 and 2 h after infusion, then at weeks 1, 2, 3, 4, 5, 6, 8, and 12 | 50.7 years |
| Absolute bioavailability study | Bioavailability | Randomized, open‐label, crossover | Multiple doses (n = 14)
|
For intravenous: predose and 1, 2, 4, and 8 h; for subcutaneous: predose and 1 and 8 h; and then 1, 3, 4, 7, 9, 14, 21, 28, 29, 31, 32, 35, 38, 42, 49, 56, 70, and 84 days after first dose for both arms | 46.8 years |
| Phase 2 dose‐regimen‐finding study, Rich et al24 | Efficacy | Randomized, double‐blind, parallel‐group, placebo‐controlled | Multiple doses (n = 404)
|
Weeks 0, 1, 2, 4, 8, 12, 16, 20, 24, 28, and 32 | 42.7–44.5 years |
| Extension of dose‐regimen‐finding study | Long‐term safety, tolerability | Open‐label extension | Multiple doses, maintenance
|
Weeks 12, 24, and 36 | 39.9–45.0 years |
| Phase 2 dose‐finding‐study, Reich et al27 | Efficacy | Randomized, double‐blind, parallel‐group, placebo‐controlled | Single and multiple doses (n = 100)
|
Predose and 2 and 4 h after infusions at weeks 1, 2, and 4; additional sampling at weeks 1, 6, 8, 10, 12, 16, 20, 24, 28, 32, 40, 48, and 56 | 43.4–45.7 years |
| Phase 2 dose‐finding‐study, Papp et al15 | Efficacy | Randomized, double‐blind, parallel‐group, placebo‐controlled | Single and multiple doses (n = 125)
|
Weeks 0, 1, 2, 4, 8, 12, 16, 20, 24, 28, 32, and 36 | 45.4–46.3 years |
| Phase 3 ERASURE study, Langley et al9 | Efficacy superiority vs. placebo | Randomized, double‐blind, parallel‐group, placebo‐controlled | Multiple doses (n = 738)
|
Predose, baseline, and weeks 4, 12, 24, and 52 | 44.9 years |
| Phase 3 FIXTURE study,b Langley et al9 | Efficacy superiority vs. placebo | Randomized, double‐blind, double‐dummy, placebo‐controlled | Multiple doses (n = 1306)
|
Predose, baseline, and weeks 4, 12, 24, and 52 | 44.5–45.4 years |
ERASURE, Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis; FIXTURE, Full‐Year Investigative Examination of Secukinumab vs. Etanercept Using 2 Dosing Regimens to Determine Efficacy in Psoriasis; PD, pharmacodynamic; PK, pharmacokinetic.
Patient numbers reported in this table are the number of enrolled patients for each study.
Results from FIXTURE were used as an external validation data set. The PK analysis set includes only the patients with evaluable PK data.
Based on exploratory modeling of phase 2 data, it was hypothesized that secukinumab PK parameters could be described using a linear 2‐compartmental distribution model with first‐order absorption for subcutaneous administration and with zero‐order infusion for intravenous administration. Therefore, a linear 2‐compartmental distribution model was used as a starting point for this analysis.
The population PK model included structural, random‐effects, covariate, and residual error models. The structural model accounted for systematic data trends. Disposition kinetics were modeled using a parameterization involving clearance, central volume, intercompartmental clearance, and peripheral volume. Subcutaneous bioavailability between 0 and 1 was modeled on a logit scale, and absorption was modeled as a first‐order process for the subcutaneous route of administration. The random‐effects model was used to account for between‐patient variability in PK parameters using multiplicative exponential random effects, and residual variability was modeled using a combined proportional/additive error model. The residual error model was used to describe discrepancies in the data that were not accounted for by the model, and the covariate model was used to explore the impact of covariates on between‐patient variability. The base model included body weight (centered at 90 kg) as a covariate on clearances and volumes of distribution; other covariates explored included age (centered at 45 years), baseline PASI score (centered at 19), sex, geographic region (Europe, United States, Taiwan, East Asia including Japan, East Asia excluding Japan, and Japan), and race (Asian vs non‐Asian). Under the assumption that changes to the typical patient parameters of more than 20% are potentially of clinical relevance, only body weight was retained in the population PK model. The small differences from the influence of age, baseline PASI score, sex, geographic region, and race were not considered clinically relevant. The reference values used for centering of age and baseline PASI score were chosen to approximately reflect the median of these covariates.
Model components were selected and assembled based on a combination of prior knowledge and data‐driven decision making, guided by statistical and heuristic rules. Decision criteria for the model were based on a combination of judgment of model plausibility and robustness, objective function value, goodness‐of‐fit diagnostics, and simulation‐based diagnostics. Predictive performance of the models was assessed in visual predictive checks, which were stratified by treatment group using 400 simulations with 10 time bins, with approximately equal observation counts. Data from the phase 3 FIXTURE study9 were used as an external validation data set to provide a rigorous assessment of the predictive performance of the model without reestimation of model parameters.
The final structural model was used to simulate PK profiles for dosing regimens of interest (secukinumab 300 and 150 mg subcutaneous regimens used in phase 3 studies). Daily PK concentrations for 1000 patients per dosing regimen were simulated, and derived exposure metrics included minimum, maximum, and average serum drug concentration (Cmin, Cmax, and Cavg), steady‐state area under the curve (AUC), time to maximum concentration in serum (tmax), and terminal half‐life. The population PK models were fitted using NONMEM 7.2 software.
Pharmacokinetics
Serum concentrations of secukinumab were measured in patients from the phase 3 ERASURE trial. In this study, patients in the active‐treatment arms received subcutaneous secukinumab 300 mg (n = 245) or 150 mg (n = 245) at baseline; at weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48.9 The last dose of secukinumab was given at week 48. At week 12, PASI 75 nonresponders receiving placebo were rerandomized to secukinumab 300 mg (n = 105) or 150 mg (n = 109), and 18 patients continued on placebo. Blood samples were taken for PK analysis at baseline; at weeks 4, 12, 24, 52, and 60 (during washout); and during unscheduled visits. Serum levels of secukinumab were determined by an enzyme‐linked immunosorbent assay method with a lower limit of quantification of 80 ng/mL.
Pharmacodynamics
The ability of secukinumab to bind and capture circulating IL‐17A was formally validated in the phase 3 JUNCTURE trial based on measurement of total serum IL‐17A. In this study, patients in the active‐treatment arms received subcutaneous secukinumab 300 mg (n = 60) or 150 mg (n = 61) at baseline; at weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48.13 The last dose of secukinumab was given at week 48. Total IL‐17A was defined as free IL‐17A plus IL‐17A complexed with secukinumab. Blood samples were taken for assessment of total IL‐17A at baseline and at weeks 4, 12, 16, 24, and 52. Total serum IL‐17A concentrations were determined by an electrochemiluminescence method with a lower limit of quantification of 20 pg/mL.
Results
Pharmacokinetics
The population PK model was built based on 10 193 secukinumab serum concentrations from 1233 patients with moderate to severe psoriasis who participated in 5 phase 1 or 2 studies and 1 phase 3 study. The mean ± standard deviation demographic and baseline characteristics used as continuous covariates included body weight (90.7 ± 23.5 kg), PASI score (21.1 ± 8.8), height (172.7 ± 9.9 cm), body mass index (30.3 ± 7.2 kg/m2), and age (44.6 ± 12.6 years). Categorical covariates included sex, race (Asian vs non‐Asian), and geographic location. The majority of patients were male (897 of 1233 [72.7%]) and non‐Asian (1027 of 1233 [83.3%]; of the non‐Asian population, 967 [94.2%] were white).
The descriptive and predictive capabilities of the population PK model were demonstrated using goodness‐of‐fit plots (Figure 1) and predictive checks. The overall fit of the 2‐compartment model to the PK data was good, with few outlying points. The weighted residuals‐versus‐time and population predictions showed no evident trends. The predictive performance of the model was confirmed based on goodness‐of‐fit plots and predictive checks using external validation data from the FIXTURE trial (Figure 2),9 because these results were similar to those of the studies included in the model. The goodness of fit for the external validation data was adequate and similar to the goodness of fit for data that were included in building the PK model. Visual predictive checks for the data used in Figure 1 and Figure 2 are shown in Supplemental Figure 1 and Supplemental Figure 2.
Figure 1.
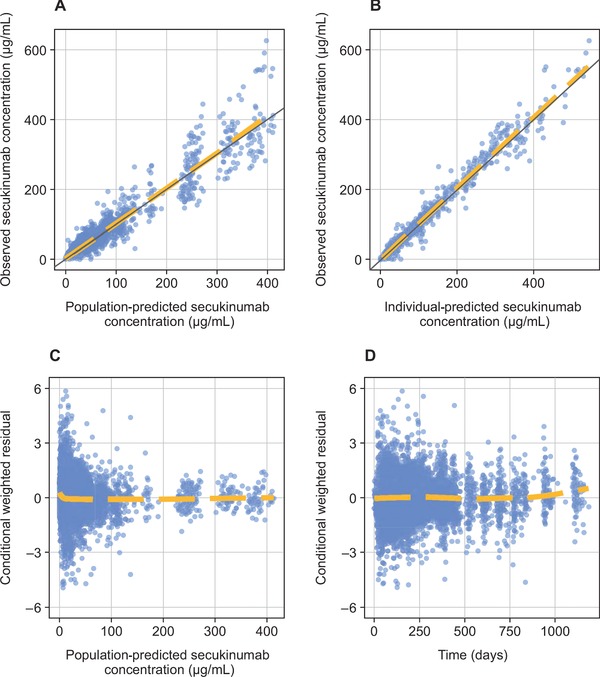
Goodness‐of‐fit diagnostics for the simulated pharmacokinetic model. (A) Observed secukinumab concentration versus population‐predicted secukinumab concentration. (B) Observed secukinumab concentration versus individual‐predicted secukinumab concentration. (C) Conditional weighted residual versus population‐predicted secukinumab concentration. (D) Conditional weighted residual versus time.
Figure 2.
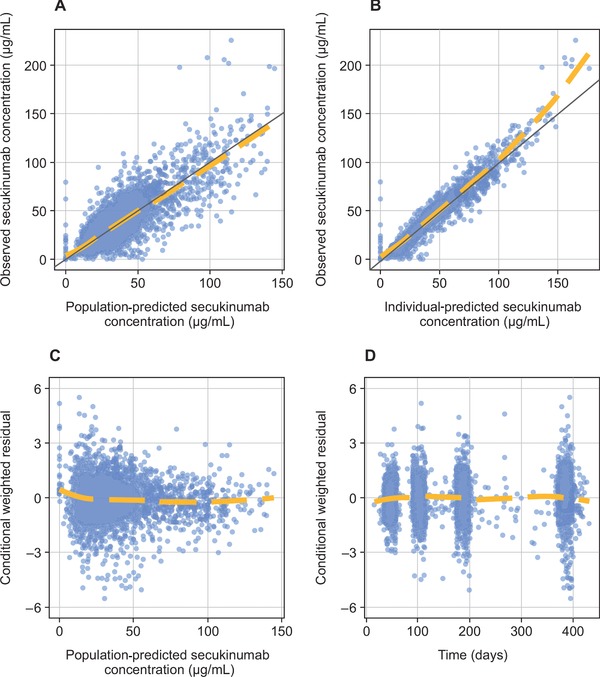
External validation of goodness‐of‐fit diagnostics for the simulated pharmacokinetic model. (A) Observed secukinumab concentration versus population‐predicted secukinumab concentration. (B) Observed secukinumab concentration versus individual‐predicted secukinumab concentration. (C) Conditional weighted residual versus population predicted secukinumab concentration. (D) Conditional weighted residual versus time.
The PK parameters estimated from the final 2‐compartment population PK model are shown in Table 2. In a typical patient with psoriasis weighing 90 kg, it was estimated that serum clearance was low (0.19 L/day with interindividual variability [IIV] of 32% coefficient of variation [CV]), and total volume of distribution was low (central compartment volume, 3.61 L with IIV of 30% CV; peripheral compartment volume, 2.87 L with IIV of 18% CV). Bioavailability after subcutaneous dosing was approximately 73%, with an absorption rate of 0.18/day with IIV of 35% CV.
Table 2.
Estimated population parameters of the final 2‐compartment PK model
| Parameter | Value | %RSE | Interindividual Variability, %CV |
|---|---|---|---|
| CL, L/day | 0.19 | 1.9 | 32 |
| V2, L | 3.61 | 2.6 | 30 |
| Q, L/day | 0.39 | 4.6 | NA |
| V3, L | 2.87 | 1.9 | 18 |
| ka, 1/day | 0.18 | 3.6 | 35 |
| F, % | 72.9 | 1.5 | NA |
| Proportional error | 0.17 | 0.8 | |
| Additive error | 371 | 0.5 |
CL, Q, V2, and V3 are given for patients with a reference weight of 90 kg. Proportional and additive errors are given as standard deviations. The additive error is given in ng/mL, the unit in which it was estimated, whereas figures are presented as μg/mL. CL, clearance; CV, coefficient of variation; F, bioavailability; ka, absorption rate; NA, not applicable (parameter estimated as a fixed effect); PK, pharmacokinetic; Q, intercompartmental clearance; RSE, relative standard error; V2, central volume of distribution; V3, peripheral volume of distribution.
The PK properties of secukinumab were linear, with no evidence of a dose dependence of clearance; there was also no evidence of a time‐dependent change in the clearance. Clearance and volume varied with body weight in an allometric relationship. For clearance and central volume of distribution, the allometric exponents were estimated to be between 0.8 and 1.0; in other words, a doubling of body weight could lead to a nearly 2‐fold increase in clearance and distribution volume. Age, sex, race (Asian vs non‐Asian), geographic region, and baseline PASI score did not have a clinically relevant effect on secukinumab clearance after adjusting for body weight. The effects of model covariates are presented in Supplemental Table 1.
Figure 3 shows simulated concentration profiles, and Table 3 shows simulated PK metrics for the subcutaneous secukinumab dosing regimens used in phase 3 studies (300 or 150 mg at baseline; at weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48). Based on these simulations, Cavg at steady state was estimated to be 44.5 μg/mL for secukinumab 300 mg and 22.2 μg/mL for secukinumab 150 mg. Cmax at steady state was 55.2 μg/mL for secukinumab 300 mg and 27.6 μg/mL for secukinumab 150 mg, and tmax at steady state occurred approximately 6 days after dosing for both regimens. Terminal half‐life averaged approximately 27 days for both doses, with an IIV of 27.2% CV. For both dosing regimens, Cmax at steady state was about 2‐fold higher than Cmax after the first dose, which was in line with an expected accumulation ratio of 2 with a 4‐week dosing interval and a terminal half‐life of approximately 4 weeks. Figure 4 shows the actual serum concentrations of secukinumab from individual patients in the phase 3 ERASURE study.
Figure 3.
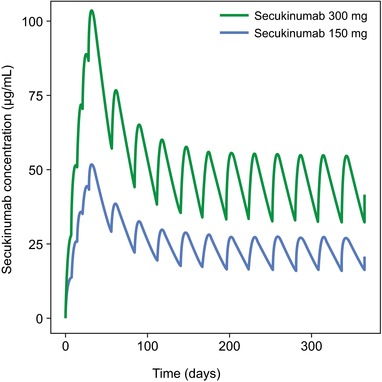
Simulated concentration profiles of secukinumab 300 and 150 mg with subcutaneous dosing regimens derived from phase 3 trials. Patients were simulated to receive secukinumab at baseline; weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48.
Table 3.
Simulated PK metrics from phase 3 studies of secukinumab
| Secukinumab 300‐mg Regimen | Secukinumab 150‐mg Regimen | |||||
|---|---|---|---|---|---|---|
| Parameter | Mean ± SD | %CV | Range (90%) | Mean ± SD | %CV | Range (90%) |
| Cmin at week 12, μg/mL | 44.2 ± 20.6 | 46.5 | 17.5–83.0 | 22.1 ± 10.3 | 46.5 | 8.7–41.5 |
| Cmin at ss, μg/mL | 32.1 ± 15.4 | 48 | 12.9–62.9 | 16.0 ± 7.7 | 48 | 6.5–31.5 |
| Cavg at ss, μg/mL | 44.5 ± 18.4 | 41.3 | 21.1–77.9 | 22.2 ± 9.2 | 41.3 | 10.5–39.0 |
| AUC at ss, μg·day/mL | 1245.0 ± 514.8 | 41.3 | 590.4–2181.7 | 622.5 ± 257.4 | 41.3 | 295.2–1090.8 |
| Cmax after first dose, μg/mL | 27.3 ± 9.5 | 34.8 | 14.2–44.6 | 13.7 ± 4.8 | 34.8 | 7.1–22.3 |
| tmax after first dose, day | 5.8 ± 0.4 | 7.6 | 5.0–6.0 | 5.8 ± 0.4 | 7.6 | 5.0–6.0 |
| Cmax at ss, μg/mL | 55.2 ± 21.5 | 38.9 | 27.5–94.8 | 27.6 ± 10.7 | 38.9 | 13.7–47.4 |
| tmax at ss, day | 6.0 ± 1.1 | 17.7 | 4.0–8.0 | 6.0 ± 1.1 | 17.7 | 4.0–8.0 |
| Cmax overall, μg/mL | 103.7 ± 33.5 | 32.3 | 59.5–165.5 | 51.8 ± 16.7 | 32.3 | 29.8–82.8 |
| tmax overall, day | 32.3 ± 1.1 | 3.3 | 31.0–34.0 | 32.3 ± 1.1 | 3.3 | 31.0–34.0 |
| Terminal half‐life, day | 26.9 ± 7.3 | 27.2 | 16.8–41.0 | 26.9 ± 7.3 | 27.2 | 16.8–41.0 |
AUC, area under the curve; Cavg, average concentration in serum; Cmax, maximum concentration in serum; Cmin, minimum concentration in serum; CV, coefficient of variation; PK, pharmacokinetic; SD, standard deviation; ss, steady state; Tmax, time to maximum concentration in serum.
Figure 4.
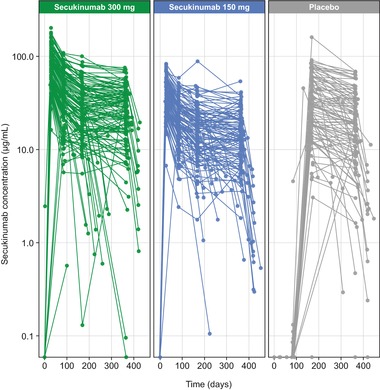
Observed serum secukinumab concentrations. Each line represents serum secukinumab concentrations from an individual patient in the phase 3 ERASURE trial.9 Secukinumab was administered at baseline; weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48. The last dose of secukinumab was given at week 48. Patients receiving placebo who did not achieve PASI 75 at week 12 were rerandomized to secukinumab 300 or 150 mg. Pharmacokinetic analysis was performed at baseline; weeks 4, 12, 24, 52, and 60; and during unscheduled visits.
Pharmacodynamics
Secukinumab binds to human IL‐17A and blocks the interaction of this cytokine with its receptor, functionally neutralizing its bioactivity. The complex of secukinumab with IL‐17A is eliminated at a slower rate than free IL‐17A, resulting in elevated levels of total IL‐17A (free + complexed) following secukinumab administration; thus, total IL‐17A is a relevant biomarker for secukinumab and is indicative of neutralization of IL‐17A by secukinumab.
Results from the phase 3 JUNCTURE study in patients with psoriasis showed that, at baseline, total (free) serum IL‐17A concentrations were below the total IL‐17A assay limit of quantification (20 pg/mL). At week 4, after weekly subcutaneous dosing with secukinumab 300 and 150 mg regimens, total IL‐17A levels increased to median values ranging from 121 to 142 pg/mL. The duration of increase in total IL‐17A was prolonged with increasing doses of secukinumab, and a plateau was reached at week 24 (Figure 5). It must be noted here that the observed increases in serum total IL‐17 were very similar for both dose levels and therefore clearly show target engagement. However, the observed time–concentration profiles for total IL‐17A do not allow for conclusions to be made about the level of suppression of free IL‐17, neither in the systemic circulation nor in the target tissue. For this purpose, evaluation of secukinumab PK and total serum concentrations of IL‐17A in a binding model would be needed.
Figure 5.
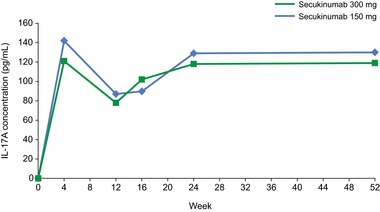
Observed serum total I‐17A concentrations. Secukinumab was administered at baseline; weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48. Pharmacodynamic analysis was performed at baseline and weeks 4, 12, 16, 24, and 52. Results represent median total IL‐17A concentrations from patients in the phase 3 JUNCTURE trial.13
Discussion
Results from this study confirm that secukinumab displays PK properties that are typical of a 150‐kDa human IgG1 antibody interacting with a soluble target (eg, long half‐life, slow serum clearance, and an allometric relationship between body weight and clearance and volume).16 The volume of distribution is low (<9 L) in patients with psoriasis following intravenous infusion, and clearance is low. At therapeutically relevant doses, exposure increases in a dose‐proportional manner. The PK properties of secukinumab are linear, with no evidence of a dose‐dependent change in clearance. The 2‐compartment population PK model adequately describes the observed secukinumab concentration–time courses, and external validation results suggest that the model will be predictive for PK outcomes of new studies (ie, studies not included in the model). When initiating treatment with secukinumab an induction phase allows for a rapid onset of effect.24 The PK parameters of secukinumab allow prescribing physicians to have confidence in obtaining desired exposure. For example, a dose of 300 mg results in approximately double the exposure compared with a 150‐mg dose. In addition, there is a clear difference in efficacy responses between secukinumab 300 mg and secukinumab 150 mg in patients with psoriasis.9 Secukinumab clearance values allow physicians to predict drug washout; assuming a washout period of 5 half‐lives, secukinumab will be washed out approximately 135 days after cessation of therapy. Distribution into skin, the target organ in psoriasis patients, appears to be fast, as skin interstitial fluid level of secukinumab is measurable after a single subcutaneous dose of 300 mg, reaching 28%–39% of the serum concentration 7 and 14 days postdose (6–8 μg/mL of secukinumab in the dermal interstitial fluid).18
These dermal concentrations of secukinumab seem largely sufficient to locally neutralize IL‐17A in the lesions. Indeed, baseline IL‐17A protein concentration was locally elevated in the dermal interstitial fluid of lesional psoriatic skin compared with nonlesional psoriatic skin and skin from healthy volunteers (9.8 vs 0.8 pg/mL), confirming the central role of IL‐17A in the pathogenesis of psoriasis.22 In contrast, IL‐17F (another IL‐17 cytokine that shares homology with IL‐17A and acts to regulate host defense and inflammatory responses)25 was not significantly increased in the skin of patients with psoriasis compared with healthy controls, and baseline IL‐17F levels did not significantly differ between psoriatic lesional and nonlesional skin. Similar to the findings for IL‐17A, and confirming its role as a disease marker, dermal baseline hBD‐2 concentrations were significantly higher in lesional skin of patients with psoriasis compared with nonlesional skin of patients with psoriasis or the skin of healthy volunteers. Because of the complex formation of IL‐17A with secukinumab, the effects of secukinumab on free IL‐17A could not be measured.22
Although the accumulation of total IL‐17A seems to plateau with the secukinumab 300‐ and 150‐mg dosing regimens, this plateau does not automatically imply a plateau in efficacy, as increasing the dose of monoclonal antibodies has been associated with further reduction of free target concentration and possible inhibition of downstream biomarkers and improved efficacy.26 To understand why the plateau in total target concentration does not imply a plateau in free target suppression and to understand how changes in the dose regimen may impact target engagement, it is useful to characterize the free and bound concentrations for the drug and target using dedicated models, which describe both target‐mediated drug disposition and the accumulation of total target during therapy. However, this was outside the scope of this article. Using such models specifically for the secukinumab–IL‐17A interaction will be described in an upcoming article.
These data suggest that secukinumab may act not only systemically but also locally in the dermis at concentrations capable of blocking the effects of IL‐17A, which in turn might explain the rapid effect on keratinocyte‐driven pathophysiology such as decreases in epidermal thickness and parakeratosis observed after 2 weeks of treatment.27 This rapid distribution of secukinumab to skin tissue may explain the rapid onset of a PD action. hBD‐2 has been shown to be upregulated in lesional psoriatic skin and in the serum of patients with psoriasis.28, 29 hBD‐2 has also been linked to keratinocyte hyperproliferation and chemoattraction of neutrophils—both important mechanisms in the pathogenesis of psoriasis.30, 31 In addition, serum and dermal interstitial fluid levels of hBD‐2 were significantly reduced to levels commonly found in healthy individuals 1–2 weeks after subcutaneous administration of a single dose of secukinumab. Results from our analysis indicate that total IL‐17A concentrations increase on initiation of secukinumab therapy, and a plateau is reached after 24 weeks for subcutaneous secukinumab doses of 150 mg and higher.
Conclusions
In summary, a 2‐compartment model can adequately describe secukinumab concentration–time courses; the PK properties of subcutaneous regimens of secukinumab 300 and 150 mg—administered at baseline; at weeks 1, 2, and 3; and then every 4 weeks from week 4 to week 48—are particularly well described by this model in terms of the central trend and variability. Secukinumab clearance and volume of distribution vary with body weight in an allometric relationship. Serum PK parameters and skin interstitial fluid concentrations of secukinumab contribute to the observed key pharmacodynamic effects (eg, increase of total IL‐17A and decrease of hBD‐2 in both serum and skin lesions). The fast and efficient distribution of secukinumab into the skin may contribute to the rapid onset of psoriatic lesion clearance. All these findings confirm the key role of IL‐17A in the pathogenesis of psoriasis.
Supporting information
Supplementary Image
Supplementary Image
Supplemental Table 1. Effects of Model Covariates on Clearance
Supplemental Figure 1. Visual Predictive Check.
Supplemental Figure 2. Visual Predictive Check for External Validation Data.
Acknowledgments
Editorial assistance was provided by Oxford PharmaGenesis, Inc., and was funded by Novartis Pharmaceuticals Corporation. The authors were fully responsible for all content and editorial decisions for this article.
Declaration of Conflicting Interests
Gerard Bruin, PhD, Christian Loesche, MD, and Oliver Sander, PhD, are full‐time employees of Novartis Pharma AG. Judit Nyirady, MD, is a full‐time employee of Novartis Pharmaceuticals Corporation.
Funding
This study was funded by Novartis Pharma AG.
References
- 1. Blauvelt A. T‐helper 17 cells in psoriatic plaques and additional genetic links between IL‐23 and psoriasis. J Invest Dermatol. 2008;128(5):1064–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71(1):141–150. [DOI] [PubMed] [Google Scholar]
- 3. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509. [DOI] [PubMed] [Google Scholar]
- 4. Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130(5):1373–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Di Cesare A, Di Meglio P, Nestle FO. The IL‐23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129(6):1339–1350. [DOI] [PubMed] [Google Scholar]
- 6. Nwe SM, Champlain AH, Gordon KB. Rationale and early clinical data on IL‐17 blockade in psoriasis. Expert Rev Clin Immunol. 2013;9(7):677–682. [DOI] [PubMed] [Google Scholar]
- 7. Lowes MA, Kikuchi T, Fuentes‐Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128(5):1207–1211. [DOI] [PubMed] [Google Scholar]
- 8. Krueger JG, Fretzin S, Suárez‐Fariñas M, et al. IL‐17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130(1):145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326–338. [DOI] [PubMed] [Google Scholar]
- 10. Leonardi C, Matheson R, Zachariae C, et al. Anti‐interleukin‐17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366(13):1190–1199. [DOI] [PubMed] [Google Scholar]
- 11. Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti‐interleukin‐17‐receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–1189. [DOI] [PubMed] [Google Scholar]
- 12. Blauvelt A, Prinz JC, Gottlieb AB, et al. Secukinumab administration by pre‐filled syringe: efficacy, safety, and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol. 2015;172(2):484–493. [DOI] [PubMed] [Google Scholar]
- 13. Paul C, Lacour JP, Tedremets L, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol. 2015;29(6):1082–1090. [DOI] [PubMed] [Google Scholar]
- 14. Mrowietz U, Leonardi CL, Girolomoni G, et al. Secukinumab retreatment‐as‐needed versus fixed‐interval maintenance regimen for moderate to severe plaque psoriasis: A randomized, double‐blind, noninferiority trial (SCULPTURE). J Am Acad Dermatol. 2015;73(1):27–36 e21. [DOI] [PubMed] [Google Scholar]
- 15. Papp KA, Langley RG, Sigurgeirsson B, et al. Efficacy and safety of secukinumab in the treatment of moderate‐to‐severe plaque psoriasis: a randomized, double‐blind, placebo‐controlled phase II dose‐ranging study. Br J Dermatol. 2013;168(2):412–421. [DOI] [PubMed] [Google Scholar]
- 16. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. [DOI] [PubMed] [Google Scholar]
- 17. Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–2668. [DOI] [PubMed] [Google Scholar]
- 18. Dragatin C, Polus F, Bodenlenz M, et al. Secukinumab distributes into dermal interstitial fluid of psoriasis patients as demonstrated by open flow microperfusion. Exp Dermatol. 2016;25(2):157–159. [DOI] [PubMed] [Google Scholar]
- 19. Gambichler T, Kobus S, Kobus A, et al. Expression of antimicrobial peptides and proteins in etanercept‐treated psoriasis patients. Regul Pept. 2011;167(2‐3):163–166. [DOI] [PubMed] [Google Scholar]
- 20. Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203(10):2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nograles KE, Zaba LC, Guttman‐Yassky E, et al. Th17 cytokines interleukin (IL)‐17 and IL‐22 modulate distinct inflammatory and keratinocyte‐response pathways. Br J Dermatol. 2008;159(5):1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolbinger F, Loesche C, Valentin M, et al. β‐Defensin‐2 is a responsive biomarker of IL‐17A‐driven skin pathology in psoriasis [published online ahead of print 2016]. J Allergy Clin Immunol. doi: 10.1016/j.jaci.2016.06.038 3 [DOI] [PubMed] [Google Scholar]
- 23. Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin‐17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2(52):52ra72. [DOI] [PubMed] [Google Scholar]
- 24. Rich P, Sigurgeirsson B, Thaci D, et al. Secukinumab induction and maintenance therapy in moderate‐to‐severe plaque psoriasis: a randomized, double‐blind, placebo‐controlled, phase II regimen‐finding study. Br J Dermatol. 2013;168(2):402–411. [DOI] [PubMed] [Google Scholar]
- 25. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin‐17 family members. Immunity. 2011;34(2):149–162. [DOI] [PubMed] [Google Scholar]
- 26. Lowe PJ, Kümmel A, Vasalou C, Matsushima S, Skerjanec A. Integrated quantitation of biotherapeutic drug–target binding, biomarkers, and clinical response to support rational dose regimen selection. Pharm Sci Encycl. 2015;13:1–21. [Google Scholar]
- 27. Reich K, Papp KA, Matheson RT, et al. Evidence that a neutrophil‐keratinocyte crosstalk is an early target of IL‐17A inhibition in psoriasis. Exp Dermatol. 2015;24(7):529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gambichler T, Skrygan M, Tomi NS, et al. Differential mRNA expression of antimicrobial peptides and proteins in atopic dermatitis as compared to psoriasis vulgaris and healthy skin. Int Arch Allergy Immunol. 2008;147(1):17–24. [DOI] [PubMed] [Google Scholar]
- 29. Jansen PA, Rodijk‐Olthuis D, Hollox EJ, et al. β‐Defensin‐2 protein is a serum biomarker for disease activity in psoriasis and reaches biologically relevant concentrations in lesional skin. PLoS One. 2009;4(3):e4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niyonsaba F, Ushio H, Nakano N, et al. Antimicrobial peptides human beta‐defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J Invest Dermatol. 2007;127(3):594–604. [DOI] [PubMed] [Google Scholar]
- 31. Niyonsaba F, Ogawa H, Nagaoka I. Human β‐defensin‐2 functions as a chemotactic agent for tumour necrosis factor‐α‐treated human neutrophils. Immunology. 2004;111(3):273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Image
Supplementary Image
Supplemental Table 1. Effects of Model Covariates on Clearance
Supplemental Figure 1. Visual Predictive Check.
Supplemental Figure 2. Visual Predictive Check for External Validation Data.