

Abstract
Ultrasmall nanoparticles (USNPs), usually defined as NPs with core in the size range 1–3 nm, are a class of nanomaterials which show unique physicochemical properties, often different from larger NPs of the same material. Moreover, there are also indications that USNPs might have distinct properties in their biological interactions. For example, recent in vivo experiments suggest that some USNPs escape the liver, spleen, and kidney, in contrast to larger NPs that are strongly accumulated in the liver. Here, we present a simple approach to study the biomolecular interactions at the USNPs bio‐nanointerface, opening up the possibility to systematically link these observations to microscopic molecular principles.
Keywords: bioinorganic chemistry, electrophoresis, nanoparticles, proteins, proteomics
In the last two decades, nanoparticles (NPs) have been intensively investigated for their potential in a wide range of applications, including biomedicine.1 Ultrasmall NPs (USNPs, considered core size 1–3 nm) can behave significantly different from “larger” NPs of the same material, sometimes presenting unique magnetic, electrical, optical, and catalytic properties.2 In the last 10 years, the biological interactions of NPs have been extensively studied, showing that, when exposed to biological media, all NPs (even with specially prepared surfaces) interact with biomolecules (proteins, lipids, etc.) that adsorb onto their surface, forming the so‐called “biomolecular corona”.3 This new biological identity can determine the final fate of NPs in living organisms.4
Most early in vivo biodistribution studies report a common tendency of NPs to accumulate extensively in the liver, with the attendant potential for toxicity.5 This tendency remains surprisingly high (30–60 %) even with specialized surface coatings and may have significant implications for long‐term toxicity and actively targeted drug delivery using NPs.6 However, several reports also suggest that USNPs accumulate less in the liver.7 It has been reported that 2 nm glutathione‐coated gold nanoparticles (GNPs) exhibit efficient renal clearance, with approximately 4 % of the NPs accumulated in the liver, 9 % in the kidney, 5 % in the lung, and 0.3 % in the spleen.8 Moreover, it has been reported that luminescent 2.7 nm (core size) tiopronin‐capped GNPs accumulate in the liver and kidneys.7b On the other hand, several examples of NPs of sizes less than 2 nm are known to exhibit much less liver accumulation.7c
We certainly expect size effects themselves to be significant in, for example, renal clearance where glomerular filtration is known to be size‐dependent and indeed appears to have a filtration‐size threshold. As the particle size becomes very small, the interactions with biomolecules are generally expected to diminish, and those that remain may lead to quite different organizations in which multiple particles interact with a single protein, rather than the reverse.9 The fact that the overall biodistribution could result from different size‐dependent effects (physical filtration and biomolecular associations), combined with the absence of any generally accessible and systematic way of studying the nature (or even presence) of these biomolecular associations, makes it difficult to progress systematic understanding of these effects. Several interesting studies have clarified aspects of the biomolecular interactions with ultrasmall nanoparticles.10
Here, in order to make contact with the in vivo studies we focus on consequences of particle size and surface on the interactions between USNPs and concentrated biological fluids. We investigate the transition regime between examples in which several proteins can bind relatively irreversibly to the particle, and those where all of the proteins are in rapid exchange, and none bind strongly. We find this transition to be quite sensitively dependent on small changes in size and surface chemistry. We chose gold as an illustrative system and note the convenience it provides in practical biodistribution studies.11 Three representative sizes have been chosen as models: 2 nm GNP (ultrasmall size range), 3 nm GNP and 5 nm GNP, the latter typically considered beyond the ultrasmall range. Moreover, an additional series of 2 nm GNPs with different surface functionalities has been included in the study in order to investigate the role of surface charge/chemistry (see Scheme 1).
Scheme 1.
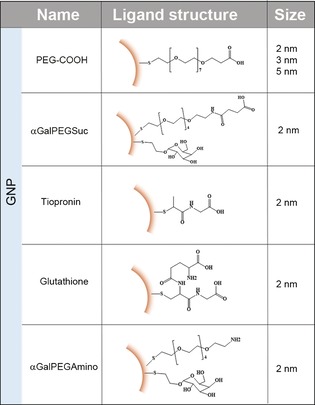
Summary of the GNPs used in this study including size and schematic representation of the ligand chemical structure.
A family of negatively charged monodisperse GNPs functionalized with SH‐PEG(7)‐COOH ligands (GNP‐PEG‐COOH) was synthesized and characterized (Figure 1, Figures S1, S2, S8a, and S10 in the Supporting Information).
Figure 1.
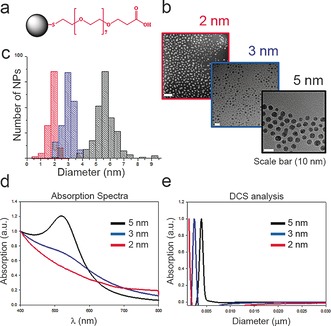
Characterization of 2, 3 and 5 nm GNP‐PEG‐COOH: a) Schematic ligand representation; b) TEM images and c) TEM size distribution; d) Absorption spectra; e) Size distribution as measured by DCS analysis.
Clearly, the approaches previously developed to study biomolecular corona formation on larger NPs (10–100 nm) are no longer appropriate for the study of USNPs which can be of comparable size, or even smaller than the biomolecules themselves. Thus the ultrasmall range is located at the edge (or below) of resolution limits of the routinely employed instruments for NPs characterization such as dynamic light scattering (DLS), differential centrifugal sedimentation (DCS) or transmission electron microscopy (TEM) and to isolate the NP–protein complexes is extremely challenging. Analytical ultracentrifugation (AUC) and fluorescence correlation spectroscopy (FCS) are very useful and sophisticated techniques that have been successfully employed to investigate USNP–protein interactions, mainly using single proteins.12
We used a series of gel assays for in situ investigation of biomolecules–USNPs interactions.13 The GNP‐PEG‐COOH samples (5 nm, 3 nm and 2 nm) were normalized by surface area and exposed to human plasma (HP). Samples were then incubated with 70 % v/v of HP to ensure protein content was in excess. The mixtures were then incubated for 30 minutes and loaded into the wells of an agarose gel next to the corresponding control (NP suspension in phosphate‐buffered saline, PBS).
We observed (Figure 2) that for 5 nm (lanes 1 and 2) and 3 nm (lanes 3 and 4) GNP‐PEG‐COOH NP–protein interactions led to significant changes in NPs mobility (as can be seen from the smear in lanes 2 and 4). In contrast, 2 nm GNP‐PEG‐COOH particles exhibit few signs of protein interaction. Different voltage and gel pore size conditions are reported in Figures S3–5 showing similar effects, leading us to conclude that larger particles form more conventional protein corona complexes (illustrated by the evident shift in mobility of the bio‐nanocomplex compared to free NPs), while smaller ones do not. In order to gain further insight into this process, we recorded a full video of NP progression through the agarose gel. For this experiment, a 3.5 % agarose gel was used in order to increase the resolution and observe smaller differences (see Figure S5). For instance, 2 nm GNP‐PEG‐COOH demonstrated slightly lower electrophoretic mobility in comparison to the NP control in PBS, at very early time points. This difference can be mainly observed when the NPs enter in the gel (Figure S5). In addition, for the gels in Figures S6–7, gold particle‐protein complexes were isolated by excising the band of interest in the gel and subjected to mass spectrometry (MS) analysis to identify the proteins involved (see Figure 2 b and Table S1). There is considerable smearing of electrophoresed gold–biomolecule complexes, possibly reflecting that, for small NP sizes, the interaction of so many different proteins (roughly 3700) generates significant heterogeneity in complex size and composition. It is, however, striking that the qualitative nature of the interactions should change so precipitously around a specific size (2 nm), with highly heterogeneous complexes forming in the vicinity of this transition.
Figure 2.
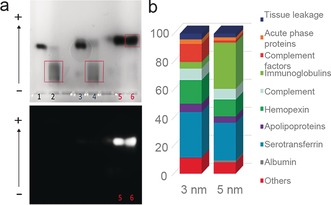
a) 1 % Agarose gel‐assay in a native buffer. The numbered lanes refer to 5 nm GNP‐PEG‐COOH in PBS (1) and in HP (2), 3 nm GNP‐PEG‐COOH in PBS (3) and in HP (4), 2 nm GNP‐PEG‐COOH in PBS (5) and in HP (6). Below, the image captured by fluorescence detection mode. Note: only 2 nm GNP‐PEG‐COOH exhibit fluorescence. b) Mass spectrometry analysis of 3 nm and 5 nm GNP‐PEG‐COOH corona complexes isolated by agarose gel electrophoresis.
The observation that corona lifetimes should decrease with size is both intuitively obvious, and also consistent with earlier theoretical observations.14 However, it is intriguing that transient particle–protein associations with lifetimes shorter than gel band resolution times still lead to modified mobility whose consequences can be observed in “cyclic capture and dissociation” (CCD) dynamics.15 As shown in Figure 3 (lane 1 NPs alone, lane 3 HP alone and lane 2 HP first run into the gel, and then “chased” by nanoparticles) while the long term (45 minutes) outcome suggests a modest effect on overall (averaged) particle mobility, the temporal evolution of the CCD dynamics is much more revealing. Thus, (see Figure 3) at early stages (before 10 minutes) the higher electrophoretic mobility of the chasing NPs allows them to overtake the first of the two major HP bands, which they appear to pass without significant retardation. However NPs reach the lower molecular weight plasma region, where they are strongly retarded (Figure 3, t=20–25 min), and follow the protein band until, after around 35 minutes, they move independently again. We have found that these effects are typical of particles (and particle surfaces) that are believed to have transient associations, and the retardations were (in molecular systems)15 interpreted as a consequence of repetitive binding and unbinding of particle and protein within the band, though in our more complex system deduction of explicit affinities will be more challenging.
Figure 3.
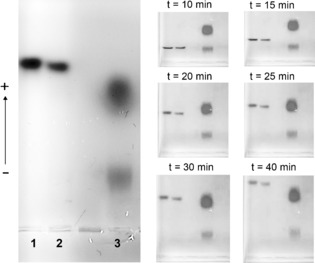
CCD experiment in 1 % agarose gel including samples: 2 nm GNP‐PEG‐COOH in PBS (1) and in HP (2), HP stained with SYPRO® orange dye (3).
We stress that the transition between the conventional long‐lived “hard corona” complexes and these non‐interacting (or “pseudo‐corona”) scenarios is a consequence of the weakening particle‐biomolecular interactions, which is parameterized by both particle size and particle surface. This surface chemistry effect may be illustrated using identical core particles but different ligands (Scheme 1, characterisation reported in Figures S8–10 and Table S2). Thus (Figure 4) tiopronin (2 nm GNP‐tiopronin, lanes 5 and 6) and glutathione ligands (2 nm GNP‐glutathione lanes 7 and 8) show significant biomolecular corona interactions, whereas PEG‐type ligands (2 nm GNP‐PEG‐COOH, lanes 1 and 2 and 2 nm GNP‐α GalPEGSuc, lanes 3 and 4) do not. That outcome is consistent with expectations and may reflect the fact that the former have shorter chain lengths and higher net charge.16
Figure 4.
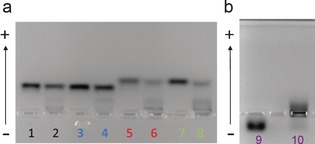
3.5 % Agarose gel‐shift assay experiment including samples: a) 2 nm GNP‐PEG‐COOH in PBS (1) and in HP (2), 2 nm GNP‐αGalPEGSuc in PBS (3) and in HP (4), 2 nm GNP‐tiopronin in PBS (5) and in HP (6), 2 nm GNP‐glutathione in PBS (7) and in HP (8); b) 2 nm GNP‐αGalPEGAmino in PBS (9) and in HP (10).
Furthermore, as expected, positively charged 2 nm GNP‐αGalPEGAmino (similar to GNP‐αGalPEGSuc but with opposite charge) also interact strongly with plasma proteins (lanes 9 and 10).
In addition to the smearing observed in Figure 4, there is evidence of a bimodal behavior (see Figure S11) in which some proteins bind and others do not (Figure 4 lanes 6 and 8). This may arise as a consequence of particle heterogeneity (for example ligand density or size), then strongly highlighted by differential protein binding. These observations are consistent with our experience, as well as being of some significance in planning the fabrication of ultrasmall nanodrugs.
When methods are applied in new ways one should exercise caution.17 For instance, we have sought to study different conditions (electrical field, pore size, etc.) to assure ourselves that this does not in itself greatly influence the exchange process (see Figure S3). With these reservations we may summarise as follows. Firstly, size (in combination with surface) may be used to control, indeed even nearly eliminate, long‐lived interactions between particle and the biomolecular environment. This suggests a transition between the regime where the particle corona identity is relatively fixed3b to one where it fluctuates rapidly. Certainly, this will lead to quite distinct biological outcomes, potentially interpolating between (non‐associating) molecular drug, and more conventional nanoparticle activity, all of which may be “pre‐screened” prior to biological or in vivo studies.
We also stress that, from a practical perspective, in this ultrasmall size range, even very small particle variations can lead to quite different biophysical interactions, reinforcing the role of “particle quality” for this regime.
Finally, the possibility, enabled by the capacity to purposefully engineer the particle corona, to form transient complex interactions, could lead to a much richer range of “particle–biomolecule” receptor target interactions, qualitatively different from molecules or larger particles. The degree to which receptor recognition paradigms could thereby be enriched is an interesting topic for further considerations.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge the UCD Conway Mass Spectrometry Facility and the Trinity College CRANN Advanced Microscopy Laboratory facility. We acknowledge Jennifer Cookman for help with the TEM analysis. We acknowledge Midatech Pharma for helpful support and scientific discussions. L.B. acknowledges the financial support of the EU H2020 Nanofacturing project (grant agreement number 646364). E.P. and K.A.D. acknowledge the Science Foundation Ireland (SFI) Principal Investigator Award 298 (agreement number 12/IA/1422). V.C. acknowledges the Irish Research Council (GOIPD/2016/128).
L. Boselli, E. Polo, V. Castagnola, K. A. Dawson, Angew. Chem. Int. Ed. 2017, 56, 4215.
Contributor Information
Dr. Luca Boselli, Email: luca.boselli@cbni.ucd.ie.
Prof. Kenneth A. Dawson, Email: kenneth.a.dawson@cbni.ucd.ie
References
- 1.
- 1a. Dreaden E. C., Alkilany A. M., Huang X., Murphy C. J., El-Sayed M. A., Chem. Soc. Rev. 2012, 41, 2740–2779; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Ferrari M., Nat. Rev. Cancer 2005, 5, 161–171; [DOI] [PubMed] [Google Scholar]
- 1c.J. Shi, P. W. Kantoff, R. Wooster, O. C. Farokhzad, Nat. Rev. Cancer 2016, advance online publication.
- 2.
- 2a. Kim B. H., Hackett M. J., Park J., Hyeon T., Chem. Mater. 2013, 26, 59–71; [Google Scholar]
- 2b. Zarschler K., Rocks L., Licciardello N., Boselli L., Polo E., Garcia K. P., De Cola L., Stephan H., Dawson K. A., Nanomedicine 2016, 12, 1663–1701. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Cedervall T., Lynch I., Foy M., Berggård T., Donnelly S. C., Cagney G., Linse S., Dawson K. A., Angew. Chem. Int. Ed. 2007, 46, 5754–5756; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5856–5858; [Google Scholar]
- 3b. Cedervall T., Lynch I., Lindman S., Berggård T., Thulin E., Nilsson H., Dawson K. A., Linse S., Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Monopoli M. P., Aberg C., Salvati A., Dawson K. A., Nat. Nanotechnol. 2012, 7, 779–786. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Salvati A., Pitek A. S., Monopoli M. P., Prapainop K., Bombelli F. B., Hristov D. R., Kelly P. M., Åberg C., Mahon E., Dawson K. A., Nat. Nanotechnol. 2013, 8, 137–143; [DOI] [PubMed] [Google Scholar]
- 4b. Walkey C. D., Olsen J. B., Song F., Liu R., Guo H., Olsen D. W. H., Cohen Y., Emili A., Chan W. C. W., ACS Nano 2014, 8, 2439–2455. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Sadauskas E., Danscher G., Stoltenberg M., Vogel U., Larsen A., Wallin H., Nanomedicine 2009, 5, 162–169; [DOI] [PubMed] [Google Scholar]
- 5b. Sonavane G., Tomoda K., Makino K., Colloids Surf. B 2008, 66, 274–280. [DOI] [PubMed] [Google Scholar]
- 6. Wilhelm S., Tavares A. J., Dai Q., Ohta S., Audet J., Dvorak H. F., Chan W. C. W., Nat. Mater. Rev. 2016, 1, 16014. [Google Scholar]
- 7.
- 7a. Liu J., Yu M., Zhou C., Yang S., Ning X., Zheng J., J. Am. Chem. Soc. 2013, 135, 4978–4981; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Poon W., Heinmiller A., Zhang X., Nadeau J. L., J. Biomed. Opt. 2015, 20, 066007; [DOI] [PubMed] [Google Scholar]
- 7c. Zhang X.-D., Luo Z., Chen J., Song S., Yuan X., Shen X., Wang H., Sun Y., Gao K., Zhang L., Sci. Rep. 2015, 5, 8669; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Zhang X.-D., Wu D., Shen X., Liu P.-X., Fan F.-Y., Fan S.-J., Biomaterials 2012, 33, 4628–4638. [DOI] [PubMed] [Google Scholar]
- 8. Zhou C., Long M., Qin Y., Sun X., Zheng J., Angew. Chem. Int. Ed. 2011, 50, 3168–3172; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3226–3230. [Google Scholar]
- 9. Deng Z. J., Liang M., Toth I., Monteiro M. J., Minchin R. F., ACS Nano 2012, 6, 8962–8969. [DOI] [PubMed] [Google Scholar]
- 10. Moyano D. F., Saha K., Prakash G., Yan B., Kong H., Yazdani M., Rotello V. M., ACS Nano 2014, 8, 6748–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Gromnicova R., Davies H. A., Sreekanthreddy P., Romero I. A., Lund T., Roitt I. M., Phillips J. B., Male D. K., PLOS ONE 2013, 8, e81043; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Khlebtsov N., Dykman L., Chem. Soc. Rev. 2011, 40, 1647–1671; [DOI] [PubMed] [Google Scholar]
- 11c. Lipka J., Semmler-Behnke M., Sperling R. A., Wenk A., Takenaka S., Schleh C., Kissel T., Parak W. J., Kreyling W. G., Biomaterials 2010, 31, 6574–6581. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Bekdemir A., Stellacci F., Nat. Commun. 2016, 7, 13121; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Carney R. P., Kim J. Y., Qian H., Jin R., Mehenni H., Stellacci F., Bakr O. M., Nat. Commun. 2011, 2, 335; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Röcker C., Potzl M., Zhang F., Parak W. J., Nienhaus G. U., Nat. Nanotechnol. 2009, 4, 577–580. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Lin C. A., Sperling R. A., Li J. K., Yang T. Y., Li P. Y., Zanella M., Chang W. H., Parak W. J., Small 2008, 4, 334–341; [DOI] [PubMed] [Google Scholar]
- 13b. Lin C. A., Yang T. Y., Lee C. H., Huang S. H., Sperling R. A., Zanella M., Li J. K., Shen J. L., Wang H. H., Yeh H., Parak W. J., Chang W. H., ACS Nano 2009, 3, 395–401. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Lacerda S. H. D. P., Park J. J., Meuse C., Pristinski D., Becker M. L., Karim A., Douglas J. F., ACS Nano 2010, 4, 365–379; [DOI] [PubMed] [Google Scholar]
- 14b. Lopez H., Lobaskin V., J. Chem. Phys. 2015, 143, 243138. [DOI] [PubMed] [Google Scholar]
- 15. Belotserkovskii B. P., Johnston B. H., Biophys. J. 1997, 73, 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Ashraf S., et al., Nanoscale 2016, 8, 17794–17800; [DOI] [PubMed] [Google Scholar]
- 16b. Pombo-García K., et al., Chem. Nano. Mater. 2016, 2, 959–971; [Google Scholar]
- 16c. Schöttler S., Becker G., Winzen S., Steinbach T., Mohr K., Landfester K., Mailänder V., Wurm F. R., Nat. Nanotechnol. 2016, 11, 372–377. [DOI] [PubMed] [Google Scholar]
- 17.C. Holm. Traffic and Granular Flow’03, 2005, 475–488.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary