

Abstract
To inhibit binding of the influenza A virus to the host cell glycocalyx, we generate multivalent peptide–polymer nanoparticles binding with nanomolar affinity to the virus via its spike protein hemagglutinin. The chosen dendritic polyglycerol scaffolds are highly biocompatible and well suited for a multivalent presentation. We could demonstrate in vitro that by increasing the size of the polymer scaffold and adjusting the peptide density, viral infection is drastically reduced. Such a peptide–polymer conjugate qualified also in an in vivo infection scenario. With this study we introduce the first non‐carbohydrate‐based, covalently linked, multivalent virus inhibitor in the nano‐ to picomolar range by ensuring low peptide‐ligand density on a larger dendritic scaffold.
Keywords: antiviral agents, in vivo studies, influenza virus, multivalency, peptides
Multivalent binding to the host cell surface is a common strategy of enveloped viruses as influenza A virus (IAV). IAV binds to the host cell surface via its trimeric spike protein hemagglutinin (HA), which recognizes sialic acids (SA) on the glycocalyx of cellular membranes.1, 2 Despite the low binding affinity of individual hemagglutinin receptor binding sites to SA (ca. 2 mm), a stable adhesion originates from multiple ligand–receptor binding events between the virus (0.02–0.04 HA nm−2) and the SA glycoconjugates on the cell surface (0.5–2 SA nm−2).3, 4
Building upon this naturally occurring multivalent binding mechanism, synthetic multivalent entry blockers were introduced in the 1990s by Whitesides and co‐workers.3, 5, 6, 7 High molecular‐weight scaffolds displaying a large number of low affinity SA derived ligands were used to achieve high HA avidity. Over the years several carrier systems were employed as scaffolds ranging from polymers,6, 8, 9 dendrimers,10, 11, 12, 13 liposomes,5, 14 proteins,15 to gold nanoparticles.16 The most affine binders reported to date consist of SA tethered to linear polyacrylamide polymers.6 Although they have a high inhibitory potential, polyacrylamide‐based inhibitors and their degradation products often show high cytotoxicity.6, 17, 18, 19, 20 Polyamidoamine (PAMAM) dendrimers have also been used for multivalent display, for example, of siallyllactose21 showing in vitro inhibition at micromolar ligand concentrations as well as protection of mice from infection. However, depending on their structure multivalent PAMAM scaffolds may also be cytotoxic.17, 22, 23
Promisingly, biocompatible polyglycerol (PG)‐based SA‐nanoparticles have been shown to reduce viral infection in vitro by up to 80 % when applied in millimolar ligand, respectively submicromolar nanoparticle concentrations.8 The importance of low receptor density and larger particle size was emphasized in this study similar to the PAMAM study.21
Alternative ligands to SA are peptides targeting HA.24, 25, 26 Peptides may have several advantages, including higher affinity and selectivity. They can be produced and conjugated to different polymeric scaffolds by straightforward synthetic methods as opposed to often synthetically challenging routes for stereochemically defined SA building blocks. Peptide affinity towards receptors can be easily optimized by phage display, microarray screening,27, 28, 29 as well as chemical modification.
Indeed, in 2006 a peptide derived from the signal sequence of fibroblast growth factor 4 (FGF 4) was shown to inhibit infection by the influenza virus.25 Thereafter, a peptide was optimized for HA binding by selection from a phage‐displayed random peptide library.24 This work also showed a multivalent improvement of its binding properties by conjugation to either an amphiphilic N‐stearoyl derivative, which formed self‐assembled multivalent structures with enhanced binding capabilities or by presenting up to six peptide copies on a carbosilane dendrimer.24, 30, 31
We recently reported on a peptide (PeB) derived from the CDR domain of a HA binding antibody which binds in proximity to the SA binding pocket of HA. The binding property of PeB was improved upon MD simulations and microarray screening, leading to the double mutant PeBGF, which binds to HA in the micromolar range.32 In line with the self‐assembly strategy, we synthesized amphiphilic stearoyl‐PeBGF to form a noncovalent multivalent receptor construct.33 Unfortunately, stearoyl‐PeBGF did not form defined micelles. We observed structures, such as sheets and fibers. As such scaffolds are not able to retain their structural integrity in a cellular (membrane) environment it challenges the general applicability of the self‐assembly strategy for the design of multivalent antiviral peptides.
Herein, we introduce covalent conjugates of peptidic ligands and a biocompatible PG‐based hydrophilic dendritic scaffold to provide uniform and structurally stable multivalent inhibitors, which turn out to be high affinity virus binders with low toxicity in cellular as well as in animal models. For synthesis of dendritic peptide‐PG scaffold we envisioned the conjugation of an alkyne equipped HA binding PeB variants to a dendritic azido‐PG34 by Cu catalyzed alkyne azide cycloaddition (CuAAC)35 (Figure 1).
Figure 1.
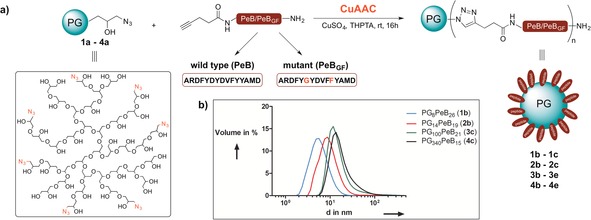
a) Scheme for CuAAC coupling between peptide and polymer (for polymer sizes see Table 1). b) DLS data for the screening of different molecular‐weight dendritic PGs conjugated with PeB peptides.
PeB and PeBGF were synthesized by solid‐phase peptide synthesis with a final N‐terminal coupling of 4‐pentynoic acid. To identify the optimal scaffold for a multivalent inhibitor, we chose dendritic PGs with different molecular weights and degrees of azide functionalization for peptide conjugation. CuAAC was carried out in basic ammonium bicarbonate buffer at room temperature overnight and the reaction mixture was pre‐purified by dialysis, followed by size exclusion chromatography.36 The conjugation products were analyzed by NMR spectroscopy and DLS to obtain the size and number of ligands for each peptide–polymer conjugate (Table 1 and Supporting Information). Particles with large polymer backbone were visualized by cryo‐TEM (Figure S19–S21). Note that the ligand number in Table 1 is an average, owing to the polymeric molecular weight distribution of polyglycerol (see Table S1).
Table 1.
Overview of PG peptide conjugates.
| No. | Compound | Mp core [kDa] | DF[b] [%] | Lig.[c] | Diameter[d] [nm] | Density[e] [Lig. nm−2] |
|---|---|---|---|---|---|---|
| 1 a | dPG8 | 7.7 | 0 | 0 | 2.4±0.1 | 0.0 |
| 1 b | dPG8PeB26 | 7.7 | 26 | 24 | 6.4±0.7 | 1.3 |
| 1 c | dPG8PeBGF 30 | 7.7 | 30 | 31 | 10.3±0.6 | 1.7 |
| 2 a | dPG14 | 14.4 | 0 | 0 | 5.1±0.5 | 0.0 |
| 2 b | dPG14PeB19 | 14.4 | 19 | 37 | 11.3±3.1 | 0.5 |
| 2 c | dPG14PeBGF 19 | 14.4 | 19 | 36 | 11.3±1.4 | 0.5 |
| 3 a | dPG100 | 100 | 0 | 0 | 8.0±0.4 | 0.0 |
| 3 b | dPG100PeB8 | 100 | 8 | 108 | 16.5±2.7 | 0.5 |
| 3 c | dPG100PeB21 | 100 | 21 | 284 | 20.2±2.2 | 1.4 |
| 3 d | dPG100PeBGF 10 | 100 | 10 | 135 | 22.4±2.8 | 0.7 |
| 3 e | dPG100PeBGF 29 | 100 | 29 | 392 | 15.1±0.7 | 2.0 |
| 4 a | dPG340 | 340 | 0 | 0 | 11.7±0.4 | 0.0 |
| 4 b | dPG340PeB9 | 340 | 9 | 414 | 17.9±0.9 | 1.0 |
| 4 c | dPG340PeB15 | 340 | 15 | 690 | 24.8±4.3 | 1.7 |
| 4 d | dPG340PeBGF 10 | 340 | 10 | 460 | 27.6±5.9 | 1.1 |
| 4 e | dPG340PeBGF 16 | 340 | 16 | 736 | 29.8±1.4 | 1.8 |
[a] Molar mass at peak maximum (Mp) of the dendritic PG. [b] DF of all end groups determined by 1H NMR spectroscopy. [c] Average number of ligands (Lig.) calculated from the DF. [d] Diameter determined from DLS measurements in 10 mm ammonium bicarbonate. Values represent the means with the standard error of the mean (s.e.m.) of at least three experiments. [e] Ligand density based on mean diameter of a sphere divided by the amount of ligands.
All compounds were tested for their binding inhibition potential with the hemagglutination inhibition assay (HAI).6 In short, we pretreated the IAV Aichi H3N2 (X31) with different concentrations of inhibitor and incubated this suspension with human erythrocytes. Virus binding to red blood cells was indicated by agglutination, which is visible as gel in the well plate. The lowest concentration of inhibitor that is able to inhibit agglutination was defined as inhibition constant (K i HAI). With increasing size of the PG backbone, the K i HAI dropped to low nanomolar values as observed for dPGs with a molecular weight of 340 kDa (Figure 2 and Table 2). A higher degree of functionalization (DF) did not substantially improve the inhibitory potential of the conjugates. In one case higher valency even reduced the inhibitory effect (4 c compared to 4 b). In contrast to the monovalent case,32 upon multivalent peptide presentation PeBGF was not superior to PeB in the HAI assay. Notably, unspecific binding of the inhibitor to the cell membrane cannot be excluded, which would reduce the effective inhibitor concentration. Therefore, we used microscale thermophoresis (MST) to measure directly the binding of inhibitors to IAV. To this end, we chose the best inhibitors 4 b and 4 d identified by the HAI assay displaying either PeB or PeBGF at low valency. MST was conducted by titration against octadecyl rhodamine B chloride (R18) labeled X31 virus to derive apparent affinity constants (KDapp) against intact virus, free in solution. While compound dPG340PeB9 (4 b) showed a KDapp of 36.6 μm (corresponding to 88.3±13.6 nm of the multivalent nanoparticles), dPG340PeBGF 10 (4 d) bound with 10‐times higher affinity (3.1 μm peptide or 6.8±1.1 nm multivalent nanoparticles) to the virus.
Figure 2.
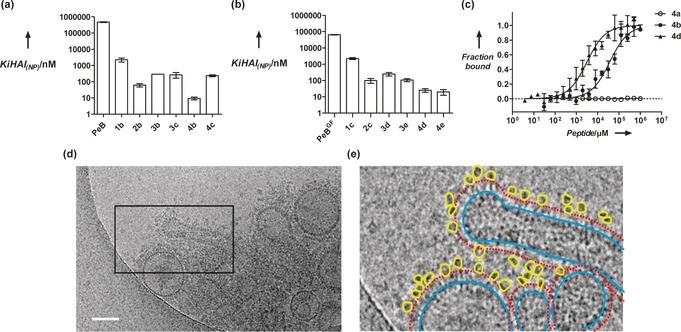
Inhibition of influenza virus X31 binding. Hemagglutination inhibition constants as a measure for binding inhibition was derived from HAI assays with a) the wildtype peptide PeB or b) the double mutant PeBGF, both in the monomeric form and compared to the multivalent form on dPG scaffolds with different sizes. Error bars correspond to the standard error of the mean (s.e.m., n≥3). Nomenclature is given in Table 2. c) Binding curves obtained from MST measurements with 340 kDa PGs (4 a=unconjugated dPG340, 4 b=PeB conjugated, 4 d=PeBGF conjugated) against fluorescently labeled X31 virus. Error bars indicate the s.e.m. (n≥3). Curves were fitted according to the function for law of mass action (see Supporting Information). d) Cryo‐TEM image of PG340PeBGF 10 (4 d) incubated with Influenza virus X31. While the best‐binding compound 4 d interacts with virus HA (compare also stereo images in Figure S21) PG340N3 (4 a) shows no binding (see Figure S20). Scale bars correspond to 100 nm. e) The marked image section of (d) (black frame) in detail, bound PG peptide conjugates are outlined in yellow, virions are outlined in blue and the corona formed by HA is marked with a red dashed line.
Table 2.
Inhibition of X31‐mediated hemagglutination of human erythrocytes and infection of MDCK II cells.
| No. | K i HAI Lig. [μm][a] | K i HAI NP [nm][b] | IC50 Lig [μm][c] | IC50 NP [nm][d] |
|---|---|---|---|---|
| 1 a | – | – | – | – |
| 1 b | 54±15 | 2250±629 | – | – |
| 1 c | 69±9 | 2218±295 | – | – |
| 2 a | – | – | – | – |
| 2 b | 2±1 | 62±14 | – | – |
| 2 c | 4±1 | 99±32 | 34.6±0.1 | 960.6±2.7 |
| 3 a | n.d. | n.d. | – | – |
| 3 b | 31±0 | 289±0 | 0.8±0.1 | 7.5±1.0 |
| 3 c | 75±29 | 264±102 | 4.4±0.6 | 15.4±0.6 |
| 3 d | 33±8 | 247±62 | 2.5±0.1 | 18.5±0.5 |
| 3 e | 42±8 | 106±21 | 17.3±0.1 | 44.2±0.2 |
| 4 a | – | – | – | – |
| 4 b | 4±1 | 9±2 | 0.3±0.1 | 0.6±0.3 |
| 4 c | 163±22 | 236±31 | 69.5±0.0 | 100.7±0.1 |
| 4 d | 12±3 | 25±6 | 0.2±0.0 | 0.4±0.1 |
| 4 e | 15±6 | 20±7 | 0.9±0.0 | 1.2±0.1 |
[a,b] For values derived from the HAI assay the s.e.m. (n≥3) is given in terms of the ligand concentration [a], or nanoparticle concentration [b]. [c,d] Ligand [c] and nanoparticle [d] IC50 values derived from a four parametric logistic fit are shown together with its asymmetric standard error (SE) of the logIC50. n.d.=not determined. (–)=no inhibition or binding.
The unconjugated compound dPG340‐azide (4 a) did not interact with the virus (Figure 2 c). This result confirms our previous findings for monomeric peptides, in which PeBGF showed higher binding affinity towards X31 than the wildtype peptide PeB.32 Interaction of dPG340PeBGF 10 (4 d) with X31 virus was also detected by cryo‐TEM (Figure 2 d,e and Figure S21), while the unconjugated inhibitor did not interact with the virus (Figure S20).
We investigated the infection‐inhibition potential of all the peptide–PG conjugates in a cell‐viability assay (Figure 3) after infecting MDCK II cells with a virus‐inhibitor mixture. Cell viability was recorded with a MTS cell proliferation assay to determine viral‐infection inhibition post‐infection. Notably, all tested peptide–PG conjugates were non‐toxic, when tested in concentrations of up to 200 μm peptide (Figure S17). In accordance with the HAI assay, peptide presentation on larger scaffolds substantially increased the inhibitory potential as evident by comparing the dissociation constants. We attribute this observation to the steric‐shielding character of the multivalent scaffolds, which can contribute additionally to the affinity of a multivalent compound.37 We found that best inhibitors were PG340 conjugates with a low DF for both, the PeB (4 b) and PeBGF (4 d). Both were able to inhibit infection at nanomolar peptide and picomolar nanoparticle concentrations, respectively (Figure 3 and Table 2) corresponding to a multivalent enhancement of inhibition by a factor of 129 for PeB and 136 for PeBGF for the multivalently presented peptides compared to the monovalent peptide (see Supporting Information). We also monitored viral nucleoprotein (NP) expression in MDCK II cells infected with X31 pretreated with the inhibitors 4 b and 4 d.
Figure 3.
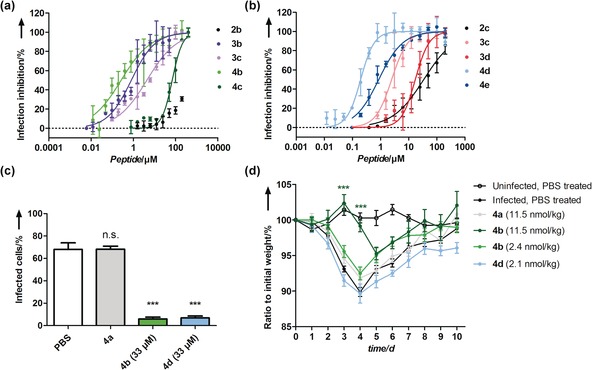
Inhibition of influenza A virus X31 infection by multivalent peptide presenting PGs. a),b) Inhibition of infection of MDCK II cells by PG‐PeB (a) and PG‐PeBGF (b) presenting variants, based on a cell viability assay (MOI 0.05). Error bars correspond to the s.e.m. (n≥3). Mean values from serial dilutions of each compound were fitted with a four parametric logistic fit. IC50 values are summarized in Table 2. c) Quantification of IAV nucleoprotein expression in MDCK II infected cells in the presence or absence of inhibitor at 33 μm peptide concentrations or equimolar nanoparticle concentrations for the non‐functionalized control 4 a. Error bars correspond to the s.e.m. (n≥3). Microscopy images are shown in Figure S18. d) In vivo infection experiments with BALB/c mice treated with pre‐incubated X31 at indicated inhibitor amounts at single‐dose intranasal administration. Error bars correspond to the s.e.m. (n≥5). Data in (c) and (d) have undergone statistical analysis using a one‐way ANOVA followed by a Dunnett's multiple comparison to the PBS treated, infected control. ***=p<0.0001.
While untreated virus at MOI 0.1 infected approximately 70 % of the cells, the virus treated with inhibitor concentrations as low as 33 μm peptide (corresponding to 79 nm 4 b and 71 nm 4 d) reduced viral NP expression significantly to less than 10 % infected cells (Figure 3 c and Figure S18). Treatment of virus with the control inhibitor dPG340N3 (4 a) did not lead to significant reduction of viral NP expression. After these promising results, we aimed to probe the peptide–PG conjugates 4 b and 4 d in in vivo experiments. Sedated 8‐weeks old BALB/c mice (body weight ca. 20 g) were infected upon an intranasal administration of an inhibitor–virus mix and their body weight monitored daily for 10 days.
After infection of mice with X31, illness is manifested, beside other symptoms, by a loss in body weight, which peaks at day 4–5 followed upon a full recovery starting on day 10 (see infected, PBS treated control, Figure 3 d).
While the control construct dPG340N3 (4 a) did not protect from infection (body weight loss), mice treated with a single dose dPG340PeB9 (4 b; 11.5 nmol kg−1) maintained their body weight until day 4 (Figure 3 d). A similar dosage with the dPG340PeBGF 10 (4 d) was not feasible owing to solubility problems at higher concentrations. We surmise that a second inhibitor dosing on day 4 would be necessary to continue protection from infection. However, such a procedure would need another sedation step, which causes additional stress to the mice. An alternative way of repetitively dosing would be the use of aerosol chambers. Lower inhibitor dosages with both inhibitors at about 2 nmol kg−1 concentrations did not significantly protect from infection.
Nevertheless, mice treated with dPG340PeB9 (4 b) were protected to a substantially higher extent than those treated with dPG340PeBGF 10 (4 d) at similar inhibitor dosage. We cannot exclude that PeBGF‐coated nanoparticles bind to respiratory structures or undergo cellular uptake in the respiratory tract, interfering with virus inhibition.
Notably, the efficiency of dPG340PeB9 (4 b; 11.5 nmol nanoparticles per kg or 4.8 μmol PeB per kg) in protecting mice from infection of was about ten times higher than multivalent siallyllactose (SL) presenting PAMAM dendrimers (50 μmol SL per kg).21
In summary, we found that a multivalent covalent display of influenza HA binding peptides PeB and PeBGF drastically enhances the antiviral potency in vitro. This potency is provided by both the high affine binding as well as the steric character of large polymers. Although PGs decorated with PeBGF showed higher affinity compared to those conjugated to PeB in various in vitro assays, we discovered that the corresponding PG–PeB conjugates are even more potent multivalent inhibitors and even active in vivo at low nmol kg−1 single‐dose administration. Thus, we present herein for the first time, non‐toxic influenza virus inhibitors based on a multivalent covalent peptide display on dendritic polymers. These findings pave the way for opportunities for peptide‐nanoparticles in future drug development against multivalent targets, since they can be obtained by straightforward synthetic methods while showing high affinity and low toxicity. Most importantly, peptide ligands can be evolved and further modified to bind to different receptors, which in concert with nanoparticle engineering, as demonstrated in this study, opens further avenues to target other disease‐relevant multivalent receptors than the one studied herein.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for financial support within the SFB765 and the SPP 1623 by the German Science Foundation (DFG) and the Focus Area Nanoscale of the Freie Universität Berlin. C.P.R.H. acknowledges funding from the Fonds der Chemischen Industrie, the Einstein‐Foundation Berlin, and the Boehringer‐Ingelheim Stiftung (Plus 3 award).
D. Lauster, M. Glanz, M. Bardua, K. Ludwig, M. Hellmund, U. Hoffmann, A. Hamann, C. Böttcher, R. Haag, C. P. R. Hackenberger, A. Herrmann, Angew. Chem. Int. Ed. 2017, 56, 5931.
Contributor Information
Prof. Dr. Christian P. R. Hackenberger, Email: hackenbe@fmp-berlin.de.
Prof. Dr. Andreas Herrmann, Email: Andreas.Herrmann@rz.hu-berlin.de
References
- 1. Russell R. J., Kerry P. S., Stevens D. J., Steinhauer D. A., Martin S. R., Gamblin S. J., Skehel J. J., Proc. Natl. Acad. Sci. USA 2008, 105, 17736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weis W., Brown J. H., Cusack S., Paulson J. C., Skehel J. J., Wiley D. C., Nature 1988, 333, 426. [DOI] [PubMed] [Google Scholar]
- 3. Mammen M., Choi S., Whitesides G. M., Angew. Chem. Int. Ed. 1998, 37, 2754; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2908. [Google Scholar]
- 4. Fasting C., Schalley C. A., Weber M., Seitz O., Hecht S., Koksch B., Dernedde J., Graf C., Knapp E. W., Haag R., Angew. Chem. Int. Ed. 2012, 51, 10472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10622. [Google Scholar]
- 5. Kingery-Wood J. E., Williams K. W., Sigal G. B., Whitesides G. M., J. Am. Chem. Soc. 1992, 114, 7303. [Google Scholar]
- 6. Mammen M., Dahmann G., Whitesides G. M., J. Med. Chem. 1995, 38, 4179. [DOI] [PubMed] [Google Scholar]
- 7. Sigal G. B., Mammen M., Dahmann G., Whitesides G. M., J. Am. Chem. Soc. 1996, 118, 3789. [Google Scholar]
- 8. Papp I., Sieben C., Sisson A. L., Kostka J., Böttcher C., Ludwig K., Herrmann A., Haag R., ChemBioChem 2011, 12, 887. [DOI] [PubMed] [Google Scholar]
- 9. Haldar J., Alvarez de Cienfuegos L., Tumpey T. M., Gubareva L. V., Chen J., Klibanov A. M., Pharm. Res. 2010, 27, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roy R., Zanini D., Meunier S. J., Romanowska A., J. Chem. Soc. Chem. Commun. 1993, 1869. [Google Scholar]
- 11. Nazemi A., Haeryfar S. M., Gillies E. R., Langmuir 2013, 29, 6420. [DOI] [PubMed] [Google Scholar]
- 12. Hidari K. I., Murata T., Yoshida K., Takahashi Y., Minamijima Y. H., Miwa Y., Adachi S., Ogata M., Usui T., Suzuki Y., Suzuki T., Glycobiology 2008, 18, 779. [DOI] [PubMed] [Google Scholar]
- 13. Ogata M., Umemura S., Sugiyama N., Kuwano N., Koizumi A., Sawada T., Yanase M., Takaha T., Kadokawa J., Usui T., Carbohydr. Polym. 2016, 153, 96. [DOI] [PubMed] [Google Scholar]
- 14. Hendricks G. L., Weirich K. L., Viswanathan K., Li J., Shriver Z. H., Ashour J., Ploegh H. L., Kurt-Jones E. A., Fygenson D. K., Finberg R. W., Comolli J. C., Wang J. P., J. Biol. Chem. 2013, 288, 8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang H., Huang W., Orwenyo J., Banerjee A., Vasta G. R., Wang L. X., Bioorg. Med. Chem. 2013, 21, 2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Papp I., Sieben C., Ludwig K., Roskamp M., Bottcher C., Schlecht S., Herrmann A., Haag R., Small 2010, 6, 2900. [DOI] [PubMed] [Google Scholar]
- 17. Reuter J. D., Myc A., Hayes M. M., Gan Z., Roy R., Qin D., Yin R., Piehler L. T., Esfand R., Tomalia D. A., J. R. Baker, Jr. , Bioconjug. Chem. 1999, 10, 271. [DOI] [PubMed] [Google Scholar]
- 18. Mangan D., Snyder I. S., Br. J. Ind. Med. 1978, 35, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlescu I., Scutaru D., Popa M., Uglea C. V., Med. Chem. Res. 2009, 18, 477. [Google Scholar]
- 20. Gambaryan A. S., Tuzikov A. B., Bovin N. V., Matrosovich M. N., Chinarev A. A., Juneja L. R., Antiviral Res. 2002, 55, 201. [DOI] [PubMed] [Google Scholar]
- 21. Kwon S. J., Na D. H., Kwak J. H., Douaisi M., Zhang F., Park E. J., Park J. H., Youn H., Song C. S., Kane R. S., Dordick J. S., Lee K. B., Linhardt R. J., Nat. Nanotechnol. 2016, 12, 48. [DOI] [PubMed] [Google Scholar]
- 22. Albertazzi L., Gherardini L., Brondi M., Sulis Sato S., Bifone A., Pizzorusso T., Ratto G. M., Bardi G., Mol. Pharm. 2013, 10, 249. [DOI] [PubMed] [Google Scholar]
- 23. Pryor J. B., Harper B. J., Harper S. L., Int. J. Nanomed. 2014, 9, 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsubara T., Onishi A., Saito T., Shimada A., Inoue H., Taki T., Nagata K., Okahata Y., Sato T., J. Med. Chem. 2010, 53, 4441. [DOI] [PubMed] [Google Scholar]
- 25. Jones J. C., Turpin E. A., Bultmann H., Brandt C. R., Schultz-Cherry S., J. Virol. 2006, 80, 11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lauster D., Pawolski D., Storm J., Ludwig K., Volkmer R., Memczak H., Herrmann A., Bhatia S., Beilstein J. Org. Chem. 2015, 11, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ng S., Lin E., Kitov P. I., Tjhung K. F., Gerlits O. O., Deng L., Kasper B., Sood A., Paschal B. M., Zhang P., Ling C. C., Klassen J. S., Noren C. J., Mahal L. K., Woods R. J., Coates L., Derda R., J. Am. Chem. Soc. 2015, 137, 5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsunaga Y., Bashiruddin N. K., Kitago Y., Takagi J., Suga H., Cell Chem. Biol. 2016, 23, 1341. [DOI] [PubMed] [Google Scholar]
- 29. Baeriswyl V., Calzavarini S., Chen S., Zorzi A., Bologna L., Angelillo-Scherrer A., Heinis C., ACS Chem. Biol. 2015, 10, 1861. [DOI] [PubMed] [Google Scholar]
- 30. Hüttl C., Hettrich C., Miller R., Paulke B. R., Henklein P., Rawel H., Bier F. F., BMC Biotechnol. 2013, 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hatano K., Matsubara T., Muramatsu Y., Ezure M., Koyama T., Matsuoka K., Kuriyama R., Kori H., Sato T., J. Med. Chem. 2014, 57, 8332. [DOI] [PubMed] [Google Scholar]
- 32. Memczak H., Lauster D., Kar P., Di Lella S., Volkmer R., Knecht V., Herrmann A., Ehrentreich-Forster E., Bier F. F., Stocklein W. F., PLoS One 2016, 11, e0159074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lauster D., Pawolski D., Storm J., Ludwig K., Volkmer R., Memczak H., Herrmann A., Bhatia S., Beilstein J. Org. Chem. 2015, 11, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roller S., Zhou H., Haag R., Mol. Diversity 2005, 9, 305. [DOI] [PubMed] [Google Scholar]
- 35. Lallana E., Fernandez-Trillo F., Sousa-Herves A., Riguera R., Fernandez-Megia E., Pharm. Res. 2012, 29, 902. [DOI] [PubMed] [Google Scholar]
- 36. Wyszogrodzka M., Haag R., Chem. Eur. J. 2008, 14, 9202. [DOI] [PubMed] [Google Scholar]
- 37. Vonnemann J., Liese S., Kuehne C., Ludwig K., Dernedde J., Bottcher C., Netz R. R., Haag R., J. Am. Chem. Soc. 2015, 137, 2572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary