

Summary
The inherited palmoplantar keratodermas (PPKs) are a heterogeneous group of genodermatoses, characterized by thickening of the epidermis of the palms and soles. No classification system satisfactorily unites clinical presentation, pathology and molecular pathogenesis. There are four patterns of hyperkeratosis – striate, focal, diffuse and punctate. Mutations in the desmoglein 1 gene (DSG1), a transmembrane glycoprotein, have been reported primarily in striate, but also in focal and diffuse PPKs. We report seven unrelated pedigrees with dominantly inherited PPK owing to mutations in the DSG1 gene, with marked phenotypic variation. Genomic DNA from each family was isolated, and individual exons amplified by polymerase chain reaction. Sanger sequencing was employed to identify mutations. Mutation analysis identified novel mutations in five families (p.Tyr126Hisfs*2, p.Ser521Tyrfs*2, p.Trp3*, p.Asp591Phefs*9 and p.Met249Ilefs*6) with striate palmar involvement and varying focal or diffuse plantar disease, and the recurrent mutation c.76C>T, p.Arg26*, in two families with variable PPK patterns. We report one recurrent and five novel DSG1 mutations, causing varying patterns of PPK, highlighting the clinical heterogeneity arising from mutations in this gene.
Short abstract
What's already known about this topic?
Twenty‐three mutations in the desmoglein 1 gene (DSG1) have been described in 25 families with palmoplantar keratoderma (PPK).
The majority have striate palmar involvement with focal plantar keratoderma, but isolated focal and diffuse PPKs have been reported.
What does this study add?
DSG1 mutations can present with variable phenotype within the same family.
Environmental factors, such as manual labour, may alter the clinical appearance of PPK.
A lower threshold should be considered for DSG1 screening in nonstriate PPK where an underlying keratin mutation has not been identified.
The inherited palmoplantar keratodermas (PPKs) are clinically and genetically heterogeneous genodermatoses, characterized by epidermal thickening of the palms and soles. Many keratodermas are restricted to these sites, but other cutaneous and extracutaneous features can occur.1 Traditionally, classification is based on the pattern of hyperkeratosis, notably diffuse, focal, striate and punctate. Identification of the genetic basis of these disorders has resulted in a molecular‐based classification;2 however, no single classification system satisfactorily unites the clinical presentation, pathology and molecular pathogenesis.
Striate PPK (SPPK) is a rare, mainly autosomal dominant disorder characterized by linear hyperkeratosis of the volar aspects of the fingers and sometimes the palms. SPPK is due to defects in desmosomes, the major epithelial intercellular adhesion junctions, which confer strength and rigidity to tissues that experience high mechanical stress. The disease displays locus heterogeneity with causative mutations in four genes – the desmosomal proteins desmoglein 1 (DSG1) and desmoplakin (DSP), keratin 1 (KRT1) and keratin 16 (KRT16) having been reported.3, 4, 5, 6
We report one American, one African and five European unrelated families with autosomal dominant PPK, owing to one recurrent and five novel DSG1 mutations. These demonstrate phenotypic variation despite a common molecular basis.
Case reports
Families 1, 2 and 7 presented to their local dermatology services. Families 3–6 were identified through the International Pachyonychia Congenita Research Registry. All reported having PPK since early childhood, with a positive family history in pedigrees 1–6. The proband in family 7 was the only known affected family member. No affected individuals had any history of skin fragility, blistering, hair or cardiac abnormalities.
In family 1, the proband had striate keratoderma of the digits and palms, with a focal pattern on the soles (Fig. 1a–c). Her sister and niece had no palmar involvement, but exhibited isolated focal keratoderma of the soles (Fig. 1d, e). In family 2, the proband and her half‐sister had focal palmar and plantar hyperkeratosis (Fig. 2a, c, d). The proband's 24‐year‐old son, a soldier, had striate hyperkeratosis of the digits, with focal palmar and plantar involvement (Fig. 2e, f). Mild hyperkeratosis of the elbows and knees was present in all three individuals. Families 3–7 had striate patterns on the palms and/or digits, with focal or diffuse plantar keratoderma (Figs. 2b, 3, 4). Furthermore, the probands in families 3 and 7 had mild hyperkeratosis over the dorsal interphalangeal joints, and knuckle (Fig. 4g) and toe pads, respectively. A biopsy of nonpalmoplantar skin for histological assessment was declined by all patients. Two individuals reported mild plantar pain when the keratoderma was very thick and unpared, but this was not a significant feature in the other cases. See Table S1 (see Supporting Information) for a clinical summary.
Figure 1.
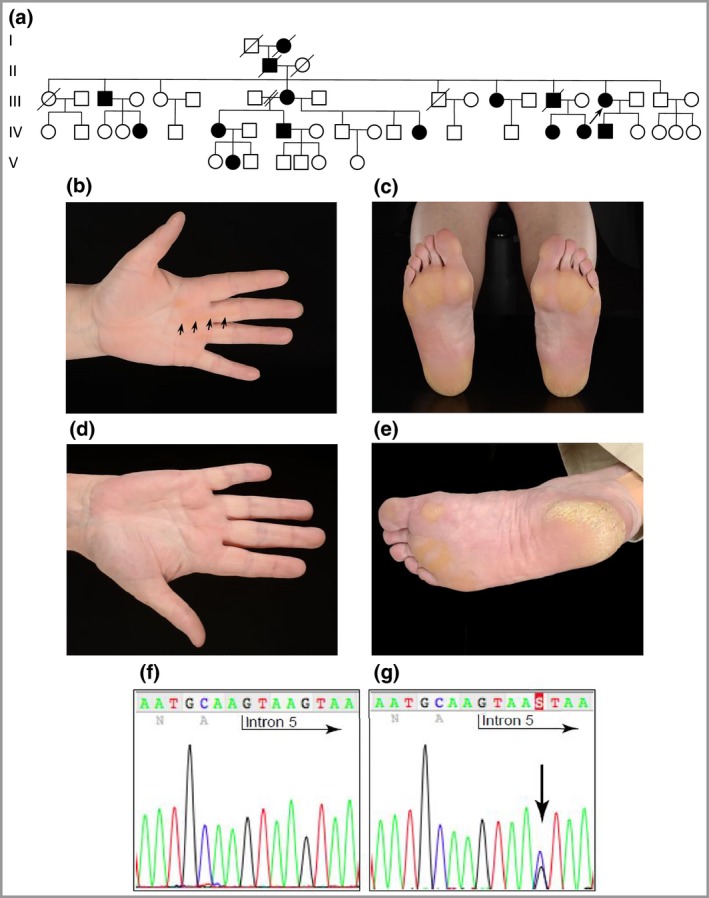
Pedigree, clinical images and mutation analysis of family 1 (Scottish). (a) Pedigree showing a history of palmoplantar keratoderma. The arrow indicates the proband. (b) Striate hyperkeratosis (arrows) of the proband's digits and palm. (c) Soles of the proband showing focal hyperkeratosis. (d) Normal palm of the proband's sister. (e) Soles of the subject in (d) showing fissuring plantar hyperkeratosis. (f) DNA sequence of the desmoglein 1 gene (DSG1) in an unaffected control sample. (g) The same region of DSG1 from the proband. The arrow indicates the novel splice‐site mutation between exon 5 and intron 5: c.517+5G>C, resulting in a frameshift and premature stop codon, p.Tyr126Hisfs*2.
Figure 2.

Pedigrees of families 2 (Scottish) and 3 (English), clinical images of family 2 and mutation analysis of families 2 and 3. (a) Pedigree of family 2 showing a history of palmoplantar keratoderma. The arrow indicates the proband. (b) Pedigree of family 3 showing a history of palmoplantar keratoderma. (c) Palm of the proband's half‐sister in family 2 showing focal hyperkeratosis (arrows). (d) Focal plantar hyperkeratosis of the proband's forefoot in family 2. (e) Fingers of the proband's son in family 2 showing linear hyperkeratosis (arrows) on the volar surface of the digits with focal palmar thickening. (f) Soles of the subject in (e) showing focal plantar hyperkeratosis. (g) DNA sequence of exon 2 of the desmoglein 1 gene (DSG1) in an unaffected control sample. (h) The same region of DSG1 from the probands of family 2 and 3. The arrow indicates a heterozygous C>T mutation at c.76 resulting in a premature termination codon at p.Arg26*.
Figure 3.

Pedigree, clinical images and mutation analysis of families 4 (Norwegian) and 5 (Northern Irish). (a) Pedigree of family 4 showing a history of palmoplantar keratoderma. The arrow indicates the proband. (b) Striate palmar hyperkeratosis (arrows) of the proband in family 4. (c) DNA sequence of exon 11 of the desmoglein 1 gene (DSG1) in an unaffected control sample. (d) The same region of DSG1 from the proband in family 4; the arrow indicates a novel mutation c.1560‐1561del resulting in a frame shift at Ser521Tyrf*2. (e) Pedigree of family 5. The arrow indicates the proband; no clinical information was available regarding her parents. (f) Striate pattern on the digits with focal palmar hyperkeratosis of the proband in family 5, who had a focal pattern on the soles of the feet. (g) DNA sequence of exon 1 of DSG1 in an unaffected control sample. (h) The same region of DSG1 from the proband of family 5. The arrow indicates a novel mutation c.8G>A, resulting in a premature termination codon at p.Trp3*.
Figure 4.

Pedigree and mutation analysis of families 6 (American) and case 7 (Ghanaian). (a) Pedigree of family 6 showing a history of palmoplantar keratoderma. The arrow indicates the proband, who had a striate digital pattern with diffuse keratoderma of the soles. (b) DNA sequence of exon 12 of the desmoglein 1 gene (DSG1) in an unaffected control sample. (c) The same region of DSG1 from the proband of family 6, the arrow indicates a novel mutation c.1771_1784del14, resulting in p.Asp591Phefs*9. (d) Pedigree of case 7. The arrow indicates the proband. (e) Striate palmar hyperkeratosis (arrows) of the proband in family 7. (f) Soles of the proband showing diffuse hyperkeratosis. (g) Knuckle pads over the proband's dorsal interphalangeal joints. (h) DNA sequence of exon 7 of DSG1 in an unaffected control sample. (i) The same region as in (g) from the proband of family 7. The arrow indicates the heterozygous mutation c.746dupT, resulting in p.Met249Ilefs*6.
Blood or saliva was obtained, following written informed consent and ethical approval by a Western Institutional Review Board that complies with the Declaration of Helsinki (File S1; see Supporting Information). Polymerase chain reaction amplification was performed and Sanger sequencing was employed to screen the exons and exon/intron boundaries of DSG1, using primers as previously described.3
Family 1 was screened directly for DSG1 mutations, as the proband presented with SPPK. Families 2–6 were initially screened for pachyonychia congenita mutations in keratin genes KRT6A, KRT6B, KRT6C, KRT16 and KRT17. In family 7, screening for KRT1 and KRT9, associated with diffuse PPK, was negative; whole‐exome sequencing of the proband and his unaffected parents was performed (File S1; see Supporting Information). In all cases, nonpathogenic variants were excluded through sequencing unaffected family members, and by reference to the Database of Single Nucleotide Polymorphisms, the 1000 Genome Project and the Exome Variant Server.
Affected members in family 1 had a novel splice‐site mutation in DSG1 between exon 5 and intron 5 (c.517+5G>C). Total RNA was extracted from a skin biopsy, and a cDNA fragment spanning exons 4–7 was amplified (File S1; see Supporting Information). DNA sequencing revealed very low levels of expression of the mutant allele, suggesting significant nonsense‐mediated mRNA decay. This mutation leads to skipping of exon 5, resulting in a frameshift and premature stop codon, p.Tyr126Hisfs*2 (Fig. 1f, g). Families 2 and 3 were heterozygous for a previously described nonsense mutation in DSG1 in exon 2 (c.76C>T, p.Arg26*) (Fig. 2g, h).3 Affected members in family 4 had a novel DSG1 mutation c.1560‐1561del in exon 11, resulting in p.Ser521Tyrfs*2 (Fig. 3c, d). A novel heterozygous nonsense mutation, c.8G>A resulting in p.Trp3* in exon 1 (Fig. 3g, h) was found in family 5. Family 6 had a novel mutation, c.1771_1784del14. This 14 base pair deletion results in p.Asp591Phefs*9 in exon 12 (Fig. 4b, c). Lastly, the proband in family 7 had a novel DSG1 duplication mutation, c.746dupT, which results in a frameshift and premature stop codon, p.Met249Ilefs*6 (Fig. 4h, i).
Discussion
There are three types of SPPK – type 1 (OMIM 148700) is the most common, caused by mutations in DSG1. SPPK types 2 (OMIM 612908) and 3 (OMIM 607654) are caused by desmoplakin and keratin‐1 mutations, respectively. Desmoplakin mutations can be associated with cardiac disease and woolly hair, but extracutaneous manifestations are not described with DSG1 mutations. Pachyonychia congenita rarely presents with striate PPK; Almutawa et al. reported a recurrent mutation in KRT16, causing striate palmar and diffuse plantar keratoderma, nail thickening and knuckle pads.6 Mutations in other DSG1 binding partners, resulting in varying PPK phenotypes, have also been described.1 To date, 23 mutations in DSG1 have been reported in 25 families with PPK; the majority had striate palmar and focal plantar keratoderma, but focal7 or diffuse8 palmar patterns have also been described. A more severe phenotype, severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome, has been described in individuals with biallelic DSG1 mutations9 and in one case with a heterozygous mutation in desmoplakin.10
We report the largest series of DSG1 mutations to date; two nonsense mutations (c.76C>T; p.Arg26* and c.8G>A; p.Trp3*), one duplication (c.746dupT; p.Met249Ilefs*6), two deletions (c.1560_1561del; p.Ser521Tyrfs*2 and c.1771_1784del14; p.Asp591Phefs*9), and one splice‐site mutation (c.517+5G>C; p.Tyr126Hisfs*2). All were autosomal dominant heterozygous mutations.
Reported DSG1 mutations causing SPPK are thought to result in haploinsufficiency through nonsense‐mediated mRNA decay. In SAM syndrome, there is a near‐total absence of DSG1 from the cell membrane,9, 10, 11 as a result of either nonsense‐mediated mRNA decay,11 failure of DSG1 localization9 or decreased DSG1 expression.10 It therefore seems reasonable to hypothesize that any reported SPPK DSG1 mutation might, if biallelic, result in SAM syndrome. Harmon et al. demonstrated that DSG1 interacts with Erbin, downregulating the Ras/MAPK pathway. Elevated Ras activity resulting from absent or insufficient DSG1 is postulated as a mechanism underlying SPPK. The high rate of PPK in disorders of the Ras/MAPK pathway supports this.12
DSG1 PPK phenotypes are restricted to areas of high pressure and abrasion.3 Keratoderma can be exaggerated by environmental factors, which may explain the striate pattern in the soldier in family 2, when other family members had isolated focal PPK. The mechanism for this is unclear, but may be a result of reduced desmosome function,3, 5 variations in desmosome configuration and protein expression,13 or a relative lack of coping with/adapting to environmental stress in desmoglein‐1 haploinsufficient individuals. Nonpalmoplantar hyperkeratotic plaques, as described in families 2 and 3, have been reported in three other families with DSG1 mutations.7, 14 The proband in family 7 is the first reported case to have knuckle and toe pads.
Our cases demonstrate phenotypic heterogeneity despite their unifying molecular basis. Intrafamilial PPK variation has been reported with DSG1 mutations,8, 15, 16 but the extent may be underestimated. In family 1, only the proband had striate palmar PPK; the two other affected relatives had no palmar disease. In family 2, the proband had focal palmoplantar disease, and it was only when another relative presented with striate palmar disease that DSG1 screening was performed, which highlights the importance of examining multiple family members. The recurrent mutation in families 2 and 3 (c.76C>T) has been reported in three other pedigrees – sporadic striate PPK,3 diffuse nonepidermolytic PPK,8 and striate palmar and focal plantar keratoderma.17 DSG1 screening has hitherto been performed primarily for striate PPK. Given the variety of phenotypes, and the difficulty in distinguishing these clinically, we suggest a low threshold for DSG1 screening in PPK where initial keratin gene screening has been negative.
Supporting information
Table S1. Summary of desmoglein 1 gene (DSG1) mutations and clinical features.
File S1. Supplementary methods.
Acknowledgments
We thank all the patients and families involved in this study. Thanks to the Genetics Team of Pachyonychia Congenita Project for useful discussions, and to Holly Evans of Pachyonychia Congenita Project for all her help with data preparation.
Funding sources F.J.D.S. and N.J.W. were supported by grants from the Pachyonychia Congenita Project (to F.J.D.S., www.pachyonychia.org). The Centre for Dermatology and Genetic Medicine at the University of Dundee is supported by a Wellcome Trust Strategic Award (098439/Z/12/Z).
Conflicts of interest None declared.
References
- 1. Itin PH, Fistarol SK. Palmoplantar keratodermas. Clin Dermatol 2005; 23:15–22. [DOI] [PubMed] [Google Scholar]
- 2. Irvine AD, Paller AS. Molecular genetics of the inherited disorders of cornification: an update. Adv Dermatol 2002; 18:111–49. [PubMed] [Google Scholar]
- 3. Hunt DM, Rickman L, Whittock NV et al Spectrum of dominant mutations in the desmosomal cadherin desmoglein 1, causing the skin disease striate palmoplantar keratoderma. Eur J Hum Genet 2001; 9:197–203. [DOI] [PubMed] [Google Scholar]
- 4. Armstrong DK, McKenna KE, Purkis PE et al Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet 1999; 8:143–8. [DOI] [PubMed] [Google Scholar]
- 5. Whittock NV, Smith FJ, Wan H et al Frameshift mutation in the V2 domain of human keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol 2002; 118:838–44. [DOI] [PubMed] [Google Scholar]
- 6. Almutawa F, Thusaringam T, Watters K et al Pachonychia congenita (K16) with unusual features and good response to acitretin. Case Rep Dermatol 2015; 7:220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Milingou M, Wood P, Masouyé I et al Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatol 2006; 212:117–22. [DOI] [PubMed] [Google Scholar]
- 8. Keren H, Gergman R, Mizrachi M et al Diffuse nonepidermolytic palmoplanta keratoderma caused by a recurrent nonsense mutation in DSG1 . Arch Dermatol 2005; 141:625–8. [DOI] [PubMed] [Google Scholar]
- 9. Samuelov L, Sarig O, Harmon RM et al Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013; 45:1244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McAleer MA, Pohler E, Smith FJ et al Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N‐terminal plakin domain of desmoplakin. J Allergy Clinc Immunol 2015; 136:1268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Has C, Jacok T, He Y et al Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br J Dermatol 2015; 172:257–61. [DOI] [PubMed] [Google Scholar]
- 12. Harmon RM, Simpson CL, Johnson JL et al Desmoglein‐1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Invest 2013; 123:1556–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wan H, Dopping‐Hepenstal PJ, Gratian MJ et al Desmosomes exhibit site‐specific features in human palm skin. Exp Dermatol 2003; 12:378–88. [DOI] [PubMed] [Google Scholar]
- 14. Nomura T, Mizuno O, Miyauchi T et al Striate palmoplantar keratoderma: report of a novel DSG1 mutation and atypical clinical manifestations. J Dermatol Sci 2015; 80:223–5. [DOI] [PubMed] [Google Scholar]
- 15. Klijuic A, Gilead L, Martinex‐Mir A et al A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol 2003; 12:523–7. [DOI] [PubMed] [Google Scholar]
- 16. Hershkovitz D, Lugassy L, Indelman M et al Novel mutations in DSG1 causing striate palmoplantar keratoderma. Clin Exp Dermatol 2008; 34:224–8. [DOI] [PubMed] [Google Scholar]
- 17. Dua‐Awereh MB, Shimomura Y, Kraemer L et al Mutations in the desmoglein 1 gene in five Pakistani families with striate palmoplantar keratoderma. J Dermatol Sci 2009; 53:192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of desmoglein 1 gene (DSG1) mutations and clinical features.
File S1. Supplementary methods.