

Abstract
Objective
Parental nutrition and lifestyle impact the metabolic phenotype of the offspring. We have reported that grandpaternal chronic high-fat diet (HFD) transgenerationally impairs glucose metabolism in subsequent generations. Here we determined whether grandpaternal diet transgenerationally impacts the transcriptome and lipidome in skeletal muscle. Our aim was to identify tissue-specific pathways involved in transgenerational inheritance of environmental-induced phenotypes.
Methods
F0 male Sprague–Dawley rats were fed a HFD or chow for 12 weeks before breeding with chow-fed females to generate the F1 generation. F2 offspring were generated by mating F1 males fed a chow diet with an independent line of chow-fed females. F1 and F2 offspring were fed chow or HFD for 12 weeks. Transcriptomic and LC-MS lipidomic analyses were performed in extensor digitorum longus muscle from F2-females rats. Gene set enrichment analysis (GSEA) was performed to determine pathways reprogrammed by grandpaternal diet.
Results
GSEA revealed an enrichment of the unfolded protein response pathway in skeletal muscle of grand-offspring from HFD-fed grandfathers compared to grand-offspring of chow-fed males. Activation of the stress sensor (ATF6α), may be a pivotal point whereby this pathway is activated. Interestingly, skeletal muscle from F1-offspring was not affected in a similar manner. No major changes were observed in the skeletal muscle lipidome profile due to grandpaternal diet.
Conclusions
Grandpaternal HFD-induced obesity transgenerationally affected the skeletal muscle transcriptome. This finding further highlights the impact of parental exposure to environmental factors on offspring's development and health.
Keywords: Skeletal muscle, Epigenetics, Unfolded protein response (UPR), Lipodome, Transcriptome, Activation of the stress sensor (ATF)
Highlights
-
•
High fat diet transgenerationally affects the skeletal muscle transcriptome.
-
•
Grandpaternal diet influences unfolded protein response pathway in skeletal muscle.
-
•
Activation of the stress sensor (ATF6α) is affected by grandpaternal diet.
-
•
Paternal high-fat diet exposure affects metabolic health of successive generations.
Abbreviations
- ATF6
Activating transcription factor 6
- Cer
Ceramide
- Chac1
ChaC Glutathione Specific Gamma-Glutamylcyclotransferase
- CHOP
CCAAT enhancer-binding protein (C/EBP) homologous protein
- DG
Diacylglyceride
- DNAJA4
DnaJ heat shock potein family (Hsp40) member A4 (Dnaja4)
- EDL
Extensor Digitorum Longus
- EIF2A
Eukaryotic translation initiation factor 2A
- Eif4a1
Eukaryotic translation initiation factor 4a1
- ER
Endoplasmic reticulum
- Gosr2
Golgi SNAP Receptor Complex Member 2
- GSEA
Gene set enrichment analysis
- GRP78
78-kilodalton glucose-regulated protein
- GRP94
94-kilodalton Glucose-Regulated Protein
- HFD
High-fat diet
- IRE1α
Inositol-requiring enzyme 1 alpha
- JNK
c-Jun N-terminal kinases
- Nolc1
Nucleolar and coiled-body phosphoprotein 1
- O2PLS
Orthogonal 2 projections to latent structures
- PC
Phosphatidylcholine
- PCA
Principal Component Analysis
- PE
Phosphatidylethanolamine
- PI
Phosphatidylinositol
- ROS
Reactive oxygen species
- Sec11a
SEC11 homolog A, signal peptidase complex subunit
- SM
Sphingomyelin
- TG
Triacylglyceride
- UPR
Unfolded protein response
- Wfs1
Wolframin ER transmembrane glycoprotein
- Wipi1
WD Repeat Domain, Phosphoinositide Interacting 1
1. Introduction
Obesity is a major world-wide public health problem that is strongly associated with increased risk of other comorbidities, such as insulin resistance, type 2 diabetes, and cardiovascular diseases [1]. While relatively simple energy balance equations have been valuable in advancing the understanding of how energy intake and energy expenditure influence body composition, obesity is now recognized as a chronic progressive disease of complex etiology, resulting from multiple environmental and genetic factors. Clinical studies reveal that parental obesity affects body weight accumulation in children and adolescents, with obesity in one or both of the parents influencing the risk of developing obesity in the offspring later in life [2], [3], [4], highlighting a heritable component to obesity. Indeed, genome-wide association studies identify more than 100 different candidate genes linked to obesity, with the majority targeted to neuroendocrine pathways influencing food intake [5]. However, environmental factors such as the nutritional status of the parent at conception or during pregnancy can influence fetal growth and development in utero, and may influence the susceptibility to obesity and obesity-associated diseases later in life through epigenetic modifications [6]. While the impact of maternal obesity on the health of subsequent generations is strongly attributed to the adverse intrauterine environment [7], [8], [9], paternal obesity can also impact metabolic health of the offspring in later life [10], [11], [12].
Grandpaternal exposure to altered food availability during the slow growth period is associated with a greater risk for obesity and cardiovascular disease in grandchildren [10], while parental diabetes at conception is associated with altered birth weight and increased risk of diabetes in the offspring [13]. When phenotypic changes in the first generation are similar or the same as the inducing stressor, this can, in turn, program the phenotype of the second generation by a process known as serial programming [14]. In rodents, paternal chronic exposure to low-protein diet increases the expression of hepatic genes involved in lipid and cholesterol metabolism [12]. Paternal chronic high fat diet (HFD) disturbs whole-body glucose metabolism, as well as pancreatic and adipose function, in F1 female offspring [12], [15]. Moreover, diet-induced paternal obesity increased adiposity and insulin resistance of two resultant generations has been shown with different degrees of penetrance to subsequent generations [16].
In rodents, high-fat diets impart a transgenerational influence on insulin sensitivity. We have shown that paternal diet-induced obesity affects the metabolic health of both F1 and F2 generations [17]. Glucose tolerance is impaired in female offspring and grand-offspring born to high-fat-fed F0-male rats indicating a paternal-transmitted epigenetic phenotype [17]. Moreover, altered expression of miRNA let-7c in sperm of F0 and F1 founders was passed down to adipose tissue of the offspring, coincident with a transcriptomic shift of the let-7c predicted targets in white adipose tissue [17]. While, adipose tissue is one of the major sites of paternal high caloric intake-induced transgenerational reprogramming [15], [16], [17], [18], [19], comparatively less is known regarding the impact of parental diet on skeletal muscle physiology and metabolism. Thus, we determined the transgenerational response of paternal diet-induced obesity on the skeletal muscle transcriptome and lipidome. Our goal was to identify novel tissue-specific pathways involved in paternal obesity-induced epigenetic transmission of disturbed metabolic phenotype to the future generations.
2. Methods
2.1. Animal care
Sprague–Dawley rats were obtained from Charles River Laboratories (Germany). The breeding strategy and diet treatment has been described previously [17]. F0 male breeders (4 weeks of age) were fed either a HFD (TD.88137/TD.08811, 42/45% energy from fat, Harlan Laboratories; USA) or chow diet (R36, Lactamin, Labfor; Sweden) for 12 weeks. The F1 offspring were generated by housing one F0 male breeder with a 12 week-old female rat. The female rats were fed chow diet during mating, gestation, and lactation. Pups were weaned from mothers at 21 days of age and fed a chow diet. At 10 weeks of age, the F1 litters were further divided into subgroups, and rats were fed either chow or HFD for 12 weeks. Chow-fed F1 males were mated with an independent line of 12 week-old female rats to generate the F2 generation. Pups were weaned to a chow diet at 21 days of age. At week 10, the F2 litters were divided into subgroups and rats were fed chow or HFD for 12 weeks (experimental layout depicted in Figure 1). Animals were housed in grouped cages throughout the entire experiment. Rats were subjected to food deprivation from 4 h prior to the termination. Animals were anesthetized with sodium pentobarbital (100 mg/kg, ip). Tissues were harvested, snap-frozen in liquid nitrogen and stored at −80 °C until use.
Figure 1.

Schematic diagram of breeding and diet strategy. F0 male Sprague–Dawley rats were fed chow or HFD for 12 weeks before breeding with chow-fed females to generate a F1 generation. Chow-fed F1 males were mated with an independent line of chow-fed females to generate F2 offspring. At 10 weeks of age, a subgroup of F1 and F2 offspring were fed HFD for 12 weeks. EDL muscle from F2-females was studied. The muscle drawing was downloaded from Servier's PowerPoint image bank (www.servier.com).
All rats were housed in a temperature-controlled environment and 12:12-h light:dark cycle at the Department of Physiology and Pharmacology, Karolinska Institutet, Stockholm, Sweden. Animals had free access to water and food. All experimental procedures were approved by the Stockholm North Ethical Committee on Animal Research (N101/12), and conducted in accordance with regulations for protection of laboratory animals.
2.2. Transcriptomic analysis
Muscle from F2 female rats was used to perform the transcriptome analysis. Total RNA from EDL muscle was extracted using RNeasy Mini Kit (Qiagen; Hilden, Germany). RNA quality was assessed and ensured using the Experion Automated Electrophoresis System (Bio-Rad laboratories; CA, USA). Affymetrix GeneChip Rat Gene 1.1 ST was used for the whole-transcriptome analysis performed at the Bioinformatics and Expression Core facility (BEA) at Karolinska Institutet, Huddinge, Sweden. The data set is deposited at the Gene Expression Omnibus (GEO accession number: GSE95490).
2.3. Lipidomic analysis
Lipidomic analysis was performed in EDL muscle from F2 female rats at the Swedish Metabolomics Centre, Umeå, Sweden. Lipids were extracted using the chloroform:MeOH (2:1 vol/vol)) phase extraction method. Input volume was first normalized to muscle weight and subjected to Liquid Chromatography-Mass Spectrometry (LC/MS) using the QTOF 6540 instrument (Agilent Technologies, USA). Lipid species were annotated to the respective lipid class, and total amount of carbons and double bonds in the fatty acid chains using ProFinder B.06 Agilent MassHunter (Agilent Technologies, USA). The lipid classes were processed separately to allow for different adducts. Peaks were controlled manually, and, if no obvious peak was observed, the feature was excluded from the analysis.
2.4. Transcriptomic and lipidomic data analyses
The transcriptomic data were analyzed using GSEA (Broad Institute; MA, USA, accessed at http://software.broadinstitute.org/gsea/). The data set was collapsed and the probe with the highest expression level for each respective gene was used for the enrichment analysis. Gene set permutation was used and a nominal p-value <0.05, and FDR <0.05 were considered significant [20], [21]. Heatmaps were created using the online source Morpheus (Broad Institute; MA, USA, accessed at https://software.broadinstitute.org/morpheus/). PCA was performed for both transcriptomic and lipidomic data using the SIMCA 14.0 software (Umetrics, Umeå, Sweden). Log2 transformed data was used for further interaction analysis of the transcriptomic and lipidomic results applying the O2PLS method [22], [23] using the SIMCA 14.0.
2.5. Quantitative real-time PCR
EDL muscle from F1 and F2 female rats was used to extract total RNA using the TRIzol® reagent (Invitrogen™, USA) according to the manufacturer's instruction. cDNA was prepared using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems; CA, USA), followed by a real-time PCR reaction using custom made SYBR green primers (ESM Table 3). Applied Biosystems StepOnePlus Real-Time PCR Systems was used for all reactions, which were performed in duplicates.
2.6. DNA methylation
Genomic DNA was extracted from EDL muscle of F2 female rats using DNeasy Blood and Tissue Kit (Qiagen), according to the manufacturer's instruction. Fragmented DNA was obtained by sonicating the samples for 10 cycles repeated for 3 times. To investigate the level of methylation, fragmented DNA was enriched by (MBD)-capture (Invitrogen; Carlsbad, CA, USA) and followed by qPCR analysis. SYBR® Green (Applied Biosystems by Life Technologies; Woolston Warrington, UK) primers were custom designed to target the regions near to the transcription start sites (−/+1000 bp relative to the TSS) (ESM Table 3).
2.7. Western blot
EDL muscle samples from F2 female rats were processed as described previously [17]. An equal amount of protein (15 μg) was separated by SDS-PAGE using Criterion XT Precast gels (Bio-Rad; Hercules, CA) and transferred to polyvinylidene difluoride membrane (PVDF) (Immobilon-P, Millipore; Billerica, MA). Protein content was determined using the following antibodies: ATF6α-p50 (#sc-22799) and CHOP (#sc-7351) from Santa Cruz Biotechnology (California, USA), GRP78 (#ab21685), GRP94 (#ab90458), p-IRE1α (#ab48187) and XBP1 (#ab37152) from Abcam (Cambridge, UK) and p-EIF2A (#9721) and p-JNK (#9251) from Cell Signaling Technology (Massachusetts, USA). Antibody dilutions were performed according to manufacturer's instructions. Ponceau S staining of the PVDF membranes was used for normalization.
2.8. Oxyblot Analysis™
The production of reactive oxygen species (ROS) was indirectly measured by performing an immunoblot detection of carbonyl groups introduced into proteins after oxidative modifications. The level of protein oxidation was determined in EDL muscle from F2 female rats using the OxyBlot Protein Oxidation Detection Kit, following manufacturer's specifications (Merck Millipore, Billerica, MA). EDL muscle (∼10 mg) was homogenized in lysis buffer (20 mM Tris pH 7.5, 6% Sodium Dodecyl Sulfate). Proteins were separated on a 10% SDS-PAGE gel and transferred to a PVDF membrane, which was exposed to the primary antibody specific to dinitrophenylhydrazone-derivatized residues (OxyBlot™) followed by incubation to a peroxidase-conjugated secondary antibody. Protein carbonylation was detected by chemiluminescence in autoradiography and quantified by densitometry utilizing the Quantity One Software (BioRad).
2.9. Glycogen content
Glycogen content was measured in EDL muscle of F2 rats using the commercial glycogen assay kit (Abcam, Cambridge, UK), following the manufacturer's instructions.
2.10. Statistics
The microarray data were normalized utilizing the expression console plier method, followed by statistical analysis using two-way ANOVA, and the Benjamini and Hochberg correction for multiple testing. Data are presented as mean ± SEM. Unpaired Student's t-test was used when appropriate. P-value ≤ 0.05 was considered significant.
3. Results
3.1. Grandpaternal HFD consumption transgenerationally affects the skeletal muscle transcriptome
We performed GSEA to first explore potential changes in the skeletal muscle transcriptomic signature arising in response to grandpaternal (Gpat) chow (CD) or HFD (HF) diet in F2 female rats, independently of the F2 diet. Four groups of rats were studied; GpatCD-CD, GpatCD-HF, GpatHF-CD, GpatHF-HF. Grandpaternal diet had no significant effect on the skeletal muscle transcriptome (ESM Table 1). When taking into consideration the diet of the grand-offspring, grandpaternal diet did not alter the skeletal muscle transcriptome of F2 rats fed chow diet (GpatCD-CD vs. GpatHF-CD) (ESM Table 2). In contrast, grandpaternal HFD induced a prominent enrichment of the unfolded protein response (UPR) pathway in skeletal muscle of F2 rats fed a HFD (GpatHF-HF vs. GpatCD-HF) (Table 1). Principal component analysis of the 101 genes in the UPR gene set showed clustering among the offspring fed chow diet (Figure 2A). GpatHF offspring fed HFD were separated into two distinct clusters, indicating a higher variation in this group (Figure 2A). The majority of the UPR-associated genes were differentially expressed in GpatHF-HF compared with GpatCD-HF rats (Figure 2B). 57 of 101 genes in the UPR gene set were part of the core enrichment (Figure 2C). Genes from the core enrichment set were selected to further validate mRNA level by RT-qPCR. We confirmed that grandpaternal HFD upregulated the expression level of several of the core genes involved in the UPR, such as the Atf6b, Eif4a1, Nolc1, Sec11a, and Wfs1 in skeletal muscle of F2 rats (Figure 2D). However, none of these targets was altered in skeletal muscle of the F1 females born to HFD-fed fathers (ESM Figure 1A), suggesting that the UPR in skeletal muscle is transgenerationally reprogrammed by grandpaternal HFD, although not evident in F1 female rats.
Table 1.
Top 20 enriched gene sets in GpatHF-HF vs GpatCD-HF retrieved from the GSEA.
| Gene set name | Size | ES | NES | NOM p-val | FDR q-val |
|---|---|---|---|---|---|
| Unfolded protein response | 101 | −0.47 | −1.60 | >0.001 | 0.012 |
| Estrogen response early | 173 | −0.39 | −1.39 | >0.001 | 0.183 |
| Notch signaling | 29 | −0.44 | −1.33 | 0.070 | 0.240 |
| Androgen response | 86 | −0.39 | −1.30 | 0.023 | 0.263 |
| Estrogen response late | 180 | −0.36 | −1.30 | 0.002 | 0.226 |
| Epithelial mesenchymal transition | 158 | −0.37 | −1.29 | 0.011 | 0.213 |
| Kras signaling up | 169 | −0.36 | −1.29 | 0.003 | 0.185 |
| IL6 JAK STAT3 Signaling | 79 | −0.38 | −1.27 | 0.041 | 0.201 |
| Angiogenesis | 32 | −0.41 | −1.27 | 0.121 | 0.184 |
| Xenobiotic metabolism | 171 | −0.35 | −1.26 | 0.015 | 0.190 |
| Cholesterol homeostasis | 61 | −0.38 | −1.26 | 0.070 | 0.177 |
| Apical junction | 177 | −0.36 | −1.26 | 0.012 | 0.166 |
| MYC targets V2 | 53 | −0.38 | −1.25 | 0.104 | 0.163 |
| Interferon gamma response | 172 | −0.35 | −1.25 | 0.012 | 0.152 |
| TGF beta signaling | 49 | −0.39 | −1.25 | 0.106 | 0.143 |
| MTORC1 Signaling | 190 | −0.35 | −1.25 | 0.013 | 0.137 |
| Interferon alpha response | 85 | −0.37 | −1.24 | 0.068 | 0.141 |
| Peroxisome | 100 | −0.35 | −1.19 | 0.115 | 0.250 |
| Adipogenesis | 186 | −0.33 | −1.18 | 0.049 | 0.247 |
| DNA repair | 124 | −0.34 | −1.17 | 0.100 | 0.264 |
ES = Enrichment Score, NES = Normalized Enrichment Score, NOM p-val = Nominal P-value, GpatHF-HF = Grandpaternal high-fat on high-fat, GpatCD-HF = Grandpaternal control diet on high-fat, GSEA = Gene Set Enrichment Analysis.
Figure 2.

Grandpaternal HFD affects the skeletal muscle transcriptome of F2 female rats. A) PCA score plot of UPR gene set in EDL muscle of GpatHF-HF vs. GpatCD-HF animals (n = 4–5). B) Enrichment plot of the UPR gene set generated by GSEA comparing GpatHF-HF vs. GpatCD-HF groups (n = 4–5). C) Heatmap showing a clustering of 57 core enriched genes in the UPR gene set in GpatCD-HF and GpatHF-HF animals (n = 4–5). Red: downregulation; Blue: upregulation. D) Expression of core enriched genes by RT-qPCR (n = 7–10) in EDL muscle of F2 rats. Geometric mean of the Beta-2-Microglobulin (B2M) and Peptidylprolyl Isomerase A (PPIA) was used as internal control. Values are means ± SEM, *p ≤ 0.05: GpatHF-HF vs. GpatCD-HF. GpatCD-HF: HFD-fed offspring from grandfathers fed a chow diet; GpatHF-HF: HFD-fed offspring from grandfathers fed a HFD.
To investigate whether DNA methylation was the epigenetic mark responsible for UPR reprogramming, we determined DNA methylation at the promoter regions of UPR genes that were altered in skeletal muscle from the GpatHF animals. DNA methylation of Atf6α, Atf6b, Chac1, Eif4a1, and Wfs1 was similar between GpatHF-HF and GpatCD-HF animals. These results suggest that either UPR gene reprogramming is not controlled by DNA methylation or that UPR genes are reprogrammed by DNA methylation of an upstream gene (ESM Figure 1B).
3.2. Role of ATF6 in the unfolded protein response in skeletal muscle
We analyzed several components of the three major signaling pathways involved in the activation of the UPR. Protein abundance of CHOP, GRP78, p-EIF2A, p-IRE1α, p-JNK, and XBP1 was unaltered between GpatHF-HF compared with GpatCD-HF offspring. However, protein abundance of the spliced and active form of ATF6α, as well as of the ER chaperone glycoprotein GRP94, was increased the GpatHF-HF compared with GpatCD-HF rats (Figure 3A,B), suggesting that this signaling pathway is activated. Thus, our results implicate ATF6α as one factor contributing to the increased UPR in skeletal muscle from GpatHF-HF rats.
Figure 3.

Activation of the ATF6 pathway in skeletal muscle of F2 female rats from HFD-fed grandfathers. A) Protein abundance of ATF6α-p50, CHOP, GR78, GRP94, p-EIF2A, p-IRE1α, p-JNK, unspliced XBP1(u), and spliced XBP1(s) in EDL muscle of F2 rats. Data are represented as fold change (FC) (n = 7–10). B) Representative blots of protein abundance of targets shown in A. C) Heatmap showing downstream targets of ATF6α retrieved from the transcriptome analysis of GpatHF-HF vs GpatCD-HF animals (n = 4–5). D) Heatmap showing the downstream targets of ATF6α found in the core enrichment of the UPR gene set in GpatHF-HF vs GpatCD-HF animals (n = 4–5). Red: downregulation; Blue: upregulation. E) Expression of ATF6α downstream targets in the core enrichment of the UPR gene set retrieved from the transcriptome analysis of F2 rats (n = 4–5). *p ≤ 0.05: GpatHF-HF vs. GpatCD-HF. GpatCD-HF: HFD-fed offspring from grandfathers fed chow diet; GpatHF-HF: HFD-fed offspring from grandfathers fed HFD.
To further explore the role of ATF6α, we further analyzed the skeletal muscle transcriptomic results of skeletal muscle from the F2 animals to evaluate whether the expression of 381 targets known to be upregulated by ATF6α were altered between GpatHF-HF and GpatCD-HF rats [24]. This analysis retrieved 294 of the 381 ATF6α targets were expressed in the EDL muscle from F2 rats and the majority were positively regulated in GpatHF-HF rats when compared with GpatCD-HF (Figure 3C). Among these targets, 16 constitute core enrichment proteins of the UPR pathway (Figure 3D, E). Wfs1, Dnaja4, Eif2ak3, and Xpb1 expression was upregulated in skeletal muscle from GpatHF-HF compared to GpatCD-HF rats (Figure 2, Figure 3, E), implicating upregulation of the UPR in skeletal muscle of offspring from HFD-fed grandfathers is coupled to activation of the ATF6α pathway.
3.3. Lipid profile, glycogen content and protein oxidation
To investigate whether grandpaternal diet is affecting UPR due to altered nutrient availability, we measured the glycogen content and lipid profile in the muscle of F2 rats on a HFD. Glycogen content was unaltered between the GpatHF-HF and GpatCD-HF groups (ESM Figure 2A). A total of 153 different lipid species from 7 lipid classes were examined in the lipidomic analysis. PCAs were performed using all the lipids species (Figure 4A, B) or specific lipid classes (data not shown). Grandpaternal diet did not affect the lipid profile in skeletal muscle from female F2 rats (Figure 4B). Cer, DG, PC, PI, SM, and TG content was unaltered between GpatHF-HF and GpatCD-HF groups (Figure 4B). PCA using all lipid species consisting of two principal components explains 65% of the variation in the lipid profiles. However, this analysis did not reveal a clear separation of the two groups studied (Figure 4A), confirming that grandpaternal diet may not directly impact in the lipid profile in skeletal muscle of F2 rats fed a HFD. TGs were retrieved as the only lipid class presenting a clear clustering compared to the other lipid classes, reflecting the fact that most of the TGs were detected to a similar abundance (Figure 4C). Additional PCAs using each of the individual lipid classes did not reveal a clustering pattern reflecting the different grandpaternal diets (data not shown).
Figure 4.

Lipid profile in skeletal muscle of F2 offspring. A) PCA score plot of lipidomic data in EDL muscle of F2 rats. The first principal component (PC1) explains 48% of the variance and PC2 explains 17% of the variance in the data set (n = 5 per group). B) Lipid abundance in EDL muscle of F2 rats. Data are presented as sum of each lipid specie in a specific lipid class (n = 5 animals per group). C) Loading plot of the PCA. Loadings are scaled as a correlation coefficient ranging from −1 to 1 (n = 153 lipid species). GpatCD-HF: HFD-fed offspring from grandfathers fed chow diet; GpatHF-HF: HFD-fed offspring from grandfathers fed HFD.
We next investigated whether grandpaternal diet affected UPR due to ER stress induced by increased reactive oxygen species (ROS). For this, we quantified protein oxidation using the OxyBlot assay as an indirect measurement of ROS production in EDL muscle. We found similar levels of protein oxidation between GpatCD-HF and GpatHF-HF rats, indicating ROS production is unlikely to account for the changes in UPR in GpatHF-HF rats (ESM Figure 2B).
3.4. Enriched UPR is associated with phosphatidylcholine levels
To investigate whether there was an association between the abundance of phosphatidylcholine and phosphatidydielthylamine phospholipids and the UPR, we performed the O2PLS analysis. O2PLS is a multivariate analysis integrates two different data sets, dividing data into three different parts: the ‘joint variance’, which describes the covariation between the two data sets; the ‘unique’ structure, which describes systematic effects that do not covariate/correlate with the other in the data set, and the ‘residual’ structures, representing data set noise. O2PLS analysis was performed using the UPR gene set (101 genes) identified in the microarray analysis and the phosphatidylcholine and phosphatidydielthylamine lipid species (49 lipid species) identified in the lipidomic analysis of EDL muscle from the F2 rats. The model consisted of 2 joint latent variables, one transcript-unique variable and one lipid-unique latent variable. O2PLS analysis revealed that 58% of the lipid data set covariates with 51% of the transcriptomic data set. The lipid-unique and the transcript-unique structures reached a variance of 15% and 20%, respectively (Figure 5A). The score plot based on the covariance between the lipid and transcript data sets (Figure 5B) showed a tendency for data from the female GpatHF-HF rats to cluster in the opposite direction from the GpatCD-HF rats (Figure 5B). Although the ratio of phosphatidylcholine to phosphatidydielthylamine lipid species was unchanged (data not shown), the enriched UPR gene set in the GpatHF-HF rats covariates with most of the phosphatidylcholine species identified (Figure 5C). A weaker covariation was observed between the gene set data and the phosphatidydielthylamine species (Figure 5C). The correlation between the phosphatidylcholine lipid species and the enriched UPR in the GpatHF-HF rats suggests that phosphatidylcholine species may indirectly trigger activation of this pathway.
Figure 5.

Enriched UPR is associated with increased phosphatidylcholine levels. A) Overview of O2PLS model representing the UPR gene set (101 genes) from the transcriptomic data and the phosphatidylcholine and PE lipid content (49 lipid species) from the lipidomic data of EDL from F2 female rats. Total joint variance shows 51% variance for the transcript analysis of the UPR data set and 58% variance for the lipidomic data including only phosphatidylcholine and phosphatidydielthylamine lipid species. The transcript-unique variance and the lipid-unique structures reached 20% and 15% variance, respectively. Residual data are 29% and 26% for the transcriptomic and lipidomic data sets, respectively. B) O2PLS score plot of the two joint latent variables. Joint latent variable (LV) 1 accounts for 40% and 17% of the transcriptomic (T) and lipidomic (L) data sets, accordingly. The joint latent variable 2 accounts for 12% and 41% of the transcriptomic and lipidomic data sets. C) Loading plot for O2PLS in B, consisting of 101 genes, 34 phosphatidylcholine and 15 phosphatidydielthylamine species. The loadings are scaled as a correlation coefficient ranging from −1 to 1 (n = 150 variables). GpatCD-HF: HFD-fed offspring from grandfathers fed chow diet; GpatHF-HF: HFD-fed offspring from grandfathers fed HFD.
4. Discussion
We explored the effect of paternal diet on skeletal muscle gene expression over several generations. Paternal transmission of metabolic dysfunction from dietary-induced obesity alters the transcriptomic profile in liver, as well as β-cell and adipose tissue [12], [15]. Here we show that paternal chronic HFD diet affects the skeletal muscle transcriptome two generations later. We identified several genes enriched in the UPR pathway that are upregulated in skeletal muscle of HFD fed female rats descending from obese grandfathers. Moreover, protein abundance of the spliced and active form of ATF6α, as well as the ER chaperone glycoprotein GRP94, is upregulated in this cohort, indicating the homeostatic control of protein production at the level of the ER may be altered as a result of transgenerational epigenetic inheritance. Of note, obesity does not appear to directly account for these changes, since expression of UPR-related genes was not altered in obese HFD-fed F1 offspring. Our findings are consistent with the notion that paternal exposure to environmental factors prior to conception contributes to the metabolic epigenetic programming over subsequent generations.
Environmental stressors can modify epigenetic programming over several generations. Thus, grandpaternal dietary-induced transgenerational epigenetic effects may be influenced by the macronutrient composition of the diet consumed by the offspring. Comparisons of the skeletal muscle transcriptome of F2 female rats fed either chow or high fat diet revealed a diet-specific response on the epigenome. We found that grandpaternal diet altered the skeletal muscle transcriptome only in F2 animals fed a high-fat diet. While several genes selected from the UPR core enrichment gene set were upregulated in skeletal muscle from HFD-fed F2 rats, they were unchanged in F1 offspring from HFD-fed fathers. This suggests that mechanisms other than transgenerational reprogramming through the male gametes regulate the skeletal muscle response to HFD between the two generations. Parental-induced phenotypes across successive generations represent either a true inheritance of an acquired metabolic phenotype [17], [25] or a serial programming [26]. For example, offspring derived from F0 mating of obese prediabetic mice exhibit defects in glucose and lipid metabolism but only upon a post-weaning dietary challenge [25]. Subsequent breeding reveals that while F1 males transmit these defects to F2 male mice in the absence of the dietary challenge, the phenotype is largely attenuated in F3 mice. In humans, a direct effect of grandparental nutrition on the grand-offspring's susceptibility to metabolic diseases has been implicated in controlling sex-specific transgenerational responses [10], [11]. Collectively, these results suggest that while metabolic phenotypes can be propagated by direct exposure to an inducing factor, additional environmental stressors may also trigger and unmask the grandpaternally-induced altered phenotype.
The accumulation of misfolded proteins in the ER leads to stress-induced activation of UPR signaling involving three parallel pathways, each characterized by the stress sensor proteins IRE-1, PERK, and ATF6 [27]. We found that only the spliced and active form of ATF6α was increased in skeletal muscle of F2 female rats from grandfathers fed a HFD, suggesting that this integral branch of the UPR pathway is active. Using transcriptomic data, we found that several ATF6α downstream targets, including Xpb1, Eif2ak3, and Wfs1, were upregulated in skeletal muscle of female grand-offspring of HFD-fed males. Interestingly, we have previously noted alterations in the hepatic methylome and transcriptome, with hypomethylation of several genes controlling glucose metabolism within the ATF-motif regulatory site in severely obese human subjects [28]. Thus, ATF-regulated genes may constitute a nexus that coordinates gene programming in response to over nutrition. ATF6α is the main mediator of transcriptional induction of ER chaperones by single or combined action with XBP1 [29] and plays a pivotal role in glucose and lipid metabolism [30], [31], [32]. In skeletal muscle, co-activation of ATF6α and PGC-1α, mediates many of the functional and metabolic benefits of exercise [33]. Wfs1 deficiency in mice causes ER stress, apoptosis, and impaired cell cycle progression, leading to progressive β-cells loss and impaired glucose tolerance [34]. Increased expression of Wfs1 prevents hyperactive ER stress signaling and dysregulation of the UPR, protecting against cell death, rather than stimulating apoptosis [35]. In order to restore ER homeostasis, a cascade of resident ER molecular chaperones that involves GRP94 and GRP78 is activated [36]. Among the genes that are induced by ATF6, Grp94 is one of the major ER chaperones [36], [37]. We found the GRP94 chaperone is upregulated in skeletal muscle of F2 female from HFD-fed grandfathers, further indicating that the ATF6α signaling pathway is reprogrammed and that GRP94 coordinates an adaptive response by the UPR in order to restore ER stress and cellular homeostasis.
Obesity-induced ER stress can lead to peripheral insulin resistance and diabetes via activation of JNK and negative feedback on insulin receptor signaling [38], [39]. Although JNK activation was unchanged by grandpaternal diet, other physiological and pathological conditions may perturb the ER function, such as nutrient availability, hypoxia and oxidative stress [27], [39], [40], [41], triggering the UPR. However, the overall lipid profile, glycogen content, and level of protein carbonyls were unchanged in skeletal muscle as result of grandpaternal HFD diet. We hypothesized that female grand-offspring of obese founders would present different lipid content in skeletal muscle, since body weight gain was reduced upon HFD challenge compared to grand-offspring of chow-fed founders [17]. However, when comparing the interaction between the skeletal muscle transcriptome and the abundance of phosphatidylcholine and phosphatidydielthylamine species, we found that the phosphatidylcholine lipid fraction covariates with the UPR gene set. ER stress and consequent activation of the UPR caused by HFD-induced obesity alter the ratio between phosphatidylcholine and phosphatidydielthylamine species, the two most abundant classes of phospholipids in the cell membranes [42], [43]. We cannot exclude the possibility that the experimental approach masked changes in the phosphatidylcholine/phosphatidydielthylamine ratio, since we used the entire muscle to evaluate the lipidomic profile, instead of using an isolated ER fraction. Collectively, our results indicate that differences in dietary fat and oxidative stress are unlikely to account for the enriched UPR in these animals. Nevertheless, our results show that the phosphatidylcholine lipid fraction covariates with the enriched UPR in the grand-offspring from obese founders, suggesting that this lipid class may be indirectly activating the UPR pathway.
In summary, transcriptomic analysis reveals that grand-offspring of diet-induced obese founders present altered UPR in skeletal muscle, possibly triggered by the activation of the stress-sensor ATF6, irrespective of changes in the lipidomic profile. These transgenerational epigenetic effects may be mitigated or aggravated by the diet composition in offspring. In conclusion, our findings advance the notion that paternal exposure to environmental factors program whole-body and tissue-specific features, affecting the development and health of successive generations.
Data availability
The transcriptome data set is publically available at Gene Ontology Omnibus GSE95490. The remaining data are available upon request from the corresponding author.
Funding
This work was funded by the Novo Nordisk Foundation (NNF14OC0009941), Swedish Research Council (2015-00165), European Research Council (ICEBERG, ERC-2008-AdG23285), Diabetes Wellness (783_2015PG), Sweden, The Swedish Diabetes Foundation (DIA2015-032), Swedish Foundation for Strategic Research (SRL10-0027), Fredrik and Ingrid Thurings Foundation, Erik and Edith Fernströms, Karolinska Institutet Fonder, and the Strategic Diabetes Research Program at Karolinska Institutet. The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk).
Contribution statement
The authors were the sole contributors to this article. The study was designed by PSA, TDCB, RB, AK, and JRZ. Data were acquired by PSA and TDCB and analyzed and interpreted by PSA, TDCB, RB, AK, and JRZ. The manuscript was written by PSA, TDCB, RB, AK, JRZ. All authors reviewed and approved the final version of the article.
Acknowledgments
We thank Ann-Marie Pettersson for technical assistance.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2017.05.009.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
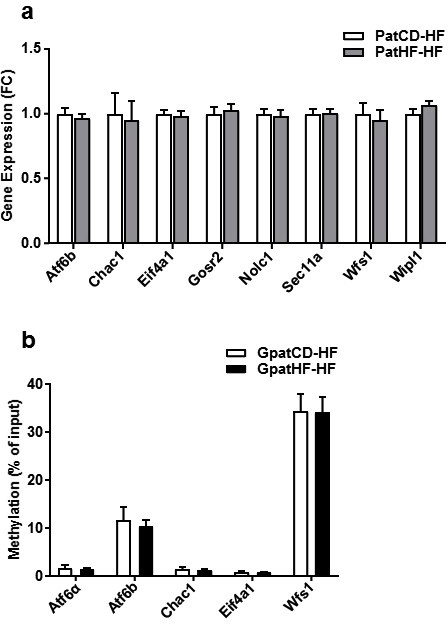
Gene expression in EDL of F1 offspring and DNA-methylation levels in EDL muscle of F2 female rats a) Expression of the core enriched genes by RT-qPCR (n = 11–12) in EDL muscle of F1-offspring. Geometric mean of the Beta-2-Microglobulin (B2M) and Peptidylprolyl Isomerase A (PPIA) was used as internal control. Data are represented as fold change (FC). PatHF-HF vs. PatCD-HF. PatCD-HF: high-fat fed offspring from fathers fed chow diet; PatHF-HF: high-fat fed offspring from fathers fed high-fat diet. b) Target-specific DNA methylation in EDL muscle of F2 rats. DNA methylation is represented as percentage of methylated DNA relative to input (total) DNA of each target measured by qPCR after MBD-capture (n = 7–9). GpatCD-HF: HFD-fed offspring from grandfathers fed chow diet; GpatHF-HF: HFD-fed offspring from grandfathers fed HFD.
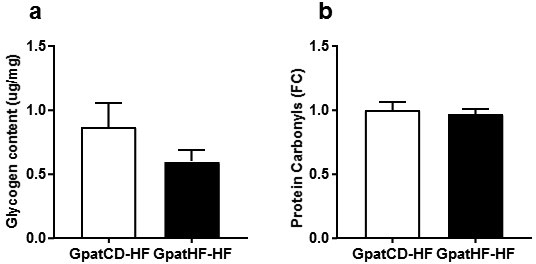
Glycogen and protein carbonyl levels in skeletal muscle of F2 female rats a) Glycogen content and b) Protein carbonyls (Oxyblot™) content in EDL muscle of F2 rats (n = 7–10). Values are means ± SEM. GpatCD-HF: HFD-fed offspring from grandfathers fed chow diet; GpatHF-HF: HFD-fed offspring from grandfathers fed HFD.
References
- 1.Nguyen T., Lau D.C. The obesity epidemic and its impact on hypertension. Canadian Journal of Cardiology. 2012;28:326–333. doi: 10.1016/j.cjca.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Lake J.K., Power C., Cole T.J. Child to adult body mass index in the 1958 British birth cohort: associations with parental obesity. Archives of Disease in Childhood. 1997;77:376–381. doi: 10.1136/adc.77.5.376. [DOI] [PubMed] [Google Scholar]
- 3.Bundred P., Kitchiner D., Buchan I. Prevalence of overweight and obese children between 1989 and 1998: population based series of cross sectional studies. British Medical Journal. 2001;322:326–328. doi: 10.1136/bmj.322.7282.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Svensson V., Jacobsson J.A., Fredriksson R., Danielsson P., Sobko T., Schiöth H.B. Associations between severity of obesity in childhood and adolescence, obesity onset and parental BMI: a longitudinal cohort study. International Journal of Obesity (London) 2011;35:46–52. doi: 10.1038/ijo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeo G.S. Genetics of obesity: can an old dog teach us new tricks? Diabetologia. 2016 doi: 10.1007/s00125-016-4187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barres R., Zierath J.R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nature Reviews Endocrinology. 2016;12:441–451. doi: 10.1038/nrendo.2016.87. [DOI] [PubMed] [Google Scholar]
- 7.Hales C.N., Barker D.J. The thrifty phenotype hypothesis. British Medical Bulletin. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- 8.Ravelli A.C.J., van der Meulen J.H.P., Michels R.P.J., Osmond C., Barker D.J., Hales C.N. Glucose tolerance in adults after prenatal exposure to famine. The Lancet. 1998;351:173–177. doi: 10.1016/s0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 9.Stein A.D., Zybert P.A., van der Pal-de Bruin K., Lumey L.H. Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: evidence from the Dutch Famine. European Journal of Epidemiology. 2006;21:759–765. doi: 10.1007/s10654-006-9065-2. [DOI] [PubMed] [Google Scholar]
- 10.Kaati G., Bygren L.O., Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents' and grandparents' slow growth period. European Journal of Human Genetics. 2002;10:682–688. doi: 10.1038/sj.ejhg.5200859. [DOI] [PubMed] [Google Scholar]
- 11.Pembrey M.E., Bygren L.O., Kaati G., Edvinsson S., Northstone K., Sjöström M. Sex-specific, male-line transgenerational responses in humans. European Journal of Human Genetics. 2006;14:159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- 12.Carone B.R., Fauquier L., Habib N., Shea J.M., Hart C.E., Li R. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyrrell J.S., Yaghootkar H., Freathy R.M., Hattersley A.T., Frayling T.M. Parental diabetes and birthweight in 236 030 individuals in the UK biobank study. International Journal of Epidemiology. 2013;42:1714–1723. doi: 10.1093/ije/dyt220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hur S.S., Cropley J.E., Suter C.M. Paternal epigenetic programming: evolving metabolic disease risk. Journal of Molecular Endocrinology. 2017;58:R159–R168. doi: 10.1530/JME-16-0236. [DOI] [PubMed] [Google Scholar]
- 15.Ng S.F., Lin R.C., Maloney C.A., Youngson N.A., Owens J.A., Morris M.J. Paternal high-fat diet consumption induces common changes in the transcriptomes of retroperitoneal adipose and pancreatic islet tissues in female rat offspring. The FASEB Journal. 2014;28:1830–1841. doi: 10.1096/fj.13-244046. [DOI] [PubMed] [Google Scholar]
- 16.Fullston T., Ohlsson Teague E.M., Palmer N.O., Palmer N.O., DeBlasio M.J., Mitchell M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. The FASEB Journal. 2013;27:4226–4243. doi: 10.1096/fj.12-224048. [DOI] [PubMed] [Google Scholar]
- 17.de Castro Barbosa T., Ingerslev L.R., Alm P.S., Versteyhe S., Massart J., Rasmussen M. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Molecular Metabolism. 2016;5:184–197. doi: 10.1016/j.molmet.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chambers T.J., Morgan M.D., Heger A.H., Sharpe R.M., Drake A.J. High-fat diet disrupts metabolism in two generations of rats in a parent-of-origin specific manner. Scientific Reports. 2016;6:31857. doi: 10.1038/srep31857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masuyama H., Mitsui T., Eguchi T., Tamada S., Hiramatsu Y. The effects of paternal high-fat diet exposure on offspring metabolism with epigenetic changes in the mouse adiponectin and leptin gene promoters. American Journal of Physiology-endocrinology and Metabolism. 2016;311:E236–E245. doi: 10.1152/ajpendo.00095.2016. [DOI] [PubMed] [Google Scholar]
- 20.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trygg J. O2-PLS for qualitative and quantitative analysis in multivariate calibration. Journal of Chemometrics. 2002;16:283–293. [Google Scholar]
- 23.Trygg J., Wold S. O2-PLS, a two-block (X-Y) latent variable regression (LVR) method with an integral OSC filter. Journal of Chemometrics. 2003;17:53–64. [Google Scholar]
- 24.Belmont P.J., Tadimalla A., Chen W.J., Martindale J.J., Thuerauf D.J., Marcinko M. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. Journal of Biological Chemistry. 2008;283:14012–14021. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cropley J.E., Eaton S.A., Aiken A., Young P.E., Giannoulatou E., Ho J.W. Male-lineage transmission of an acquired metabolic phenotype induced by grand-paternal obesity. Molecular Metabolism. 2016;5:699–708. doi: 10.1016/j.molmet.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei Y., Yang C.R., Wei Y.P., Zhao Z.A., Hou Y., Schatten H. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proceedings of the National Academy of Sciences United States of America. 2014;111:1873–1878. doi: 10.1073/pnas.1321195111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature Reviews Molecular Cell Biology. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 28.Kirchner H., Sinha I., Gao H., Ruby M.A., Schönke M., Lindvall J.M. Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Molecular Metabolism. 2016;5:171–183. doi: 10.1016/j.molmet.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto K., Sato T., Matsui T., Sato M., Okada T., Yoshida H. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Developmental Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y., Vera L., Fischer W.H., Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. doi: 10.1038/nature08111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto K., Takahara K., Oyadomari S., Okada T., Sato T., Harada A. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Molecular Biology of the Cell. 2010;21:2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X., Zhang F., Gong Q., Cui A., Zhuo S., Hu Z. Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator-activated receptor alpha. Diabetes. 2016;65:1904–1915. doi: 10.2337/db15-1637. [DOI] [PubMed] [Google Scholar]
- 33.Wu J., Ruas J.L., Estall J.L., Rasbach K.A., Choi J.H., Ye L. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metabolism. 2011;13:160–169. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamada T., Ishihara H., Tamura A., Takahashi R., Yamaguchi S., Takei D. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Human Molecular Genetics. 2006;15:1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- 35.Fonseca S.G., Ishigaki S., Oslowski C.M., Lu S., Lipson K.L., Ghosh R. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. The Journal of Clinical Investigation. 2010;120:744–755. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu G., Lee A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. Journal of Cellular Physiology. 2015;230:1413–1420. doi: 10.1002/jcp.24923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rutkowski D.T., Kaufman R.J. A trip to the ER: coping with stress. Trends in Cell Biology. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Ozcan U., Cao Q., Yilmaz E., Lee A.H., Iwakoshi N.N., Ozdelen E. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 39.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cullinan S.B., Diehl J.A. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. The International Journal of Biochemistry & Cell Biology. 2006;38:317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 41.Kars M., Yang L., Gregor M.F., Mohammed B.S., Pietka T.A., Finck B.N. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59:1899–1905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Meer G., Voelker D.R., Feigenson G.W. Membrane lipids: where they are and how they behave. Nature Reviews Molecular Cell Biology. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu S., Yang L., Li P., Hofmann O., Dicker L., Hide W. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The transcriptome data set is publically available at Gene Ontology Omnibus GSE95490. The remaining data are available upon request from the corresponding author.