

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is an inherited myopathy affecting approximately 1 in 7500 individuals worldwide. It is a progressive disease characterised by skeletal muscle weakness and wasting. A genetic mutation on the 4q35 chromosome results in the expression of the double homeobox 4 gene (DUX4) which drives oxidative stress, inflammation, toxicity, and atrophy within the skeletal muscle. FSHD is characterised by oxidative stress, and there is currently no cure and a lack of therapies for the disease. Antioxidants have been researched for many years, with investigators aiming to use antioxidants therapeutically for oxidative stress-associated diseases. This has included both natural and synthetic antioxidants. The use of antioxidants in preclinical or clinical models has been largely successful with a plethora of research reporting positive results. However, when translated to clinical trials, the use of antioxidants as a therapeutic intervention for a variety of disease has been largely unsuccessful. Moreover, specifically focusing on FSHD, limited research has been conducted on the use of antioxidants as a therapy in either preclinical or clinical models. This review summarises the current state of antioxidant use in the treatment of FSHD and discusses their potential avenue for therapeutic use for FSHD patients.
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is an inherited myopathy characterised by progressive muscle weakness and wasting. FSHD is the third most common muscular dystrophy [1–3], estimated to affect 1 in 7500 individuals worldwide. As indicated by the name of the dystrophy, the facial, humeral, and shoulder girdle muscles are most significantly affected; however, the foot extensors, rectus abdominis muscles, and pelvic-girdle muscles can also become affected towards the latter stage of the disease progression [4]. Individuals with FSHD experience muscle weakness and atrophy, which is often asymmetric [5]. Unlike other muscular dystrophies, the pharyngeal muscles and cardiac muscles are rarely affected in FSHD [4]. Cardiac involvement is found in approximately only 5% of individuals with FSHD and when present manifests as a predisposition to atrial arrhythmias [6]. Around 1% of individuals with FSHD experience restrictive respiratory disease which requires an intervention; however, this is typically seen in those FSHD patients with severe muscle weakness [7]. Patients can also present with additional symptoms such as severe muscular inflammation, subclinical hearing loss, and abnormalities of the peripheral retinal capillary [8–12]. Research has also suggested that FSHD can result in changes to the central nervous system. For instance, Quarantelli et al. [13] reported that patients with FSHD demonstrated a lower volume of grey matter, which was slightly correlated to clinical severity (P < 0.05); however, no correlation was found between brain tissue volume and D4Z4 repeat size. Research has also shown links between FSHD, mental retardation, and epilepsy, with patients presenting with a larger deletion in the FSHD region having the greatest possibility of displaying severe clinical phenotypes with central nervous system abnormalities [14, 15].
2. Genetics of FSHD
FSHD is the result of an epigenetic mechanism which results in disturbances to the transcriptional control of multiple genes [16]. There are two different forms of FSHD, FSHD1 and FSHD2. Of those who are diagnosed with FSHD, 95% of the individuals are diagnosed with FSHD1 with the remainder being FSHD2. FSHD1 is due to deletions in the subtelomeric region chromosome 4q35 within an array of D4Z4 repeat elements. FSHD2, which presents with the same symptoms as FSHD1, is due to a mutation on chromosome 18 resulting in deficiency of the protein SMCHD1 which normally interacts with the D4Z4 repeat on chromosome 4 [17]. As FSHD1 is the most common form of FSHD, the scope of this review is limited to the FSHD1 condition only. FSHD1, which hereafter will be referred to as FSHD, is characterised by a shortened D4Z4 repeat array which consists of 1–10 repeats, whereas in healthy individuals the repeat units range from 11 to 150 [18]. Individuals can present with a similar D4Z4 shortening which is located on chromosome 10q; however, no contractions of the 10qter have been reported to result in FSHD [19, 20].
The D4Z4 repeat array within FSHD individuals contains the double homeobox 4 gene (DUX4), and contractions of the D4Z4 repeat array results in epigenetic changes leading to the expression of DUX4 [21, 22]. DUX4 is active during early development but is silenced by hypermethylation of the D4Z4 region, such that in healthy adult cells, DUX4 is not produced. Within FSHD, the shortened region leads to hypomethylation which allows the expression of DUX4. However, hypomethylation of the D4Z4 region only results in FSHD when it occurs with a permissive chromosome 4. There are two allelic variants of the 4q subtelomere which are both just as common as each other in a healthy population; these are named 4qA and 4qB [23, 24]. FSHD is only associated with the 4qA allele which contains a shortened D4Z4 repeat and working pLAM sequence which allows the abnormally shortened DUX4 gene to produce protein [23]. A similar repeat on the 4qB-type allele will not cause FSHD [25]. The main difference between the 4qA- and 4qB-type alleles is the interstitial (TTAGGG)n array adjacent to the 68 bp satellite DNA within 4qA [24]; (Figure 1). Furthermore, in some instances, the 4q D4Z4 repeat array has been shown to contain an extra 2 kb of D4Z4 sequence, which results in a longer repeat unit in cis with a 4qA allele; this is commonly referred to as the 4qA-L allele [26]; ( Figure 1).
Figure 1.

The molecular signatures of chromosome 4 in both FSHD and healthy individuals. Healthy unaffected individuals carry 11–100 repeat units, whereas individuals with FSHD carry less than 15 repeat units (green triangles represent repeat units). Contraction of the D4Z4 repeat in FSHD results in the protein DUX4 being expressed but only in conjunction with a 4qA allele and working pLAM. Individuals with FSHD have an increased amount of hypomethylated CpGs which also contributes to the transcription of DUX4.
3. Clinical and Molecular Pathophysiology
As previously mentioned, contractions of the D4Z4 repeat array results in DUX4 expression. The DUX4 protein is a transcription factor which drives expression of gene products that mediate the cell inflammatory response ([10]; Figure 2). It is also responsible for a large gene deregulation cascade and inducing genes involved in oxidative stress, inflammation, toxicity, and skeletal muscle atrophy [27–29]. Beyond its direct role as a transcription factor, DUX4 has an indirect role on gene expression because it regulates pituitary homeobox 1 (PITX1; [22, 30–32]). Importantly, PITX1 induces expression of the tumour protein 53 gene (P53; [29, 32]). Overexpression of P53 is orchestral in FSHD as it promotes skeletal muscle atrophy by increasing inflammatory cytokine levels, which subsequently exacerbate further oxidative stress [28, 29, 32]. In addition to the increase in inflammation and oxidative stress, DUX4 expression results in a decrease in MyoD expression (myogenic differentiation), which plays a major role in regulating skeletal muscle differentiation [33].
Figure 2.
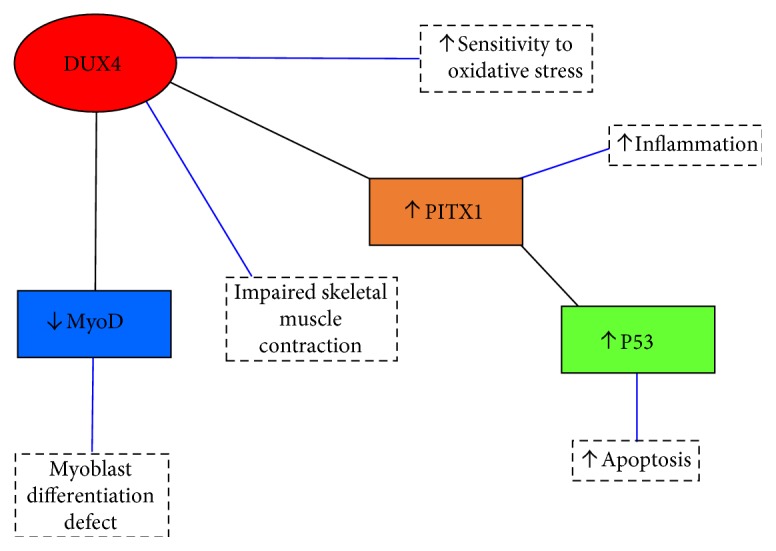
Schematic of the transcriptional cascade effect of DUX4 in which it increases sensitivity to oxidative stress, inflammation, and apoptosis; impairs skeletal muscle contraction; and results in a myoblast differentiation defect. PITX1 = pituitary homeobox 1; P53 = tumour protein 53; MyoD = myogenic differentiation.
4. Oxidative Stress and FSHD
Oxidative stress is often referred to as a condition where the increased production of free radicals or reactive oxygen species (ROS) and oxidative-related reactions results in cellular damage [34]. Examples of free radicals include superoxide (O2·−), hydroxyl (·OH), nitric oxide (NO·), and peroxynitrite (ONOO−). O2·− is the most common free radical in the body, and it occurs as a result of an oxygen molecule obtaining an unpaired electron, thus becoming negatively charged and unstable (Figure 3). Multiple processes can result in an oxygen molecule becoming charged and unstable, with examples being the oxidation of the catecholamines and the activation of arachidonic acid cascade [35]. The cellular enzymes NADPH oxidase and xanthine oxidase (XO) are known to be major contributors in the production of O2·−. Although O2·− is a radical, it is not very reactive [34] and the antioxidant enzyme, superoxide dismutase (SOD), converts O2·− to the weaker oxidant, hydrogen peroxide (H2O2). H2O2 is relatively stable in its natural state; however, in the presence of transition ions such as iron and copper, it has the ability to rapidly diffuse across cell membranes and convert into ·OH radicals [35]. ·OH radicals have an extremely short half-life and are one of the most potent oxidants in the body. Notably, they react at their site of formation and attack most biological molecules, which in turn propagates free radical chain reactions [34, 35].
Figure 3.
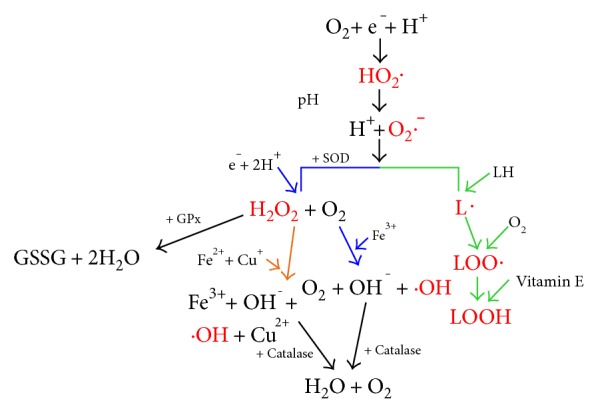
A schematic of the reactions which lead to the formation of free radicals. Red text indicates free radicals. Green arrows represent lipid peroxidation, orange arrows represent the Fenton reactions, and the blue arrows represent the Haber-Weiss reactions. SOD is the enzyme superoxide dismutase, and GSSG refers to glutathione disulphide. HO2· = hydroperoxyl radical; O2·− = superoxide radical; H2O2 = hydrogen peroxide; ·OH = hydroxyl radical; L· = lipid radical; LOO· = fatty acid peroxyl radical; LOOH = lipid hydroperoxide.
The production of O2·− is not just limited to enzymatic reactions. Under certain conditions, mitochondria and the electron-transport chain will produce ROS [36]. When the concentration of oxygen within the mitochondria decrease, O2·− and ·OH radicals are produced [36]. Moreover, the production of O2·− is not just limited to oxygen concentration; it is also reliant on the concentration of potential electron donors [37]. Complex I is known to drive significant O2·− production within the mitochondria, particularly in two conditions: firstly, when there is a high proton motive force due to insufficient ATP production and a reduced coenzyme Q (CoQ) pool [38, 39] and secondly, when the mitochondrial matrix has a high NADH/NAD+ ratio [40, 41]. The production of mitochondrial ROS can disturb cellular homeostasis through driving redox signalling, mitochondrial dysfunction, and apoptosis/necrosis.
Antioxidant enzymes such as SOD, glutathione peroxidase (GPx), and catalase are involved in protecting cells from being damaged by ROS [42]. As previously stated, SOD converts O2·− to the weaker radical H2O2. H2O2 can then be broken down by GPx in the mitochondria and cytosol in a reaction where glutathione (GSH) is oxidised producing glutathione disulphide and 2H2O [43]. H2O2 can also be broken down into H2O and O2 by the enzyme, catalase (Figure 3). Overproduction of ROS can pose a serious problem to bodily homeostasis because of oxidative damage to tissues [44]. These adverse effects of ROS can be limited by the natural antioxidant pathways represented by SOD, GPx, and catalase. Moreover, as will be discussed in more detail later, under certain circumstances, if ROS becomes too high, natural antioxidant pathways can become overwhelmed, allowing excess ROS to cause cellular damage [44].
Individuals with FSHD have been reported to have an altered oxidative stress response, in part due to differential expression of proteins [33, 45–49]. FSHD myoblasts were shown to have increased levels of the protein p21, when compared to healthy myoblasts under normal growth conditions [49]. Increased levels of p21 have previously been shown to be upregulated in human and murine fibroblasts in response to oxidative stress [50]. p21 is known to induce cell cycle arrest [51], and it has been reported that p21 expression correlates with the onset of muscle differentiation in myoblasts growing under normal conditions [52]. Winokur et al. [49] concluded that due to the increased p21, FSHD myoblasts wrongly signal a state of oxidative stress to their nucleus, resulting in cell cycle arrest, hindering the formation of new myotubes. Furthermore, Tassin et al. [53] reported that SOD levels were increased in FSHD-affected muscle, along with two other proteins involved in the oxidative stress response, glutathione S-transferase and thioredoxin peroxidase. These three proteins act to protect against oxidative stress, and it is hypothesised that within FSHD, the antioxidant defence system is increased to try and alleviate the increased oxidative stress. In addition to changes in the antioxidant defence system and oxidative stress signalling, the oxygen storage in FSHD muscle is also affected [47]. It was reported that levels of myoglobin (a protein found in the muscle which binds to iron and oxygen, thus storing oxygen in the muscles) in FSHD muscle were 25% lower than those in healthy muscle. The increases reported in oxidative stress response proteins are likely a compensatory mechanism for the increased ROS levels and decreased oxygen storage capacity in FSHD muscle.
FSHD muscle shows marked changes in mitochondrial metabolism through a decrease in cytochrome c oxidase and increases in NADH-dehydrogenase flavoprotein and NADH-ubiquinone oxidoreductase [47]. This is expected to be the results of the decreased oxygen storage capacity of the muscle; thus, mitochondrial dysfunction occurs resulting in an increase in oxidative stress. Turki et al. [54] reported that mitochondria organisation in FSHD human skeletal muscle biopsies was at times markedly altered when compared to healthy controls, with large mitochondria pools in the intermyofibrillar and subsarcolemmal compartments. Furthermore, Turki et al. [54] reported that FSHD patients have increased oxidative stress, increased inflammatory response, and abnormal mitochondrial function when compared to healthy controls. Both mitochondrial dysfunction and oxidative stress correlated with function muscle impairment [54].
Excessive oxidative stress and inflammation of the skeletal muscle can interfere with muscular contractions. In mammalian skeletal muscle, excitation contraction coupling depends on motor neuron-induced cell depolarization and the subsequent interaction between dihydropyridine receptor (DHPR) and the ryanodine receptor (RyR1), which results in Ca2+ being released from the terminal cisternae of the sarcoplasmic reticulum [55]. RyR1s are redox-sensitive channels; therefore, alterations in redox state can result in their activation [56, 57] or inactivation [58]. Furthermore, Stoyanovsky et al. [56] reported that in under oxidative stress, RyR1s can become unstable resulting in an increased open probability that subsequently causes Ca2+ leak. Along with the oxidative modification of RyR1, pro-oxidant states lead to the activation of CaMKII [59]. The activation of CaMKII has previously been shown by Rodney et al. [60] to promote the phosphorylation of RyR1, which consequently results in the leaky release of Ca2+ from the sarcoplasmic reticulum [55].
More specifically, in regard to muscular dystrophy, Bellinger et al. [61] reported that mdx mouse muscle (a mouse model of Duchenne muscular dystrophy (DMD)) experiences Ca2+ leak as a result of cysteine-nitrosylation, which is likely to contribute to muscle weakness within the disease. DMD is the result of a deficiency in dystrophin which severely disrupts the dystrophin glycoprotein complex (DPG), leading to muscle damage as the result of pathological Ca2+ signalling [62–64]. The mdx mouse model has a nonsense point mutation on the dystrophin gene, from glutamine to threonine, resulting in the production of a nonfunctioning dystrophin protein [65]. Although the mdx mouse is the most commonly used model of DMD, it is often seen as a “milder” form of DMD as they present minimal clinical symptoms and a lifespan reduced by 25% [66] in comparison to a reduction of 75% in humans. Moreover, Andersson et al. [67] reported that RyR1 channels in a murine model of limb-girdle muscular dystrophy (LGMD; a deficiency in β-sarcoglycan—a protein involved in the DPG) muscle are oxidised, cysteine nitrosylated, phosphorylated, and depleted of calastabin1 which results in leaky channels along with a decreased fast twitch muscle force and impaired capacity for exercise. The same authors [67] went on to suggest that leaky RyR1 channels may underline multiple forms of muscle dystrophy, particularly those which have a disrupted DPG and suggested that pharmaceutical targeting of RyR1 channels may be a potential therapeutic treatment in DMD and LGMD. To date there is no known link between Ca2+ leak and FSHD; however, based on the data presented in DMD and LGMD, it is hypothesised that Ca2+ leak may be a pathological mechanism in FSHD.
5. Synergy between Oxidative Stress and Inflammation
In addition to oxidative stress, FSHD individuals experience increased skeletal muscle inflammation which can also interfere with muscular contraction. Muscle atrophy as a result of chronic inflammation is associated with increased proinflammatory cytokine production, such as tumour necrosis factor alpha (TNF-α), interleukin 1 (IL-1), interleukin 6 (IL-6), and interferon gamma (IFN-γ; 59). The overexpression of TNF-α has been shown to result in significant body and muscle mass loss in mice [68, 69]. Moreover, Li et al. [70] reported that cultured C2C12 skeletal muscle myotubes show protein degradation following prolonged exposure to TNF-α (1–6 ng · ml−1). It was reported that TNF-α stimulates protein degradation through binding to surface receptor (TNFR1), which induced mitochondrial production of ROS resulting in a rapid activation of the ubiquitin-proteasome pathway and the degradation of I-κBα, the protein responsible for inhibiting the key inflammatory response mediator NF-κB [70]. As a result, NF-κB is activated, translocates to the nucleus to switch on expression of its target genes, including a subset that controls muscle proteolysis. Linking together oxidative stress and inflammation, Kosmidou et al. [71] reported that C2C12 myotubes exposed to either XO or H2O2 for 24 hours showed a concentration-dependent increase in IL-6 production. In regard to FSHD specifically, Turki et al. [54] reported that patients with FSHD presented with significantly increased levels of the proinflammatory cytokines TNF-α (P < 0.001), IFN-α2 (interferon alpha-2; P < 0.001), RANTES (regulated on activation, normal T-cell expressed and secreted; P < 0.001), MCP1 (monocyte chemotactic protein-1; P < 0.01), and IL-6 (P < 0.01), along with a significant positive correlation between TNF-α and glutathione disulphide (GSSG) levels (P = 0.02). To date, this is the only known data which shows a synergistic relationship between oxidative stress and inflammation in FSHD; thus, it remains to be defined in any real detail.
In vivo, similar mechanisms have been shown to operate with proinflammatory cytokines inducing muscle wasting via the activation of the ubiquitin-proteasome pathway and apoptosis [72–74]. Baracos et al. [72] reported that the soleus and extensor digitorum of young male CD-strain mice incubated in IL-1 for two hours experienced a significant increase in protein degradation. The authors reported that proteolysis increased; however, the rate of protein synthesis remained the same. Furthermore, the incubation of muscles in IL-1 also resulted in the synthesis of prostaglandin E2, which is known to augment muscle protein breakdown.
In addition to these biochemical changes to protein, oxidative stress can lead to impaired muscle contractions because of effects on muscle force. Callahan et al. [75] reported that O2·− and ·OH significantly decreased maximum calcium-activated force by 14.5% and 43.9%, respectively, in rat diaphragm fibres. Furthermore, Edwards et al. [76] reported that mouse extensor digitorum longus (EDL) untreated muscle exposed to O2·− significantly decreased maximum Ca2+-activated specific force (kN/m2; P < 0.001); however, this was attenuated when the muscle was pretreated with the potent synthetic antioxidant, Tempol (4-hydroxy-2,2,2,2-tetramethylpiperidine-1-oxyl). When the muscles were incubated at 37 °C with 1 mM Tempol, they showed no significant reduction in contractile capacity, suggesting that antioxidants such as Tempol can reduce the levels of oxidative stress. These results explain that the contractile capacity of mouse muscle is impaired by the superoxide anion radical, likely due to oxidation of various contractile chemical groups, particularly those located on the myosin heads. Together, these findings show the vulnerability of FSHD muscle to oxidative stress, leading to a reduction in contractile capacity and ultimately muscle weakness in addition to showing the potential of antioxidants to help alleviate the problems caused in FSHD.
6. Antioxidants and FSHD
As oxidative stress underlies FSHD, antioxidant treatment is a potential therapeutic option. El Haddad et al. [77] reported that healthy and FSHD muscles pretreated with retinoic acid (RA; a metabolite of vitamin A) presented lower ROS levels and apoptosis than when exposed to H2O2 overnight (both P < 0.05). Additionally, myoblast adhesion was restored by pretreatment with RA and associated with a higher IC50 (the H2O2 concentration at which half of the cells adhere to the culture dish). The underlying protective mechanism involved glutathione peroxidase 3 (GPx3) showed significantly higher GPx3 mRNA levels (P < 0.01) in healthy human myoblasts pretreated with RA, when compared to untreated human myoblasts. In addition, GPx3 activity was significantly increased following exposure to RA (P < 0.05). Limited clinical trials have been performed studying the effects of vitamin A supplementation; thus, it is unclear if it would be useful clinically, especially for FSHD patients.
Synthetic antioxidants have been shown to reduce oxidative stress in FSHD muscle, along with slowing disease processes. Bosnakovski et al. [78] identified 52 compounds which can inhibit DUX4-induced toxicity in myoblasts. Several of these compounds were known to be antioxidants, with the authors reporting that a large amount of the other compounds lacked any obvious reducing activity; therefore, it was hypothesised that these antioxidants protect cells from oxidative stress in an indirect manner. Furthermore, Dmitriev et al. [79] reported that FSHD myoblasts present increased DNA damage. This is evident by high levels of the DNA damage marker protein γH2AX (P < 0.05; the phosphorylation of histone H2A variant H2AX at Ser139) and increased apurinic/apyrimidinic sites (abasic site; a location of the DNA which has neither a purine or pyrimidine base, this may occur spontaneously or are a result of DNA damage). If DUX4 levels are knocked down via siRNA in FSHD primary myoblasts, the DNA damage was significantly reduced (P < 0.05). Together, these findings provide clear evidence that DUX4 induces DNA damage. If the FSHD myoblasts were treated with Tempol, there was reduced DNA damage (P < 0.05). However, DNA damage was not reduced completely. Tempol was also shown to improve myotube formation of FSHD myoblasts. Similar findings were obtained with the synthetic antioxidant NAC (N-acetylcysteine). Therefore, antioxidant treatment may reduce DNA damage in FSHD myotubes, thus improving muscle health.
To date, a plethora of research has shown that exercise can induce antioxidant effects and alleviate oxidative stress. It is known that exercise results in an acute increase in ROS; however, this is shortly followed by a compensatory increase in antioxidant defences [80]. Gomez-Cabrera et al. [81] reported that exhaustive exercise conducted in rats results in the activation of mitogen-activated protein (MAP) kinases and the NF-κB pathway which in turn stimulates the production of antioxidant defences such as SOD. When rats were administered with allopurinol (an inhibitor of XO which is involved in O2·− production following exhaustive exercise), the adaptive changes in antioxidant defences and muscle adaptations were abolished, thus confirming that exercise can be considered as a potent antioxidant. Furthermore, Karabulut et al. [82] reported that following a 12-week aerobic exercise program, the lipid peroxidation marker malondiadehyde (MDA) significantly decreased whereas the antioxidant parameters SOD and GSH significantly increased. More recently, a systematic review and meta-analysis conducted by de Sousa et al. [83] reported that physical activity induced antioxidant effects and decreased pro-oxidant effects regardless of the type of exercise, volume, intensity, and population studied. Consequently, it can be concluded that exercise is antioxidant in nature and can balance the redox state to elicit positive health outcomes.
Antioxidants act to decrease oxidative stress and reduce the threat free radicals pose to bodily homeostasis. However, in certain situations, antioxidants can become pro-oxidants and will elicit the opposite effect to what is desired. Pro-oxidants can induce oxidative stress via two pathways, firstly through the production of free radicals and secondly, through inhibiting the antioxidant defence systems. For example, vitamin C is primarily considered a potent antioxidant, but when it is combined with iron and copper, it promotes a reaction which results in iron or copper being reduced from Fe3+ to Fe2+ or Cu3+ to Cu2+, respectively. These augment the reduction of H2O2 to ·OH thereby increasing ROS. Similarly, α-tocopherol (vitamin E), another well-known antioxidant, can become pro-oxidant when used at high concentrations. This occurs because upon reacting with a free radical, α-tocopherol becomes the tocopheroxyl radical, and without the presence of ascorbic acid, it will remain in this reactive state. It has been shown that the tocopheroxyl radical promotes the autoxidation of linoleic acid [84].
The well described antioxidants, carotenoids, can also become pro-oxidants in certain situations. At high concentrations, carotenoids exposed to oxidative stress undergo oxidative breakdown which drives pro-oxidative effects through the formation of ·OH radicals [85, 86]. Similarly, flavonoids can act as pro-oxidants and when they are in the presence of both transition ions such as copper, iron or zinc, and oxygen. In such an environment, flavonoids can be catalysed resulting in the formation of ROS and free radicals such as the phenoxyl radical [87]. Phenoxyl radicals primarily result in DNA and lipid damage.
Some synthetic antioxidants have also been reported to become pro-oxidant and contribute to oxidative stress. Under high concentrations (10−4–10−2 M), Tempol is known to contribute to oxidative stress [88, 89]. NAC also has a similar response and, in certain situations, can enhance the production of free radicals, such as ·OH [90]. Furthermore, it has been reported that some antioxidants can elicit a harmful effect on the differentiation of skeletal muscle progenitor cells. Ding et al. [91] reported that the inhibition of nuclear factor erythroid 2-related factor 2 (Nrf2), Nrf2-gultamate-cysteine ligase (GCL), and glutathione reductase (GR) through small interfering RNA (siRNA) blocked muscle differentiation in C2C12 cells. Lee et al. [92] reported that inhibition of complex I (a driver of O2·− production) via siRNA, resulted in suppressed differentiation in H9c2 rat cardiac myoblasts. Moreover, this research was supported by Won et al. [93] who reported that peroxiredoxin-2 (Prx-2), which protects against oxidative stress, is upregulated during muscle differentiation, and treatment with NAC prevents Prx-2 upregulation, thus subduing myotube formation. Consequently, determining the correct balance between ROS level and antioxidant therapy is of great importance, as it is clear from what is discussed above, too much of either can have detrimental effects on bodily homeostasis.
7. Clinical Trials
Whilst there are many positives for the use of antioxidants, they have their limitations. There has been a small amount of studies investigating the effects of antioxidants on FSHD muscle and the potential benefits antioxidant therapies may have. Passerieux et al. [94] reported that dietary supplementation of vitamin C, vitamin E, zinc, and selenium did not improve the two-minute walk test in FSHD patients. However, at a mechanistic level, it did improve maximum voluntary contraction and endurance of the quadriceps. The contradictory findings may be due to the small population size of the study. In another study, van der Kooi et al. [95] reported that supplementation of folic acid and methionine (which can lead to an increase in DNA methylation) had no effect on DNA methylation in FSHD patients and subsequently showed no positive effect on the disease state or muscle health. More rigorous research is needed therefore before a conclusion can be made on the benefits of antioxidant supplementation in FSHD. Whilst the in vitro research is showing positive results, it would be naïve to extend conclusions to clinical settings.
With the antioxidant effects of exercise being noted previously, it is interesting to examine the literature on exercise interventions in FSHD. Bankole et al. [96] reported that a combined strength and interval cycling exercise-training program resulted in significant increases (P < 0.05) in VO2 peak, mean arterial pressure, muscle endurance, maximum voluntary contraction of the quadriceps, and 6-minute walking distance after 24 weeks. In addition, the amount of fatigue the patient experienced decreased (P < 0.01) with no exacerbation of muscle tissue damage. It is clear from the literature that exercise can be a potent antioxidant and can also elicit positive results in FSHD individuals, however, is it unclear whether antioxidant treatment would be beneficial or detrimental to the skeletal muscle adaptations in response to exercise [97]. It would be interesting to investigate the effects a combination of antioxidant and exercise therapy could have on FSHD muscle as this may shed light on whether antioxidants have a positive or negative effect on the exercise-induced skeletal muscle adaptations.
8. Conclusion
In conclusion, there is a vast amount of research which shows the beneficial effects antioxidants can have on reducing oxidative stress and associated cell damage. Whilst this research is primarily positive, there exists enough contradictory data to highlight that our understanding of what determines when antioxidants are beneficial or detrimental is not yet completely understood. Even less well understood are the effects of antioxidants on the disease process of FSHD. Current evidence suggests that antioxidants should suppress the oxidative stress response in myoblasts, thereby helping to preserve muscle quality and function. Overall, the results discussed throughout this review are strong indicators that supplementation of antioxidants may be beneficial for improving muscle health, particularly the functional and contractile properties in addition to alleviating some of the oxidative stress witnessed within FSHD patients; however, as of yet, there have been no large clinical trials that have been successful.
Acknowledgments
This work was supported by a FSHD Global Grant-in-Aid (Alison Kay Heather).
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- 1.Emery A. E. H. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromuscular Disorders. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-U. [DOI] [PubMed] [Google Scholar]
- 2.Pandya S., King W. M., Tawil R. Facioscapulohumeral dystrophy. Physical Therapy. 2008;1:105–113. doi: 10.2522/ptj.20070104. [DOI] [PubMed] [Google Scholar]
- 3.Deenen J. C. W., Arnts H., van der Maarel S. M., et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83(12):1056–1059. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Statland J., Tawil R. Neurologic Clinics. 3, . Vol. 32. Elsevier B.V; 2014. Facioscapulohumeral muscular dystrophy; pp. 721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tawil R. Facioscapulohumeral muscular dystrophy. Neurotherapeutics. 2008;5(4):601–606. doi: 10.1016/j.nurt.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laforet P., de Toma C., Eymard B., et al. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology. 1998;51(5):1454–1456. doi: 10.1212/WNL.51.5.1454. [DOI] [PubMed] [Google Scholar]
- 7.Wohlgemuth M., van der Kooi E. L., van Kesteren R. G., van der Maarel S. M., Padberg G. W. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology. 2004;63(1):176–178. doi: 10.1212/01.WNL.0000133126.86377.E8. [DOI] [PubMed] [Google Scholar]
- 8.Taylor D. A., Carroll J. E., Smith M. E., Johnson M. O., Johnston G. P., Brooke M. H. Facioscapulohumeral dystrophy associated with hearing loss and coats syndrome. Annals of Neurology. 1982;12(4):395–398. doi: 10.1002/ana.410120414. [DOI] [PubMed] [Google Scholar]
- 9.Voit T., Lamprecht A., Lenard H. G., Goebel H. H. Hearing loss in facioscapulohumeral dystrophy. European Journal of Pediatrics. 1986;145(4):280–285. doi: 10.1007/BF00439401. [DOI] [PubMed] [Google Scholar]
- 10.Ishihara T., Orimo S., Lee J. E. H., Goto K., Nonaka I. Necrotic fibers was paralleled by an increased number of inflammatory cells. Muscle and Nerve. 1995;(Supplementary 2):56–66. [Google Scholar]
- 11.Padberg G. W., Frants R. R., Brouwer OF, Wijmenga C., Bakker E., Sandkuijl L. A. Facioscapulohumeral muscular dystrophy in the Dutch population. Muscle & Nerve. 1995;18(S13):S81–S84. doi: 10.1002/mus.880181315. [DOI] [PubMed] [Google Scholar]
- 12.Padberg G. W., Brouwer OF, de Keizer R. J., et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle & Nerve. 1995;18(S13):S73–S80. doi: 10.1002/mus.880181314. [DOI] [PubMed] [Google Scholar]
- 13.Quarantelli M., Lanzillo R., Del Vecchio W., et al. Modifications of brain tissue volumes in facioscapulohumeral dystrophy. NeuroImage. 2006;32(3):1237–1242. doi: 10.1016/j.neuroimage.2006.04.226. [DOI] [PubMed] [Google Scholar]
- 14.Funakoshi M., Goto K., Arahata K. Epilepsy and mental retardation in a subset of early onset 4q35-facioscapulohumeral muscular dystrophy. Neurology. 1998;50(6):1791–1794. doi: 10.1212/WNL.50.6.1791. [DOI] [PubMed] [Google Scholar]
- 15.Sharma S., Namrata S. Association of schizophrenia and mental retardation with facio-scapulohumeral muscular dystrophy. Neurology India. 2004;52:104–105. [PubMed] [Google Scholar]
- 16.De Greef J. C., Wohlgemuth M., Chan O. A., et al. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology. 2007;69(10):1018–1026. doi: 10.1212/01.wnl.0000271391.44352.fe. [DOI] [PubMed] [Google Scholar]
- 17.Lemmers R. J., Tawil R., Petek L. M., et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nature Genetics. 2012;44(12):1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards M., Coppee F., Thomas N., Belayew A., Upadhyaya M. Facioscapulohumeral muscular dystrophy (FSHD): an enigma unravelled? Human Genetics. 2012;131:325–340. doi: 10.1007/s00439-011-1100-z. [DOI] [PubMed] [Google Scholar]
- 19.Lemmers R. J., De Kievit P., Van Geel M., et al. Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Annals of Neurology. 2001;50(6):816–819. doi: 10.1002/ana.10057. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y., Forner J., Fournet S., Jeanpierre M. Improved characterization of FSHD mutations. Annales de Génétique. 2001;44(2):105–110. doi: 10.1016/S0003-3995(01)01075-9. [DOI] [PubMed] [Google Scholar]
- 21.Hewitt J. E., Lyle R., Clark L. N., et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Human Molecular Genetics. 1994;3(8):1287–1295. doi: 10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 22.Gabriëls J., Beckers M. C., Ding H., et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236(1):25–32. doi: 10.1016/S0378-1119(99)00267-X. [DOI] [PubMed] [Google Scholar]
- 23.Lemmers R. J., de Kievit P., Sandkuijl L., et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nature Genetics. 2002;32(2):235–236. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 24.Van Geel M., Dickson M. C., Beck A. F., et al. Genomic analysis of human chromosome 10q and 4q telomeres suggest a common origin. Genomics. 2002;79(2):210–217. doi: 10.1006/geno.2002.6690. [DOI] [PubMed] [Google Scholar]
- 25.Lemmers R. J., Wohlgemuth M., Frants R. R., Padberg G. W., Morava E., van der Maarel S. M. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. American Journal of Human Genetics. 2004;75(6):1124–1130. doi: 10.1086/426035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemmers R. J., van der Vliet P. J., Klooster R., et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329(5999):1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bosnakovski D., Xu Z., Gang E. J., et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. The EMBO Journal. 2008;27(20):2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarzkopf M., Coletti D., Marazzi G. Chronic p53 activity leads to skeletal muscle atrophy and muscle stem cell perturbation. Basic Applied Myology. 2008;18(5):131–138. [Google Scholar]
- 29.Wallace L. M., Garwick S. E., Mei W., et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Annals of Neurology. 2011;69(3):540–552. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Östlund C., Garcia-Carrasquillo R. M., Belayew A., Worman H. J. Intracellular trafficking and dynamics of double homeodomain proteins. Biochemistry. 2005;44(7):2378–2384. doi: 10.1021/bi047992w. [DOI] [PubMed] [Google Scholar]
- 31.Dixit M., Ansseau E., Tassin A., et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proceedings of the National Academy of Sciences. 2007;104(46):18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pandey S. N., Cabotage J., Shi R., et al. Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biology Open. 2012;1(7):629–639. doi: 10.1242/bio.20121305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Celegato B., Capitanio D., Pescatori M., et al. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics. 2006;6(19):5303–5321. doi: 10.1002/pmic.200600056. [DOI] [PubMed] [Google Scholar]
- 34.Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet. 1994;344(8924):721–724. doi: 10.1016/S0140-6736(94)92211-X. [DOI] [PubMed] [Google Scholar]
- 35.Betteridge D. J. What is oxidative stress? Metabolism. 2000;49(2) Supplement 1:3–8. doi: 10.1016/S0026-0495(00)80077-3. [DOI] [PubMed] [Google Scholar]
- 36.Liochev S. I., Fridovich I. The role of O2·− in the production of HO. Free Radical Biology & Medicine. 1994;16(1):29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 37.Murphy M. P. How mitochondria produce reactive oxygen species. Journal of Biochemistry. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korshunov S. S., Skulachev V. P., Starkov A. A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters. Federation of European Biochemical Societies. 1997;416(1):15–18. doi: 10.1016/S0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 39.Lambert A. J., Brand M. D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. The Biochemical Journal. 2004;382(Part 2):511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudin A. P., Bimpong-Buta N. Y. B., Vielhaber S., Elger C. E., Kunz W. S. Characterization of superoxide-producing sites in isolated brain mitochondria. The Journal of Biological Chemistry. 2004;279(6):4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 41.Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(20):7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Afonso V., Champy R., Mitrovic D., Collin P., Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint, Bone, Spine. 2007;74:324–329. doi: 10.1016/j.jbspin.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Chance B., Sies H., Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharyya A., Chattopadhyay R., Mitra S., Crowe S. E. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiological Reviews. 2014;94(2):329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barro M., Carnac G., Flavier S., Mercier J., Vassetzky Y., Laoudj-Chenivesse D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. Journal of Cellular and Molecular Medicine. 2010;14(1-2):275–289. doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheli S., François S., Bodega B., et al. Expression profiling of FSHD-1 and FSHD-2 cells during myogenic differentiation evidences common and distinctive gene dysregulation patterns. PLoS One. 2011;6(6, article e20966) doi: 10.1371/journal.pone.0020966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laoudj-Chenivesse D., Carnac G., Bisbal C., et al. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. Journal of Molecular Medicine. 2005;83(3):216–224. doi: 10.1007/s00109-004-0583-7. [DOI] [PubMed] [Google Scholar]
- 48.Macaione V., Aguennouz M., Rodolico C., et al. RAGE-NF-κB pathway activation in response to oxidative stress in facioscapulohumeral muscular dystrophy. Acta Neurologica Scandinavica. 2007;115(2):115–121. doi: 10.1111/j.1600-0404.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 49.Winokur S. T., Barrett K., Martin J. H., et al. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscular Disorders. 2003;13(4):322–333. doi: 10.1016/S0960-8966(02)00284-5. [DOI] [PubMed] [Google Scholar]
- 50.Lander H. M., Ogiste J. S., Kenneth K., Novogrodsky A., Chem J. B., Teng K. K. p21ras as a common signaling target of reactive free radicals and cellular redox stress. The Journal of Biological Chemistry. 1995;270(36):21195–21198. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 51.Otten A. D., Firpo E. J., Gerber A. N., Brody L. L., Roberts J. M., Tapscott S. J. Inactivation of MyoD-mediated expression of p21 in tumor cell lines. Cell Growth & Differentiation. 1997;8(November):1151–1160. [PubMed] [Google Scholar]
- 52.Wang J., Walsh K. Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science. 1996;273(5273):359–361. doi: 10.1126/science.273.5273.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., et al. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? Journal of Cellular and Molecular Medicine. 2013;17(1):76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turki A., Hayot M., Carnac G., et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radical Biology & Medicine. 2012;53(5):1068–1079. doi: 10.1016/j.freeradbiomed.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 55.Rudolf R., Magalhães P. J., Pozzan T. Direct in vivo monitoring of sarcoplasmic reticulum Ca2+ and cytosolic cAMP dynamics in mouse skeletal muscle. The Journal of Cell Biology. 2006;173(2):187–193. doi: 10.1083/jcb.200601160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoyanovsky D., Murphy T., Anno P. R., Kim Y. M., Salama G. Cell Calcium. 1. Vol. 21. Pearson Professional ltd; 1997. Nitric oxide activates skeletal and cardiac ryanodine receptors; pp. 19–29. [DOI] [PubMed] [Google Scholar]
- 57.Eager K. R., Dulhunty A. F. Activation of the cardiac ryanodine receptor by sulfhydryl oxidation is modified by Mg2+ and ATP. Journal of Membrane Biology. 1998;163:9–18. doi: 10.1007/s002329900365. [DOI] [PubMed] [Google Scholar]
- 58.Marengo J. J., Hidalgo C., Bull R. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophysical Journal. 1998;74:1263–1277. doi: 10.1016/S0006-3495(98)77840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erickson J. R., Joiner M. L., Guan X., et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodney G. G., Williams B. Y., Strasburg G. M., Beckingham K., Hamilton S. L. Regulation of RYR1 activity by Ca2+ and calmodulin. Biochemistry. 2000;39(26):7807–7812. doi: 10.1021/bi0005660. [DOI] [PubMed] [Google Scholar]
- 61.Bellinger A. M., Reiken S., Carlson C., et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nature Medicine. 2009;15(3):325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glesby M. J., Rosenmann E., Nylen E. G., Wrogemann K. Serum CK, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle & Nerve. 1988;11(8):852–856. doi: 10.1002/mus.880110809. [DOI] [PubMed] [Google Scholar]
- 63.Turner P. R., Westwood T., Regen C. M., Steinhardt R. A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 64.Fong P. Y., Turner P. R., Denetclaw W. F., Steinhardt R. A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science. 1990;250(4981):673–676. doi: 10.1126/science.2173137. [DOI] [PubMed] [Google Scholar]
- 65.Sicinski P., Geng Y., Ryder-Cook A., Barnard E., Darlison M., Barnard P. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 66.Chamberlain J. S., Metzger J., Reyes M., Townsend D., Faulkner J. A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. The FASEB Journal. 2007;21(9):2195–2204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- 67.Andersson D. C., Meli A. C., Reiken S., et al. Leaky ryanodine receptors in beta-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skeletal Muscle. 2012;2(1):1–9. doi: 10.1186/2044-5040-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Llovera M., Lopez-Soriano F. J., Argiles J. M. Effects of tumor necrosis factor-a on muscle-protein turnover in female Wistar rats. Journal of the National Cancer Institute. 1993;85(16):1334–1339. doi: 10.1093/jnci/85.16.1334. [DOI] [PubMed] [Google Scholar]
- 69.Ling P. R., Schwartz J. H., Bistrian B. R. Mechanisms of host wasting induced by administration of cytokines in rats. The American Journal of Physiology. 1997;272(3:1):E333–E339. doi: 10.1152/ajpendo.1997.272.3.E333. [DOI] [PubMed] [Google Scholar]
- 70.Li Y. P., Schwartz R. J., Waddell I. D., Holloway B. R., Reid M. B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. The FASEB Journal. 1998;12(10):871–880. doi: 10.1096/fasebj.12.10.971. [DOI] [PubMed] [Google Scholar]
- 71.Kosmidou I., Vassilakopoulos T., Xagorari A., Zakynthinos S., Papapetropoulos A., Roussos C. Production of interleukin-6 by skeletal myotubes: role of reactive oxygen species. American Journal of Respiratory Cell and Molecular Biology. 2002;26(5):587–593. doi: 10.1165/ajrcmb.26.5.4598. [DOI] [PubMed] [Google Scholar]
- 72.Baracos V., Rodemann H. P., Dinarello C. A., Goldberg A. L. Stimulation of muscle protein degradation and prostaglandin E2 release by leukocytic pyrogen (interleukin-1). A mechanism for the increased degradation of muscle proteins during fever. The New England Journal of Medicine. 1983;308(10):553–558. doi: 10.1056/NEJM198303103081002. [DOI] [PubMed] [Google Scholar]
- 73.Saini A., Nasser A. S., Stewart C. E. H. Waste management-cytokines, growth factors and cachexia. Cytokine and Growth Factor Reviews. 2006;17:475–486. doi: 10.1016/j.cytogfr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 74.Reid M. B., Li Y. P. Tumor necrosis factor-alpha and muscle wasting: a cellular perspective. Respiratory Research. 2001;2(5):269–272. doi: 10.1186/rr67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Callahan L. A., She Z. W., Nosek T. M. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. Journal of Applied Physiology. 2001;90:45–54. doi: 10.1152/jappl.2001.90.1.45. [DOI] [PubMed] [Google Scholar]
- 76.Edwards J. N., Macdonald W. A., Van Der Poel C., Stephenson D. G., Jn E., Wa M. O2·− production at 37°C plays a critical role in depressing tetanic force of isolated rat and mouse skeletal muscle. American Journal of Physiology-Cell Physiology. 2007;293(2):650–660. doi: 10.1152/ajpcell.00037.2007. [DOI] [PubMed] [Google Scholar]
- 77.El Haddad M., Jean E., Turki A., et al. Glutathione peroxidase 3, a new retinoid target gene, is crucial for human skeletal muscle precursor cell survival. Journal of Cell Science. 2012;125(24):6147–6156. doi: 10.1242/jcs.115220. [DOI] [PubMed] [Google Scholar]
- 78.Bosnakovski D., Choi S. H., Strasser J. M., Toso E. A., Walters M. A., Kyba M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skeletal Muscle. 2014;4(1):p. 4. doi: 10.1186/2044-5040-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dmitriev P., Bou Saada Y., Dib C., et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radical Biology and Medicine. 2016;99:244–258. doi: 10.1016/j.freeradbiomed.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 80.Fisher-Wellman K., Bloomer R. J. Acute exercise and oxidative stress: a 30 year history. Dynamic Medicine. 2009;8(1):1–25. doi: 10.1186/1476-5918-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gomez-Cabrera M. C., Domenech E., Viña J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radical Biology & Medicine. 2008;44(2):126–131. doi: 10.1016/j.freeradbiomed.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 82.Karabulut A. B., Emin Kafkas M., Kafkas A. S., Onal Y., Kiran T. R. The effect of regular exercise and massage on oxidant and antioxidant parameters. Indian Journal of Physiology and Pharmacology. 2013;57(4):378–383. [PubMed] [Google Scholar]
- 83.de Sousa C. V., Sales M. M., Rosa T. S., Lewis J. E., de Andrade R. V., Simões H. G. The antioxidant effect of exercise: a systematic review and meta-analysis. Sports Medicine. 2017;47:277–293. doi: 10.1007/s40279-016-0566-1. [DOI] [PubMed] [Google Scholar]
- 84.Cillard J., Cillard P., Cormier M., Girre L. α-Tocopherol prooxidant effect in aqueous media: increased autoxidation rate of linoleic acid. Journal of the American oil Chemists Society. 1980;57(8):252–255. doi: 10.1007/BF02668254. [DOI] [Google Scholar]
- 85.Young A. J., Lowe G. M. Antioxidant and prooxidant properties of carotenoids. Archives of Biochemistry and Biophysics. 2001;385(1):20–27. doi: 10.1006/abbi.2000.2149. [DOI] [PubMed] [Google Scholar]
- 86.Siems W., Wiswedel I., Salerno C., et al. β-Carotene breakdown products may impair mitochondrial functions—potential side effects of high-dose β-carotene supplementation. The Journal of Nutritional Biochemistry. 2005;16(7):385–397. doi: 10.1016/j.jnutbio.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 87.Galati G., O’Brien P. J. Potential toxicity of flavonoids and other dietary phenolics: significance for their chemopreventive and anticancer properties. Free Radical Biology & Medicine. 2004;37(3):287–303. doi: 10.1016/j.freeradbiomed.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 88.Alpert E., Altman H., Totary H., et al. 4-Hydroxy tempol-induced impairment of mitochondrial function and augmentation of glucose transport in vascular endothelial and smooth muscle cells. Biochemical Pharmacology. 2004;67(10):1985–1995. doi: 10.1016/j.bcp.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 89.May J. M., Qu Z.-C., Juliao S., Cobb C. E. Ascorbic acid decreases oxidant stress in endothelial cells caused by the nitroxide tempol. Free Radical Research. 2005;39(2):195–202. doi: 10.1080/10715760400019661. [DOI] [PubMed] [Google Scholar]
- 90.Sagristá M. L., García A. E., Africa De Madariaga M., Mora M. Antioxidant and pro-oxidant effect of the thiolic compounds N-acetyl-L-cysteine and glutathione against free radical-induced lipid peroxidation. Free Radical Research. 2002;36(3):329–340. doi: 10.1080/10715760290019354. [DOI] [PubMed] [Google Scholar]
- 91.Ding Y., Choi K. J., Kim J. H., et al. American journal of pathology. 6. Vol. 172. American Society for Investigative Pathology; 2008. Endogenous hydrogen peroxide regulates glutathione redox via nuclear factor erythroid 2-related factor 2 downstream of phosphatidylinositol 3-kinase during muscle differentiation; pp. 1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee S., Tak E., Lee J., et al. Cell Research. 5. Vol. 21. Nature Publishing Group; 2011. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation; pp. 817–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Won H., Lim S., Jang M., et al. Peroxiredoxin-2 upregulated by NF-κB attenuates oxidative stress during the differentiation of muscle-derived C2C12 cells. Antioxidants & Redox Signaling. 2012;16(3):245–261. doi: 10.1089/ars.2011.3952. [DOI] [PubMed] [Google Scholar]
- 94.Passerieux E., Hayot M., Jaussent A., et al. Free Radical Biology and Medicine. Vol. 81. Elsevier; 2015. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: a double-blind randomized controlled clinical trial; pp. 158–169. [DOI] [PubMed] [Google Scholar]
- 95.van der Kooi E. L., de Greef J. C., Wohlgemuth M., et al. No effect of folic acid and methionine supplementation on D4Z4 methylation in patients with facioscapulohumeral muscular dystrophy. Neuromuscular Disorders. 2006;16(11):766–769. doi: 10.1016/j.nmd.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 96.Bankolé L.-C., Millet G. Y., Temesi J., et al. Safety and efficacy of a 6-month home-based exercise program in patients with facioscapulohumeral muscular dystrophy: a randomized controlled trial. Medicine (Baltimore) 2016;95(31, article e4497) doi: 10.1097/MD.0000000000004497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Radak Z., Ishihara K., Tekus E., et al. Redox Biology. Vol. 12. Elsevier B.V; 2017. Exercise, oxidants, and antioxidants change the shape of the bell-shaped hormesis curve; pp. 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]