

Abstract
An IL-4 antagonist was designed based on structural and biochemical analysis of unbound IL-4 and IL-4 in complex with its high-affinity receptor (IL-4Rα). Our design strategy sought to capture a protein–protein interaction targeting the high affinity that IL-4 has for IL-4Rα. This strategy has impact due to the potential relevance of IL-4Rα as a drug target in the treatment of asthma. To mimic the IL-4 binding surface, critical side chains for receptor binding were identified, and these side chains were transplanted onto a previously characterized, de novo-designed four-helix protein called designed helical protein 1 (DHP-1). This first-generation design resolved the ambiguity previously described for the connectivity between helices in DHP-1 and resulted in a protein capable of binding to IL-4Rα. The second-generation antagonist was based upon further molecular modeling, and it succeeded in binding IL-4Rα better than the first-generation. This protein, termed DHP-14-AB, yielded a protein with a cooperative unfolding transition (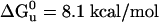 ) and an IC50 of 27 μM when in competition with IL-4 whereas DHP-1 had no affinity for IL-4Rα. The crystal structure of DHP-14-AB was determined to 1.9-Å resolution and was compared with IL-4. This comparison revealed how design strategies targeting protein–protein interactions require high-resolution 3D data and the incorporation of orientation-specific information at the level of side-chains and secondary structure element interactions.
) and an IC50 of 27 μM when in competition with IL-4 whereas DHP-1 had no affinity for IL-4Rα. The crystal structure of DHP-14-AB was determined to 1.9-Å resolution and was compared with IL-4. This comparison revealed how design strategies targeting protein–protein interactions require high-resolution 3D data and the incorporation of orientation-specific information at the level of side-chains and secondary structure element interactions.
Keywords: de novo protein design, helical bundle, IL-4 receptor, structure-based design
We sought to design an antagonist to compete with the high-affinity interaction between IL-4 and its receptor, IL-4Rα. This protein–protein interaction is an important drug development target in the treatment of acute allergic asthma and other atopic conditions such as seasonal allergies, urticaria, and eczema (1, 2). Our purpose was to create a method for antagonist generation that could be instructive for curing these diseases and other diseases resulting from rouge protein–protein interactions. IL-4·IL-4Rα forms a heterotrimeric receptor complex along with another cytokine receptor of either IL-13 receptor α chain 1 (IL-13Rα1) or γ common chain (γC) to activate signaling across cell membranes in the immune system (3). IL-4 is in the short-chain helical cytokine family; this family lacks any significant sequence homology between members (4, 5). The IL-4 four-helix bundle has an up–up–down–down order and orientation between major helices (6–8).
The IL-4·IL-4Rα complex formation (Kd ≈ 160 pM) is one of the highest affinity interactions of a cytokine for its receptor (9). IL-4Rα is a member of the hematopoietic receptor superfamily (10). Its association rate constant, kon ≈ 1.8 × 107 M–1·s–1, for IL-4 to IL-4Rα is nearly 100-fold as fast as the presumably diffusion-limited association rate constant for cytokine interactions with their first contact single receptor chain, such as human growth hormone·hGHR (kon ≈ 3 × 105 M–1·s–1) (9, 11). This fast and high-affinity interaction has been attributed to “electrostatic steering” and to the complementary interface between the positively charged IL-4 and negatively charged IL-4Rα surfaces; the opposing charges attract each other, resulting in associate rates faster than diffusion, and coordinate each other in the complex (9).
The platform selected for IL-4 antagonist design was our own de novo-designed, four-helix bundle protein, designed helical protein (DHP) 1; previously, we showed that DHP-1 has stability like that of a natural protein and a structure exactly as intended (12). That design was based upon conjugation of four 24-aa amphipathic helical peptides that were built from a reduced set of the natural amino acids. The DHP-1 crystal structure did not resolve the connectivity between the helices, which resulted in a structural ambiguity. Both bundle topology and superhelical twist are described as “right-handed” or “left-handed.” The helical bundle topology refers to the macroscopic handedness (chirality) and is based on the backbone connectivity and unit direction vector of the helices (13). In contrast, the superhelical twist of a bundle refers to the wrapping of helices around the protein core axis regardless of connectivity between helices, for which most frequently bundles are left-handed (14). The DHP-1 helices have a left-handed superhelical twist. The DHP-1 structure without the loops had an ambiguity in the bundle topology that was resolved here by creating IL-4Rα binding function.
The DHP-1 structure showed that it had a pair of neighboring helices of approximately similar antiparallel orientation, size, and spacing as IL-4 (Fig. 1). This structural similarity was sufficient to initiate designing an antagonist to IL-4 onto the DHP-1 helices. The residues on IL-4, which interact with IL-4Rα, were analyzed to select a subset for transfer. These residues on IL-4 were then modeled in silico onto DHP-1 to create the first-generation antagonists. Adding these residues was the first step toward resolving the topology question about DHP-1 by breaking its symmetry.
Fig. 1.
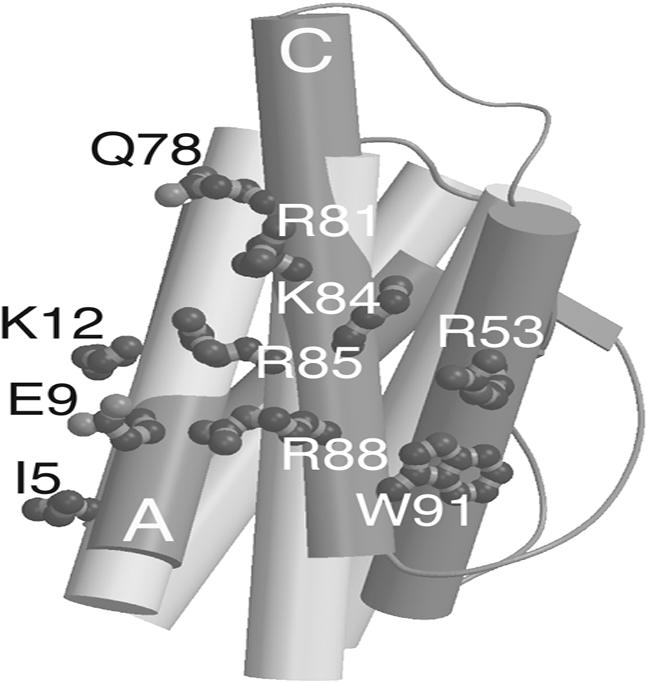
DHP-1 (light gray) helices superimposed on unbound IL-4 (dark gray). IL-4 side chains are displayed in atom representation for mutations made onto DHP-1 in the first round design. Side chains fit onto the DHP-1 backbone structure except for R53, because the third helix of DHP-1 did not overlap well on the corresponding IL-4 B helix.
After the first generation of antagonist development, the so-called DHP-10 bundles, our proteins were assessed for stability in solution and functional binding to IL-4Rα to ascertain which designed topology was represented. At the time when IL-4·IL-4Rα coordinates became available, we were able to integrate the topology design results with analysis of steric clash-points revealed on IL-4Rα that were suboptimal in the context of our first design. This observation led to the second-generation design to remove potential clashes from our IL-4 antagonist and to fit the target complex. This second-generation design, DHP-14-AB, was solved and compared with IL-4. The analysis of this structure along with IL-4 indicated that the crossing angle (Ω) between helices is likely to be as important as the overlap of the Cα positions within the binding site and the energetic value of the side chain interactions with the receptor.
Materials and Methods
Molecular Modeling. The o suite software lsqman (15) was used to superimpose the molecules with 36 Cα atoms included. Side chains were mutated by using the 3D modeling software moloc (16). Rotamers were selected from the library based on which side chain could fit on DHP-1 and be consistent with IL-4 unbound and later IL-4 bound to IL-4Rα. First-generation models were subjected to energy minimization with cns (17).
Gene Synthesis. Two genes were designed, synthesized (Integrated DNA Technologies, Coralville, IA), and subcloned into cloning and expression plasmids. The genes were based on the 108-aa sequence of DHP-1 (12) (Table 4, which is published as supporting information on the PNAS web site). Each gene was sequenced in the forward and reverse directions in both vectors.
Protein Expression and Purification. DHPs were expressed in DH-5α Escherichia coli by using the pMal-c2x vector, and levels were similar to DHP-1, with a yield of >25 mg/liter purified protein (12). DHP concentration was determined by absorption of ultra-violet light at 280 nm due to one tryptophan and the molar extinction coefficient of 5,500 M–1·cm–1 (18).
Helical Structure Monitored by CD. CD measurements were made by using a J-715 spectropolarimeter (Jasco, Easton, MD) with a temperature controlled holder and a 2-mm path length cuvette.
Unfolding Free-Energy (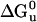 ) Calculation. The unfolding transition was monitored at the α-helical signal minimum 222 nm by CD as a function of the denaturant, guanidinium hydrochloride (Gdn·HCl), concentration (M). The buffer was 10 mM sodium phosphate (pH 7.0) at 25°C; each component of the solution was weighted out in order to eliminate protein concentration variation. Samples were equilibrated overnight in the dark at 25°C. Gdn·HCl concentration was determined by using the difference in refractive index (ΔN) between the buffer and the solutions with Gdn·HCl added (19). The data were fit to a two-state transition by using a nonlinear free-energy curve-fitting method (20) and the software kaleidagraph (Synergy Software, Reading, PA).
) Calculation. The unfolding transition was monitored at the α-helical signal minimum 222 nm by CD as a function of the denaturant, guanidinium hydrochloride (Gdn·HCl), concentration (M). The buffer was 10 mM sodium phosphate (pH 7.0) at 25°C; each component of the solution was weighted out in order to eliminate protein concentration variation. Samples were equilibrated overnight in the dark at 25°C. Gdn·HCl concentration was determined by using the difference in refractive index (ΔN) between the buffer and the solutions with Gdn·HCl added (19). The data were fit to a two-state transition by using a nonlinear free-energy curve-fitting method (20) and the software kaleidagraph (Synergy Software, Reading, PA).
Dynamic Light Scattering (DLS). Monodispersity, hydrodynamic radius (Rh), and molecular weight of DHPs were determined by DLS at 22°C by using the DynaPro MS/X Instrument and dynamics 5.25.44 software (Proterion, Piscataway, NJ). The regularization algorithm was used for analyzing the experimental autocorrelation function (channels 1–80), yielding the translational diffusion coefficient (Dt), Rh, and intensity distributions of particle diameter. Solvent refractive index and solvent viscosity were 1.333 and 1.019, respectively. Approximately 70% of the measurements were averaged and used to estimate Rh and molecular weight based on an empirical curve of known proteins. Before DLS analysis, DHPs at 3–5 mg/ml were dialyzed overnight against buffer (PBS, 10 mM sodium phosphate, pH 7.0, or 10 mM Tris, pH 7.4) and filtered.
Competitive Inhibition Assay. Each protein was incubated for 1 h at room temperature on a 96-well plate fixed with biotinylated IL-4Rα and horseradish peroxidase (HRP)-conjugated IL-4. The loss of HRP activity due to displacement of HRP–IL-4 was monitored as a function of competing protein concentration. The curve-fit data Kis were converted to IC50s: IL-4, 2.3 ± 0.16 nM; DHP-14-AB, 64 ± 2.3 μM; DHP-AB, 1.1 ± 0.20 mM; and DHP-AD, 2.2 ± 0.29 mM. The IC50 for IL-4 was 2.3 nM, although it was typically 1 nM; therefore, division by 2.3 normalized each IC50 for assay condition variation (Table 1).
Table 1.
ΔGu0 and unfolding midpoint (Gdn·HCl) was calculated by nonlinear extrapolation (20)
| Protein | ΔGu0, kcal/mol | Unfolding midpoint, M | IC50, μM |
|---|---|---|---|
| DHP-1 | 13.1 | 4.8 | N.B. |
| DHP-10-AB | 13.5 | 4.6 | 495 |
| DHP-10-AD | 4.3 | 4.4 | 970 |
| DHP-14-AB | 8.1 | 3.2 | 27 |
IC50 for each protein were determined by competition with IL-4·HRP for IL-4Rα. DHP-1 did not bind (N.B.) IL-4Rα under these conditions.
Crystallization and X-Ray Diffraction Data Collection. Crystals were grown of DHP-14-AB, as an maltose-binding protein (MBP) fusion, by the hanging drop vapor diffusion method, with 1:1 ratio drops of protein solution (10 mg/ml) in 20 mM Tris (pH 7.4) with a well solution of 2 M ammonium sulfate, 50 mM sodium acetate (pH 4.6) at room temperature. Crystalline needles were used for seeding growth of rod-like crystals, which diffracted to 1.9 Å at Advanced Light Source (ALS) beamline 8.3.1 using glycerol in artificial mother liquor for cryoprotection. Diffraction data were processed by using elves (21) with mosflm (22) and ccp4 (23).
Structure Determination and Refinement. A molecular replacement solution was found by using epmr 2.5 (24) with MBP as the search model. Rigid body refinement and semiautomated building using arp/warp (25) produced interpretable maps with 77% of the asymmetric unit MBP and 23% DHP-14-AB (Table 2). Subsequent rounds of building were performed with moloc (16) and o (15), and refinement was done with ccp4 (23) and cns suites (17).
Table 2. Summarized data collection and structure refinement statistics.
| Diffraction resolution, Å | 1.9 |
| Space group | P212121 |
| Unit cell, Å | a = 69.56, b = 74.69, c = 103.69 |
| Final R-factor* (Rfree), % | 20.2 (22.4) |
| No. of residues† | |
| Protein | 494 (14) |
| Waters | 284 |
| Maltose | 1 |
| rmsd angles, ° | 1.1 |
| rmsd bonds, Å | 0.0049 |
Complete data statistics are available in Table 5, which is published as supporting information on the PNAS web site. rmsd, rms deviation.
R-factor = Σhkl∥Fobs| — |Fcalc∥/Σhkl| Fobs|. Rfree is computed in the same manner as the R-factor with the test set of reflections (5%).
The values in parentheses are for number of missing residues.
Results
Transplantation of IL-4Rα Binding Site onto DHP-1. Hage et al. (3) determined the structure of IL-4·IL-4Rα and compared the atomic coordinates of IL-4 from the free IL-4 with those in the IL-4·IL-4Rα complex. They found that the IL-4Rα binding site is compact and composed exclusively of side-chains, most of which were derived from IL-4 helices A and C, and provided a predominantly positively charged interfacial surface of ≈800 Å2 of solvent accessible surface area on IL-4. Upon interaction with IL-4Rα, IL-4 binding site helices shifted on average by <1Å, and IL-4 accommodated by adjusting amino acid side-chains and tilting helices to form the interaction. For example, the distance between Cα positions on IL-4 helices A and C within the binding interface decreased by 0.4–0.6 Å. More significant contractions in the range of 1–1.5 Å occurred between Cα positions on IL-4 helices A and D. Helices A and D support the putative binding site for the second receptor (26). Our four-helix bundle design mimics only the high-affinity receptor binding site on IL-4 helices A and C, because it does not incorporate the putative second receptor binding site.
To design the interactions for binding to IL-4Rα, the Cα positions for two helices of DHP-1 were superimposed on IL-4 helices A and C. Upon superposition of these positions, the rms deviation between the structures is 0.96 Å using the binding site portion of the IL-4 structure and the analogous residues from two DHP-1 helices (Fig. 1). The crossing angle (Ω) between IL-4 A and C helices was broader by 2.6° than the DHP-1 crossing angle between helices A and B and broader by 5° than that for DHP-1 helices A and D. The inter-helix distances were within 1 Å between IL-4 and either unique pair of DHP-1 helices (27) (Table 3). Because the backbone helices superimposed within less than an angstrom and DHP-1 could place two helices facing toward the receptor in a similar orientation and position as IL-4 is to IL-4Rα (Fig. 1), the first generation of mutations was designed onto the first two neighboring DHP-1 helices in the amino acid sequence.
Table 3. Crossing angle (Ω) and closest approach distance between target helices for IL-4 antagonist template peptides and proteins and IL-4.
| Structure | Helix pair | Ω, ° | Distance, Å |
|---|---|---|---|
| GCN4 | 1, 2 | 24.3 | 9.4 |
| PD1 | 1, 2 | 18.0 | 8.6 |
| 1, 4 | 17.4 | 10.2 | |
| DHP-1 | A, B | 21.0 | 8.5 |
| A, D | 18.6 | 9.9 | |
| DHP-14-AB | A, B | 18.1 | 8.2 |
| A, D | 9.0 | 9.9 | |
| IL-4 free | A, C | 23.6 | 9.6 |
| A, D | 31.1 | 9.8 | |
| IL-4 bound | A, C | 27.6 | 9.6 |
| A, D | 38.1 | 10.4 |
Helix packing interactions were calculated with webmol (27) on these coordinates: IL-4 (1RCB), IL-4·IL-4Rα (1IAR), DHP-1 (4HB1), GCN4 (2ZTA), and DHP-14-AB. Crossing angle values were added to 180° in order to make all values relative to parallel equal 0°. In order to represent the four-helix bundle of DHP-1, the DHP-1 asymmetric unit of two helices (A and B) had a twofold crystallographic symmetry operation applied to it.
To select critical binding site residues to incorporate onto DHP-1, the IL-4·IL-4Rα structural analysis (3) and associated mutagenesis (28) were used to create a three-component scoring function to identify side-chains for transplantation onto DHP-1. Wang et al. characterized mutants in IL-4, which provided the relative changes in binding energy (ΔΔG) associated with the loss of a side chain (28). The scoring function was based on three criteria: decrease in solvent accessible surface area (ΔSASA) upon complex formation of >50 Å2 (2 points), ΔΔG upon mutation to alanine of >0.3 kcal/mol (2 points), and a positively charged side chain (1 point). In total, fifteen residues were scored from IL-4 helices A and C: I5, T6, Q8, E9, K12, T13, Q78, R81, F82, K84, R85, R88, N89, W91, and G92. Side chain positions, which scored higher than 3 of the total 5 points, were selected for transplantation onto DHP-1.
Based on their score, seven residues were designed into DHP-1 corresponding to the following IL-4 residues: I5, E9, K12, K84, R81, R85, and R88. These residues represent 47% of the possible residues in the binding site on IL-4. W91 did not meet the three-point cut off, but was added to the design because it had a larger ΔΔG of 0.73 kcal/mol and it rearranged upon binding IL-4Rα. Q78 was added to the design to remove a hydrophobic leucine side chain from the binding surface on DHP-1. Nine first-round mutations, 60% of the possible binding site residues, were made in silico onto two adjacent helices on DHP-1, hence termed DHP-9.
IL-4 helix B, the least interacting helix for IL-4Rα binding, contributed one critical residue, R53, based upon a ΔSASA (decrease in solvent accessible surface area) upon binding of 50 Å2, ΔΔG of 0.84 kcal/mol upon mutation to glutamine, and being positively charged (3, 28). R53 contributed to the receptor-binding interface through interaction with F41 of IL4-Rα, which completes a hydrophobic collar around IL-4 R88 in the complex. Removal of IL-4 R88 resulted in a ΔΔG of ≈3.75 kcal/mol, which indicated its importance in binding and suggested that coordination of its position should be included in our design. Furthermore, IL-4 Y56, W91, and R53 rearrange upon binding to IL-4Rα to bury IL-4 R88 in the interface. To simplify incorporation of R53, this side chain was placed in the model as close in 3D space as feasible, because DHP-1 did not have a helix that overlapped IL-4 helix B (Fig. 1). We expected that adding this approximation of R53 to our design of the binding interaction would improve mimicking of the most critical residue IL-4 R88. Thus, DHP-10 was designed to have a total of 10 IL-4Rα binding mutations (Table 4).
Protein Design for Helical Bundle Topology. The goal of de novo protein design is to create a 3D model for a target fold with a corresponding linear amino acid sequence, synthesize it, and then determine its exact structure. The ambiguity of whether DHP-1 had right- or left-handed topology is due in part to the glycine linkers not being defined in the crystal structure and compounded by the repeating amino acid sequence of each helix. To resolve this topological uncertainty, two functional enantiomers were created toward a target with a specified fold, which was IL-4Rα. DHP-1 and IL-4 both have the same four-helix core structures. Thus, IL-4 is compatible with our design efforts to resolve topology because wrapping around the DHP-1 and IL-4 four-helical bundle, each structure has the same inter-helical anti-parallel relationship. The IL-4Rα binding function will require the side-chains to be displayed only on a unique set of adjacent helices in the DHP-1 template structure.
Two molecules were designed to address the two-way ambiguity in the topology between the helices of DHP-1 (Fig. 2). The spacing between helices in DHP-1, along with the designed length of loops between helices, targeted it to be, but did not confirm, a right-handed topology. DHP-1 itself was assembled from helices [termed PD1 (29)], where short linkers of 3, 4, and 3-glycine residues were inserted between the four helices to maintain the inter-helical spacing observed in the PD1 structure. The DHP-1 structure was maintained by its hydrophobic protein core and supported by “knobs into holes” side-chain packing interactions between these helices.
Fig. 2.

Helical bundle scheme illustrates two possible topologies and the difference in the binding site depending upon the topology. As a matter of convention, orienting from the N-terminal helix, a left-handed bundle has the second helix in the bundle to the left; a right-handed bundle has the second helix in the bundle to the right (13). To imagine formation of a binding site for productive interaction with IL-4Rα, two circle halves are split between neighboring helices in two different arrangements. When the circle is continuous and merging, the functional alignment of binding interactions is presented. When the circle is broken and diverging, the nonfunctional alignment of the binding interactions is presented. (A) The left-handed model forms a productive binding site when the circle halves could merge across the first and last helices. (B) The right-handed model forms a productive binding site when the circle halves could merge across the first and second helices. (C) The left-handed model does not form a productive binding site when the circle halves diverge across the first and last helices. (D) The right-handed model does not form a productive binding site when the circle halves diverge across the first and second helices.
Creating IL-4Rα binding function broke the DHP-1 sequence redundancy and structural pseudo 222-symmetry. DHP-10-AD carried the binding site on the adjacent helices A and D to represent the left-handed helical bundle (Fig. 2 A); DHP-10-AB carried the binding site on the adjacent helices A and B to represent the right-handed helical bundle (Fig. 2B). The logic follows that, depending on which protein had stability and bound to IL-4Rα, the DHP topology could be deduced. The protein with better stability and function would indicate that the binding site conformed to the restrictions of inter-helical packing interactions stabilizing the protein and that it was intact on one surface between adjacent helices.
First Generation: Stability and Function. To determine stability of the designed proteins in solution, the free energy of unfolding (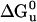 ) was measured by monitoring the loss of helical structure as a function of increasing denaturant concentration (Fig. 3). DHP-10-AB had stability similar to DHP-1 with
) was measured by monitoring the loss of helical structure as a function of increasing denaturant concentration (Fig. 3). DHP-10-AB had stability similar to DHP-1 with 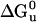 equal to 13.5 and 13.1 kcal/mol, respectively. These values of
equal to 13.5 and 13.1 kcal/mol, respectively. These values of 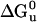 are considered equivalent because the data, collected previously and reanalyzed, for DHP-1 had a narrower concentration range for the unfolded baseline >6 M Gdn·HCl as compared with DHP-10-AB and the unfolding transition overlaps. DHP-10-AD was less stable in solution, with
are considered equivalent because the data, collected previously and reanalyzed, for DHP-1 had a narrower concentration range for the unfolded baseline >6 M Gdn·HCl as compared with DHP-10-AB and the unfolding transition overlaps. DHP-10-AD was less stable in solution, with 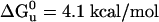 (Table 1).
(Table 1).
Fig. 3.

Unfolding transition plot for each protein used to calculate 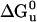 . DHP-10-AB (open squares), DHP-10-AD (open circles), and DHP-14-AB (filled diamonds) unfolding transitions were analyzed to determined free energy of unfolding,
. DHP-10-AB (open squares), DHP-10-AD (open circles), and DHP-14-AB (filled diamonds) unfolding transitions were analyzed to determined free energy of unfolding, 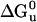 . DHP-1 (filled circles) data were from Schafmeister et al. (12) and were reanalyzed along with the new data for comparison.
. DHP-1 (filled circles) data were from Schafmeister et al. (12) and were reanalyzed along with the new data for comparison.
To access the aggregation state in solution, dynamic light scattering was used to measure the Rh and calculate the molecular mass for each protein. DHP-1 was mono-disperse with an average Rh of 1.79–1.80 nm and a molecular mass of 11.9–12 kDa, in agreement with the measured DHP-1 Rh determined by analytical ultracentrifugation (12). DHP-10-AB, which turned out to be the expected right-handed topology, was mono-disperse with an Rh of 1.78–1.95 nm and a calculated molecular mass of 11.9–12.7 kDa. In contrast, DHP-10-AD was poly-disperse at room temperature, which prevented an accurate determination of Rh and molecular mass.
To determine the IL-4Rα binding activity, each protein was subject to competitive binding versus IL-4. DHP-1 did not compete with IL-4 for binding to IL-4Rα over a concentration range from μM to tens of mM. The IC50 for DHP-10-AB was 495 μM. The IC50 for DHP-10-AD was 970 μM (Table 1). Thus DHP-10-AB bound with approximately twice the affinity of DHP-10-AD but was still 5 × 105 times lower affinity than IL-4. This result is consistent with the right-handed topology.
Second Generation: Additional Mutations, Stability, and Function. When the coordinates became available for the IL-4·IL-4Rα complex, superimposing our model of DHP-10-AB with IL-4·IL-4Rα, a number of residues in DHP-10-AB were found to overlap residues in IL-4Rα (Fig. 6, which is published as supporting information on the PNAS web site). Therefore, additional mutations were added to the DHP-10-AB template to create DHP-14-AB. IL-4 residues T13, D87, N89, and G92 were incorporated into DHP-14-AB (Table 4). IL-4 G92 was mimicked with G92A within our model because glycine has the potential to destabilize the helix and alanine did not seem to overlap with IL-4Rα in our model. On DHP-10-AB helix C, IL-4 R53 mimic was moved three residues toward the binding site, because the first R53 mimic pointed away from the rest of the IL-4Rα binding site and its key residue analogous to IL-4 R88. DHP-14-AB was mono-disperse like DHP-10-AB with an Rh of 1.79–1.82 nm and a calculated molecular mass of 11.9–12.4 kDa. The unfolding free energy (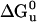 ) decreased from 13.5 kcal/mol to 8.1 kcal/mol, whereas the functional activity increased to IC50 27 μM, 18 times the affinity of DHP-10-AB. DHP-14-AB was progressively improved as an IL-4 antagonist from DHP-1, which was stable and functionless, although still 2.7 × 104 times lower in affinity than IL-4 itself. Structure Determination for DHP-14-AB. To facilitate structure determination, we purified DHP-14-AB as a C-terminal fusion to MBP. Electron density for four helices was obvious in the maps calculated with phases from MBP. DHP-14-AB was wedged against MBP within one asymmetric unit (Fig. 4A). An omit map verifies continuous density of a previously unobserved glycine loop (Fig. 4B). This structure allows for the definitive assignment of topology to the designed DHP-14-AB, which was the expected right-handed bundle.
) decreased from 13.5 kcal/mol to 8.1 kcal/mol, whereas the functional activity increased to IC50 27 μM, 18 times the affinity of DHP-10-AB. DHP-14-AB was progressively improved as an IL-4 antagonist from DHP-1, which was stable and functionless, although still 2.7 × 104 times lower in affinity than IL-4 itself. Structure Determination for DHP-14-AB. To facilitate structure determination, we purified DHP-14-AB as a C-terminal fusion to MBP. Electron density for four helices was obvious in the maps calculated with phases from MBP. DHP-14-AB was wedged against MBP within one asymmetric unit (Fig. 4A). An omit map verifies continuous density of a previously unobserved glycine loop (Fig. 4B). This structure allows for the definitive assignment of topology to the designed DHP-14-AB, which was the expected right-handed bundle.
Fig. 4.

DHP-14-AB·MBP and experimental electron density map. (A) Structure of DHP-14-AB (red) fusion with MBP (blue through orange). (B) DHP-14-AB “omit” map contoured at 1 σ for the loop between helix A and B. Figures were made with pymol (33), molscript (34, 35), and raster3d (36).
To compare our design with IL-4, the differences in helical packing interactions among the designed proteins and the target structure IL-4 were calculated (27) (Table 3). The distance between helices is measured at the nearest inter-helical approach. The crossing angle (Ω), also called the inter-helical angle, is the angle between the helix axes when projected onto their plane of contact (30). Classical knobs-into-holes packing between α helices has a calculated crossing angle of 20° (31). PD1 helices have crossing angles of slightly less than 20°, which are nearly classical. Upon linking those helices together in DHP-1, the putative helices A and B Ω value decreased by 3° whereas the inter-helical distance decreased by 0.1 Å. However, the pairing of DHP-1 helices was ambiguous and may be the reverse, i.e., helices A and B could be helices A and D. Where DHP-1 helices A and B and DHP-14-AB helices A and B are compared, DHP-14-AB functional mutations decreased Ω by 2.9°. Where DHP-1 helices A and D are compared with DHP-14-AB helices A and D, the mutations decreased Ω by 9.6°.
Helix packing analysis of IL-4 highlighted how the crossing angles increased significantly upon binding to IL-4Rα (Table 3). The crossing angle between helices A and C increased by 4°. The crossing angle between helices A and D increased by 7°. The difference in crossing angle between IL-4 (helices A and C) from the complex and DHP-14-AB binding site helices (helices A and B) is –9.5°. Because GCN4 peptides were also used to mimic IL-4, Ω was measured at 24.3°, which is intermediate between the crossing angles of helices A and C IL-4 free (0.7°) and IL-4 bound (–3.3°). The DHP-14-AB helices are farther from the target of IL-4 bound to IL-4Rα than the GCN4 peptides model.
Discussion
At 108 aa in length, the proteins we created remain the largest stable and functional de novo-designed proteins. Our goal was to incorporate IL-4Rα receptor binding function into a stable precursor design, thereby creating a template for antagonist development based on its crystal structure and an analysis of the published data on IL-4·IL-4Rα. The proteins were characterized for stability in solution and competition with IL-4 for IL-4Rα. The ambiguity in the topological handedness of our precursor DHP-1 has been resolved, and two generations of design based on incorporation of positive elements and avoidance of negative elements of the interaction led to an antagonist of Ki ≈ IC50 = 27 μM.
Domingues et al. (32) modified a GCN4 leucine zipper, a parallel helix dimer, to design a set of peptides that bind to IL-4Rα. Their results suggested that a stable structure, which preserves the spatial geometric relationship of the binding site of IL-4, was required to reconstitute binding function. The IL-4 mimetic peptides required modification to include stabilizing disulfide bonds between helices (32). They achieved affinities in the range between 2 mM and 5 μM even though GCN4 helices are parallel rather than anti-parallel like IL-4. The affinities were measured directly by surface plasmon resonance rather than by competition binding for IL-4Rα. By comparison, our platform DHP-1 derives its stability from a well packed hydrophobic core whereas GCN4 peptides were disulfide cross-linked. We focused primarily on generating IL-4Rα binding activity within our stable four-helix bundle protein and observed an increased affinity along with decreased stability.
Competitive binding activity toward IL-4Rα was the functional assay used to resolve the ambiguity in the previous protein design, DHP-1 (Table 1). DHP-10-AB, the correct topology as seen in the DHP-14-AB structure, had higher affinity for IL-4Rα than DHP-10-AD by a factor of two. This result suggested that the right-handed topology model, DHP-10-AB, merged the binding surface between the first and second helices to form a productive binding site (Fig. 2B). Based on the right-handed model, the DHP-10-AD interaction surface on the first and last helices must diverge and the surface displays only half the binding site for IL-4Rα (Fig. 2D). This result of a relatively weaker IC50 at this stage is consistent with DHP-10-AD being unstable in solution and also at best only presenting half the binding site on one helix rather than conjugating it on two neighboring helices.
Measurement of stability of DHP-10-AB and DHP-10-AD also separated models in the design scheme for helical bundle topology. The design aimed at the right-handed configuration DHP-10-AB was (at 13.5 kcal/mol) as stable as its parent DHP-1 but much more stable than DHP-10-AD (at 4.1 kcal/mol). DHP-10-AB is a homogeneous monomer in solution whereas DHP-10-AD was relatively unstable and had a heterogeneous aggregation state. This result suggested that the binding site disrupted the DHP-10-AD protein core design and the knobs-into-holes packing of hydrophobic side-chains between the helices.
Based on modeling with the complex coordinates available, two additional areas were targeted for second-generation mutations to remove what were perceived as clashes with IL-4Rα (Fig. 6). Larger side-chains were replaced with IL-4-mimetic residues within the interface. The mimic of IL-4 R53 was moved closer to the middle of the putative IL-4Rα binding site to create a smaller interaction surface. These mutations applied to DHP-10-AB grafted most of the binding site for IL-4Rα and improved the affinity for IL-4Rα to an IC50 of 27 μM, which was surprisingly below the IC50 for IL-4 in the assay at 1 nM.
Because DHP-1 helices overlapped the IL-4Rα binding helices on free IL-4 within a 1-Å rms deviation, we thought it would be straightforward to create a reasonably high-affinity antagonist for IL-4. However, calculation of the crossing angle between helices highlighted another feature of the IL-4 interaction with IL-4Rα. DHP-1 helices A and B had a measured inter-helix crossing angle that was nearly 3° smaller than for free IL-4. If the proteins are considered rigid bodies, this 3° crossing angle difference meant that some residues would be in nearly perfect overlap whereas others would be significantly displaced from the binding site. Upon binding, changes in crossing angle between the IL-4 binding site helices (A and C) increased by 4° (Table 3), yet IL-4 A and C helices were observed to shift by ≈0.5 Å upon binding. DHP-14-AB had a crossing angle between binding site helices (A and B) of 18.1°, which is 5.5° smaller than free IL-4 and 9.5° smaller than bound IL-4. The DHP-14-AB helices have short glycine linkers between helices and symmetric knobs-into-holes packing interactions due to alternating leucine and alanine residues. DHP-14-AB may be structurally restricted by inter-helix packing that prevented the helices from shifting to accommodate binding to IL-4Rα. In contrast, the other peptide mimic of IL-4 based on GCN4 helices had a crossing angle of 24.3°, which is a closer match to either free or bound IL-4 than DHP-14-AB (Table 3). Thus, DHP-14-AB may not bind IL-4Rα like IL-4 because the helices are not interacting with one another as they do in IL-4 and it may not be able to adapt to the binding site movements required for the high-affinity interaction.
The extraordinarily high IL-4 affinity for IL-4Rα seemed to be a result of “electrostatic steering” by highly charged surfaces as well as coordinated movements between helices presenting the charged interface (Fig. 5). In addition to increased crossing angles between IL-4 helices A and C, the distribution of the charge on IL-4 shifts from a cluster to a specific arrangement coordinated by hydrogen bonds to IL-4Rα. Even though we designed the same number of charges into the IL-4Rα binding interface, more atomic precision is required to match the IL-4 affinity than what we designed into DHP-14-AB.
Fig. 5.

Molecular surfaces for IL-4 free and bound to IL-4Rα and DHP-14-AB superimposed with electrostatic potential. The rearrangement of charge on IL-4 upon binding IL-4Rα may be broader than what DHP-14-AB could access on its smaller binding interface “footprint.” Electrostatic surfaces were calculated with grasp (37) with a blue-to-red color scale between –15 and +15 J and illustrated with pymol (33).
De novo design principles were applied to mimic the presentation of the functional binding site on IL-4. By designing two enantiomers and testing their stability and function, we resolved the ambiguity in the four-helix design template topology as right-handed and achieved an IC50 of 495 μM for IL-4Rα. A subsequent design was based on the later structure of the IL-4·IL-4Rα complex analysis to remove potentially detrimental interactions that could not be foreseen when using the structure of IL-4 alone. The resulting designed protein, DHP-14-AB (the largest designed protein to date), is well ordered, yields an instructive crystal structure, and has an IC50 of 27 μM. The apparent affinity of DHP-AB-14 is ≈104 lower than IL-4, which may be due to transplantation of only a subset of IL-4 binding residues as well as the geometry of the helices interacting with IL-4Rα. De novo protein design of function requires atomic precision in the binding site as well as incorporating the entire structure. We observed a difference in crossing angle between DHP-14-AB and IL-4 free and bound of 5.5° to 9.5° for the binding site helices, respectively. Accounting for secondary structure interactions and dynamics between those elements as measured in the differences in crossing angle may increase the precision of and ability to optimize protein–protein interactions.
Supplementary Material
Acknowledgments
We thank Julian Chen, Andrew Shiau, Janet Finer-Moore, and Robert Keenan for feedback on the manuscript. We thank the Sandler Family Foundation for Asthma Research at the University of California at San Francisco (to R.M.S.) and the American Cancer Society (to S.L.L.) for funding this project.
Author contributions: S.L.L. designed research; S.L.L., B.C.C., and L.J.M. performed research; S.L.L., C.M.F., and D.A. contributed new reagents/analytic tools; S.L.L., B.C.C., L.J.M., and R.M.S. analyzed data; and S.L.L. wrote the paper.
Abbreviations: IL-4Rα, IL-4 receptor α chain; IL-13Rα1, IL-13 receptor α chain 1; γC, γ common chain; DHP, designed helical protein; MBP, maltose-binding protein; Rh, hydrodynamic radius; ΔΔG, relative change in binding energy.
Data deposition: The atomic coordinates and structure factors for DHP-AB-14-MBP have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1Y4C).
References
- 1.Grunig, G., Warnock, M., Wakil, A. E., Venkayya, R., Brombacher, F., Rennick, D. M., Sheppard, D., Mohrs, M., Donaldson, D. D., Locksley, R. M. & Corry, D. B. (1998) Science 282, 2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corry, D. B. & Kheradmand, F. (1999) Nature 402, B18–B23. [DOI] [PubMed] [Google Scholar]
- 3.Hage, T., Sebald, W. & Reinemer, P. (1999) Cell 97, 271–281. [DOI] [PubMed] [Google Scholar]
- 4.Nicola, N. A. (1989) Annu. Rev. Biochem. 58, 45–77. [DOI] [PubMed] [Google Scholar]
- 5.Nicola, N. A. & Hilton, D.J. (1998) Adv. Protein Chem. 52, 1–65. [DOI] [PubMed] [Google Scholar]
- 6.Powers, R., Garrett, D. S., March, C. J., Frieden, E. A., Gronenborn, A. M. & Clore, G. M. (1992) Science 256, 1673–1677. [DOI] [PubMed] [Google Scholar]
- 7.Wlodaver, A., Pavlovsky, A. & Gustchina, A. (1992) FEBS Lett. 309, 59–64. [DOI] [PubMed] [Google Scholar]
- 8.Walter, M. R., Cook, W. J., Zhao, B. G., Cameron, R. P., Jr., Ealick, S. E., Walter, R. L., Jr., Reichert, P., Nagabhushan, T. L., Trotta, P. P. & Bugg, C. E. (1992) J. Biol. Chem. 267, 20371–20376. [DOI] [PubMed] [Google Scholar]
- 9.Shen, B. J., Hage, T. & Sebald, W. (1996) Eur. J. Biochem. 240, 252–261. [DOI] [PubMed] [Google Scholar]
- 10.Bazan, J. F. (1990) Proc. Natl. Acad. Sci. USA 87, 6934–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunningham, B. C. & Wells, J. A. (1993) J. Mol. Biol. 234, 554–563. [DOI] [PubMed] [Google Scholar]
- 12.Schafmeister, C. E., LaPorte, S. L., Miercke, L. J. & Stroud, R. M. (1997) Nat. Struct. Biol. 4, 1039–1046. [DOI] [PubMed] [Google Scholar]
- 13.Presnell, S. R. & Cohen, F. E. (1989) Proc. Natl. Acad. Sci. USA 86, 6592–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harbury, P. B., Plecs, J. J., Tidor, B., Alber, T. & Kim, P. S. (1998) Science 282, 1462–1467. [DOI] [PubMed] [Google Scholar]
- 15.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 16.Mueller, K., Amman, H. K., Doran, D. M., Gerber, P. R., Gubernator, K. & Schrepfer, G. (1988) Bull. Soc. Chim. Belg. 97, 655–667. [Google Scholar]
- 17.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 18.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G. & Gray, T. (1995) Protein Sci. 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton, T. E. (1989) in Practical Approach Series (Oxford Univ. Press, Oxford).
- 20.Peters, R. J., Shiau, A. K., Sohl, J. L., Anderson, D. E., Tang, G., Silen, J. L. & Agard, D. A. (1998) Biochemistry 37, 12058–12067. [DOI] [PubMed] [Google Scholar]
- 21.Holton, J. & Alber, T. (2004) Proc. Natl. Acad. Sci. USA 101, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie, A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography, No. 26.
- 23.Collaborative Computational Project No. 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763.15299374 [Google Scholar]
- 24.Kissinger, C. R., Gehlhaar, D. K. & Fogel, D. B. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 484–491. [DOI] [PubMed] [Google Scholar]
- 25.Morris, R. J., Perrakis, A. & Lamzin, V. S. (2003) Methods Enzymol. 374, 229–244. [DOI] [PubMed] [Google Scholar]
- 26.Letzelter, F., Wang, Y. & Sebald, W. (1998) Eur. J. Biochem. 257, 11–20. [DOI] [PubMed] [Google Scholar]
- 27.Walther, D. (1997) Trends Biochem. Sci. 22, 274–275. [DOI] [PubMed] [Google Scholar]
- 28.Wang, Y., Shen, B. J. & Sebald, W. (1997) Proc. Natl. Acad. Sci. USA 94, 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schafmeister, C. E., Miercke, L. J. & Stroud, R. M. (1993) Science 262, 734–738. [DOI] [PubMed] [Google Scholar]
- 30.Chothia, C., Levitt, M. & Richardson, D. (1981) J. Mol. Biol. 145, 215–250. [DOI] [PubMed] [Google Scholar]
- 31.Crick, F. H. C. (1953) Acta Crystallogr. 6, 689–697. [Google Scholar]
- 32.Domingues, H., Cregut, D., Sebald, W., Oschkinat, H. & Serrano, L. (1999) Nat. Struct. Biol. 6, 652–656. [DOI] [PubMed] [Google Scholar]
- 33.DeLano, W. L. (2002) The PyMOL Molecular Graphics System (DeLano Scientific, San Carlos, CA).
- 34.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946–950. [Google Scholar]
- 35.Esnouf, R. M. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 938–940. [DOI] [PubMed] [Google Scholar]
- 36.Merritt, E. A. & Bacon, D. J. (1997) Methods Enzymol. 277, 505–524. [DOI] [PubMed] [Google Scholar]
- 37.Petrey, D. & Honig, B. (2003) Methods Enzymol. 374, 492–509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}