

Abstract
Synthetic prions were produced in our laboratory by using recombinant mouse prion protein (MoPrP) composed of residues 89-230. The first mouse synthetic prion strain (MoSP1) was inoculated into transgenic (Tg) 9949 mice expressing N-terminally truncated MoPrP(Δ23-88) and WT FVB mice expressing full-length MoPrP. On first and second passage in Tg9949 mice, MoSP1 prions caused disease in 516 ± 27 and 258 ± 25 days, respectively; numerous, large vacuoles were found in the brainstem and gray matter of the cerebellum. MoSP1 prions passaged in Tg9949 mice were inoculated into FVB mice; on first and second passage, the FVB mice exhibited incubation times of 154 ± 4 and 130 ± 3 days, respectively. In FVB mice, vacuolation was less intense but more widely distributed, with numerous lesions in the hippocampus and cerebellar white matter. This constellation of widespread neuropatho-logic changes was similar to that found in FVB mice inoculated with Rocky Mountain Laboratory (RML) prions, a strain derived from a sheep with scrapie. Conformational stability studies showed that the half-maximal GdnHCl (Gdn1/2) concentration for denaturation of MoSP1 prions passaged in Tg9949 mice was ≈4.2 M; passage in FVB mice reduced the Gdn1/2 value to ≈1.7 M. RML prions passaged in either Tg9949 or FVB mice exhibited Gdn1/2 values of ≈1.8 M. The incubation times, neuropathological lesion profiles, and Gdn1/2 values indicate that MoSP1 prions differ from RML and many other prion strains derived from sheep with scrapie and cattle with bovine spongiform encephalopathy.
Keywords: neurodegeneration
Prions consist solely of a disease-causing isoform of prion protein (PrPSc), a β-sheet-rich conformer of cellular PrP. Accumulation of PrPSc in the central nervous system of animals and humans leads to fatal maladies collectively known as prion diseases (1). Examples of prion diseases are Creutzfeldt-Jakob disease in humans, bovine spongiform encephalopathy in cattle, scrapie in sheep, and chronic wasting disease in cervids (1).
Limited protease digestion of PrPSc produces PrP 27-30 that retains prion infectivity and polymerizes into rod-shaped structures with the ultrastructural and tinctorial properties of amyloid (2, 3). Moreover, a synthetic prion consisting of 55 residues of PrP with the analogous mutation (P101L) causing Gerstmann-Sträussler-Scheinker disease in humans produced prion disease in transgenic (Tg) 196 mice expressing mouse PrP (MoPrP, P101L). In the brains of these mice, there was a profound deposition of PrP amyloid as well as protease-sensitive PrPSc(P101L) (4). On this background, we produced recombinant (rec) MoPrP composed of residues 89-230 expressed in Escherichia coli and folded the purified protein into a β-sheetrich state that polymerized into amyloid fibrils (5).
After producing both seeded and unseeded amyloid fibrils composed of recMoPrP(89-230) (6), we inoculated Tg mice expressing MoPrP(Δ23-88) (7). These Tg(MoPrP,Δ23-88)9949/Prnp0/0 mice, referred to as Tg9949 mice, developed neurologic disease between 382 and 660 days after inoculation. The seeded amyloid-inoculated mice showed slightly shorter incubation times compared with those inoculated with unseeded amyloid (5).
The shortest incubation time for a Tg9949 mouse inoculated with seeded amyloid was 382 days. On second passage as described here, the mean incubation time in Tg9949 mice was 258 days. Prions in the brains of Tg9949 mice also were inoculated into FVB and Tg4053 mice expressing full-length MoPrP, with incubation times of 154 and 90 days, respectively (5). By Western blot analysis, this strain of mouse synthetic prion1 (MoSP1) exhibited protease-resistant PrPSc.
Because many strains of prions are resistant to limited proteolysis, we asked whether the neuropathologic profiles of Tg9949 mice inoculated with MoSP1 remained distinct from those inoculated with the Rocky Mountain Laboratory (RML) strain derived from sheep with scrapie. On both first and second passage, MoSP1 produced vacuolation of the neuropil in the brainstem and cerebellum, and intense PrP immunostaining along the rims of large vacuoles. Such PrP deposits have been found in the brains of patients with Creutzfeldt-Jakob disease but not in WT mice inoculated with a variety of prion strains.
Using resistance to limited protease digestion as a marker for nondenatured PrPSc, we examined the conformational stabilities of PrPSc in the brains of mice inoculated with MoSP1. The half-maximal GdnHCl (Gdn1/2) concentration for denaturation of MoSP1 passaged in Tg9949 mice was ≈4.2 M compared with RML prions in Tg9949 mice, for which the Gdn1/2 value was ≈1.8 M (Table 1, which is published as supporting information on the PNAS web site). Passage of MoSP1 in FVB mice reduced the Gdn1/2 value to ≈1.7 M. The conformational stability of MoSP1 passaged in Tg9949 mice differed substantially from that of more than a dozen other strains of prions derived from sheep with scrapie and cattle with bovine spongiform encephalopathy.
Materials and Methods
Mice. Tg(MoPrP,Δ23-88)9949/Prnp0/0 mice used in this study have been described (7). FVB mice were obtained from Charles River Laboratories.
RML Prions and Bioassays for Prion Infectivity. The full-length murine RML prion strain propagated in Swiss mice was originally provided by W. Hadlow (Rocky Mountain Laboratory, Hamilton, MT) and was passaged in Swiss CD-1 mice obtained from Charles River Laboratories (8). Brain homogenates were diluted 1:10 into sterile, calcium- and magnesium-free PBS plus 5 mg/ml BSA. Weanling mice were inoculated intracerebrally with 30 μl of diluted samples. Inoculations were carried out with a 26-gauge, disposable hypodermic needle inserted into the right parietal lobe. After inoculation, mice were examined daily for neurologic dysfunction. Standard diagnostic criteria were used to identify animals affected by prion disease (9, 10). Animals whose deaths were imminent were killed, and their brains were removed for histological and biochemical analysis.
Preparation of Brain Homogenates. Crude brain homogenates [10% (wt/vol)] in calcium- and magnesium-free PBS were prepared by repeated extrusion through syringe needles of successively smaller size as described (11). Nuclei and debris were removed by centrifugation at 1,000 × g for 5 min. The brain homogenates prepared from diseased mice were used for serial passage and for studies designed to characterize PrPSc.
Neuropathology. Brains were removed rapidly at the time of death, immersion-fixed in 10% buffered formalin, and embedded in paraffin. Eight-micrometer-thick sections were stained with hematoxylin and eosin for evaluation of prion disease. Peroxidase immunohistochemistry with antibodies to glial fibrillary acidic protein was used to evaluate the degree of reactive astrocytic gliosis. Hydrolytic autoclaving was performed as described (12), using the rec human-mouse (HuM)-R2 Fab to detect PrPSc. Vacuolation scores, which are semiquantitative estimates of an area of a brain region occupied by vacuoles (13), were determined by a single rater (S.J.D.).
HuM Chimera Fab Preparation. Sequences obtained from a phage display library (14) were used for the HuM-D18 and HuM-R2 clones. Preparation of rec HuM-D18 and HuM-R2 Fabs was completed as described (15).
PrPSc Detection by Western Immunoblotting. PrPSc was measured by limited proteinase K (PK) digestion and immunoblotting as described (16). Limited protease digestion was performed with 20 μg/ml PK (Invitrogen) for 60 min at 37°C. Digestions were terminated by the addition of 8 μl of 0.5 M PMSF in absolute ethanol (Roche, Indianapolis). Digested samples then were mixed with equal volumes of 2× SDS sample buffer. All samples were boiled for 5 min before electrophoresis. SDS/PAGE analyses were performed on 1.5-mm 12% polyacrylamide gels (17). After electrophoresis, Western blotting was performed as described (11). Membranes were blocked with 5% nonfat milk protein in PBST (calcium- and magnesium-free PBS plus 0.1% Tween 20) for 1 h at room temperature. Blocked membranes were incubated with 1 μg/ml recHuM-D18. After incubation with the primary Fab, membranes were washed three times for 10 min in PBST, incubated with horseradish peroxidase-labeled, anti-human Fab secondary antibody (ICN) diluted 1:5,000 in PBST for 45 min at room temperature, and washed again, four times for 10 min in PBST. After enhanced chemiluminescence (ECL) detection (Amersham Pharmacia Bioscience) for 1-5 min, blots were sealed in plastic covers and exposed to ECL Hypermax film (Amersham Pharmacia). Films were processed automatically in a Konica (Tokyo) film processor.
Conformational Stability Assay. To 50 μl of a 10% brain homogenate, 50 μl of a GdnHCl mix, using a range of concentrations from 0 to 8 M GdnHCl, was added. After dilution, samples were equilibrated at 22°C for 2 h. Using these dilutions up to a final concentration of 4 M GdnHCl could be tested; for higher concentrations of GdnHCl, smaller amounts of brain homogenates were used.
At the end of the denaturation period, samples were diluted by addition of 850 μl of Tris buffer composed of 10 mM Tris·HCl (pH 8.0), 150 mM NaCl, 0.5% Nonidet P-40, and 0.5% deoxycholate. Then, an additional 50 μl of GdnHCl to 0.4 M was added to each sample. Samples were digested with 20 μg/ml PK at 37°C for 60 min. Reactions were terminated by the addition of PMSF (final concentration 1 mM) and subsequently centrifuged in a Beckman Tabletop Ultracentrifuge for 1 h at 103,219 × g. From each sample, the supernatant was decanted and the pellet was solubilized in 100 μl of Tris buffer. SDS/PAGE and Western blot analyses were carried out as described above.
Densitometric Analysis of Western Blots. Densitometric analysis was performed by using a Kodak Digital Science Image Station 440CF equipped with Kodak 1d image analysis software.
Results
Transmission of MoSP1 to Tg9949 Mice. In the initial isolation of MoSP1, Tg9949 mice were inoculated with amyloid produced from recMoPrP(89-230) (5). The mean incubation time was 516 ± 27 days, and the first mouse became ill at 382 days. When the brain extract prepared from this ill Tg9949 mouse was passaged into additional Tg9949 mice, the mean incubation time was 258 ± 25 days (Fig. 1A). When the same brain extract prepared from the ill Tg9949 mouse was passaged into FVB mice, the mean incubation time was 154 ± 4 days. On second passage in FVB mice, the incubation time for MoSP1 decreased to 130 ± 3 days (Fig. 1B).
Fig. 1.

Survival times of mice inoculated with synthetic prions. (A and B) First (○) and second (▪) passage of MoSP1 in Tg9949 (A) and FVB (B) mice. Mice were inoculated intracerebrally with 30 μl of a 1% (wt/vol) crude brain extract. (C) First (○) and second (▪) passage of RML prions in Tg9949 mice and a typical passage (shaded triangles) in FVB mice.
Neuropathology of Tg9949 Mice Inoculated with Prions. Transmission of MoSP1 to Tg9949 mice was characterized by formation of large vacuoles in the cerebellar granule cell layer (Fig. 2E), tegmentum of the pons, selected nuclei of the midbrain, and septal nuclei. They were conspicuously absent from the hippocampus (Fig. 2 A), neocortex, thalamus, hypothalamus, and caudate nucleus (Fig. 3A). These large vacuoles appeared in well delineated clusters. In the cerebellar granule cell layer, clusters of large vacuoles were confined to relatively small areas and did not occupy >10-20% of the total granule cell area in a tissue section. This pattern also was observed in the midbrain, where vacuoles were confined to structures such as the red nucleus and lateral geniculate nucleus. Virtually all of the vacuoles were large and their interior walls were often lined with PrPSc (Fig. 2I). Between the vacuoles, immunohistochemistry revealed small deposits of PrPSc measuring ≈10 μm in diameter and finer punctate deposits of 3-5 μm in diameter.
Fig. 2.

Neuropathology of Tg9949 mice inoculated with MoSP1 (A, B, E, F, I, and J) or RML (C, D, G, H, K, and L) prions. First (A, E, and I) and second (B, F, and J) serial transmissions of MoSP1 are shown. First (C, G, and K) and second (D, H, and M) serial transmissions of RML prions are shown. Two brain regions stained by the hematoxylin/eosin method are shown: hippocampal CA1 region (A-D) and cerebellum (E-H). No significant neuropathologic changes occur in the hippocampus after inoculation with MoSP1 (A and B), whereas small and large vacuoles as well as varying degrees of pyramidal neuron loss occur after RML inoculation (C and D). (E and F) Well delineated clusters of large vacuoles occur in the granule cell layer of the cerebellar cortex. (G and H) Few or no vacuoles appear in the granule cell layer; in contrast, both large and small vacuoles formed in the white matter. (I-M) Immunoperoxidase staining for PrPSc.(I and J) PrPSc is found mostly rimming the interior of large vacuoles. In addition, small numbers of PrPSc deposits measuring ≈10 and 3-5 μm are found in the neuropil of the cerebellar granule cell layer. (K-M) Large vacuoles and a few small vacuoles lined by PrPSc are found. Densely packed, finely granular PrPSc deposits fill the gray matter neuropil as shown for the hippocampal CA1 region (K and M). (L) Small numbers of punctate PrPSc deposits appear in the cerebellar white matter while more densely packed punctate deposits occur in the granule cell and molecular layers of the cerebellar cortex. m, molecular layer of the cerebellar cortex; p, pyramidal cell layer of the hippocampal CA1 region; wm, white matter of cerebellum. (Bar in H represents 100 μm and also applies to A-G; bar in L represents 60 μm and also applies to I-K and M.)
Fig. 3.
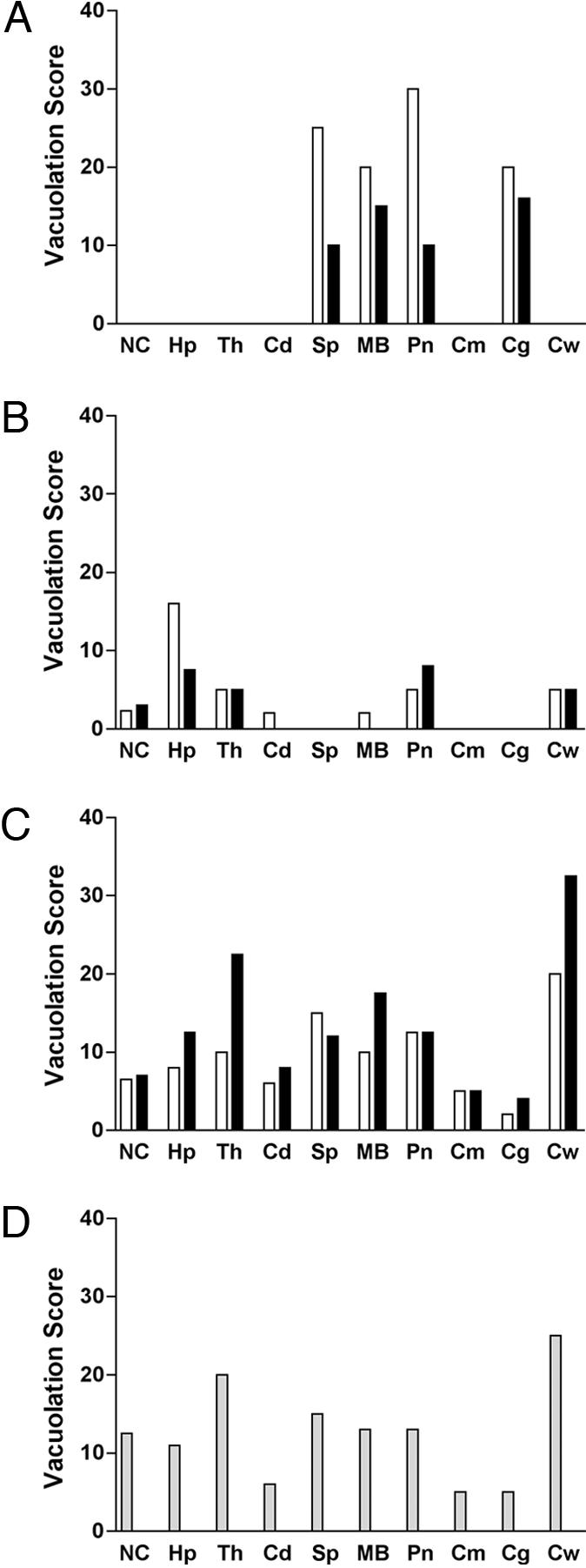
Vacuolation profiles in the brains of Tg9949 (A and B) and FVB (C and D) mice inoculated with MoSP1 or RML prions. Vacuolation scores of first (open bars) and second (filled bars) passages of MoSP1 (A and C) and RML prions (B and D). Vacuolation scores are semiquantitative estimates of the area of a brain region occupied by vacuoles. (A) The scores of 20 and 15 for the first and second transmissions, respectively, represent the area occupied by vacuoles locally in the cluster of vacuoles not for the entire granule cell layer. During the second passage of MoSP1 in Tg9949 mice, vacuoles were distributed to the same brain regions in the two Tg9949 mice that were examined. (C) MoSP1 serially transmitted in FVB mice show a distribution of vacuolation different from that in Tg9949 mice (A). (D) RML prions passaged in FVB mice. Because RML has been passaged numerous times in FVB and CD1 mice, both of which are WT mice expressing cellular PrP-A, the bars are shaded gray. NC, neocortex; Hp, hippocampus; Th, thalamus; Cd, head of caudate nucleus; Sp, septal nuclei; MB, midbrain nuclei; Pn, pontine tegmentum; Cm, cerebellar molecular layer; Cg, cerebellar granule cell layer; Cw, cerebellar white matter.
Second passage of MoSP1 in Tg9949 mice produced similar clusters of large vacuoles in the granule cell layer of the cerebellar cortex (Figs. 2 B and F). Serial sections, 80 μm apart, through the full thickness of the brain blocks showed that the clusters of vacuoles are ≈100 μm in diameter and are distributed to the same brain regions as from the first passage in Tg9949 mice (Fig. 3A). Immunohistochemistry also showed PrPSc lining the interior walls of the vacuoles (Fig. 2J).
In contrast, the neuropathologic phenotype of RML prions during both the first and second passages in Tg9949 mice was substantially different from that of MoSP1 in the same mouse line. First, smaller vacuoles outnumbered larger vacuoles in all brain regions (Fig. 2 C and D). Second, vacuolation occurred in the cerebellar white matter (Fig. 2 G, H, and L) with RML prions, whereas none was found with MoSP1 (Fig. 3A). Vacuolation of the cerebellar white matter was associated with punctate deposits of PrPSc (Fig. 2L). Third, vacuolation and PrPSc deposition were widely distributed in cerebral hemispheres, including the neocortex, hippocampus, and thalamus (Fig. 3B). Fourth, vacuoles and PrPSc deposition resulting from RML infection were diffusely distributed within a brain region, in contrast to their focal occurrence with MoSP1. Finally, dense, finely granular PrPSc deposits filled the gray matter neuropil between larger vacuoles that were lined with PrPSc (Fig. 2 K-M).
Notably, the deposition of PrPSc along the rims of vacuoles in Tg9949 mice inoculated with MoSP1 or RML prions has not been seen in any other rodents with experimental prion disease. However, such vacuoles lined with PrPSc have been seen in sporadic Creutzfeldt-Jakob disease patients (18).
MoSP1 Passaged in FVB Mice. Inoculation of MoSP1 from first passage in Tg9949 mice into FVB mice had a profound effect on the neuropathologic phenotype, which was maintained on second passage to FVB mice (Fig. 3C). Small vacuoles and finely granular PrPSc deposits were observed in most regions of the CNS compared with their restricted locations in Tg9949 mice (Fig. 3A). Remarkably, the vacuolation score histogram (area of a brain region occupied by vacuoles) of MoSP1 in FVB mice was indistinguishable from that of RML prions continuously passaged in FVB mice (Fig. 3D).
Conversely, RML prions passaged from FVB mice expressing full-length MoPrP into Tg9949 mice expressing MoPrP(Δ22-88) showed a recognizable change in the neuropathologic phenotype (compare Fig. 3 B and D). In Tg9949 mice, vacuolation scores for RML prions were markedly decreased in all brain regions compared with those seen from passage of RML prions in FVB mice. In addition, some large vacuoles lined by PrPSc were formed in Tg9949 mice inoculated with RML prions, which are not present in FVB mice inoculated with RML prions.
Western Blots of PrPSc Comprising MoSP1. To examine the molecular characteristics of PrPSc in the brains of Tg9949 mice, Western immunoblotting was performed. Brain extracts were either examined directly or subjected to limited digestion catalyzed by PK (20 μg/ml) at 37°C for 60 min. After limited digestion of the crude brain extracts, three PrP bands were evident (Fig. 4). The upper band of ≈30 kDa is the diglycosylated form of PrP 27-30, the middle band is the monoglycosylated form, and the lower band of ≈21 kDa is the unglycosylated form. The protease-resistant unglycosylated band in the Tg9949 mouse brain inoculated with MoSP1 is a doublet (Fig. 4A, lane 4); this finding contrasts with the single protease-resistant, unglycosylated band in the Tg9949 mouse brain inoculated with RML prions (Fig. 4A, lane 2). The molecular basis of this doublet remains to be determined. Additionally, the origin of fastest-migrating PrP band in the undigested samples of Tg9949 brain extracts is unclear (Fig. 4A, lanes 1 and 3).
Fig. 4.

Western immunoblotting of PrPSc in the brains of Tg9949 mice (A) and FVB mice (B) inoculated with RML prions (lanes 1 and 2) and MoSP1 (lanes 3 and 4). In lanes 2 and 4, samples were digested with 20 μg/ml PK for 60 min at 37°C. Apparent molecular masses based on migration of protein standards are indicated in kDa.
The profiles of the protease-resistant glycoforms in the brains of FVB mice inoculated with either MoSP1 or RML prions were indistinguishable and showed the expected shift in molecular weight caused by the digestion of ≈70 residues from the N terminus (Fig. 4B, lanes 2 and 4).
Conformational Stability Studies. To analyze the strain-specific properties of MoSP1 in the brains of Tg9949 mice, we measured the conformational stability of PrPSc. Aliquots of Tg9949 mouse brain extract were exposed to increasing concentrations of GdnHCl, followed by PK digestion and Western immunoblotting of PrP 27-30 (the protease-resistant fragment of PrPSc) using the rec HuM-D18 Fab (15). Unexpectedly, PrPSc in brain homogenates prepared from Tg9949 mice inoculated with MoSP1 resisted denaturation at concentrations of GdnHCl up to 4 M (Fig. 5A). Densitometric scans of Western blots showed that the Gdn1/2 for denaturation of MoSP1 is ≈4.2 M (Fig. 5C).
Fig. 5.
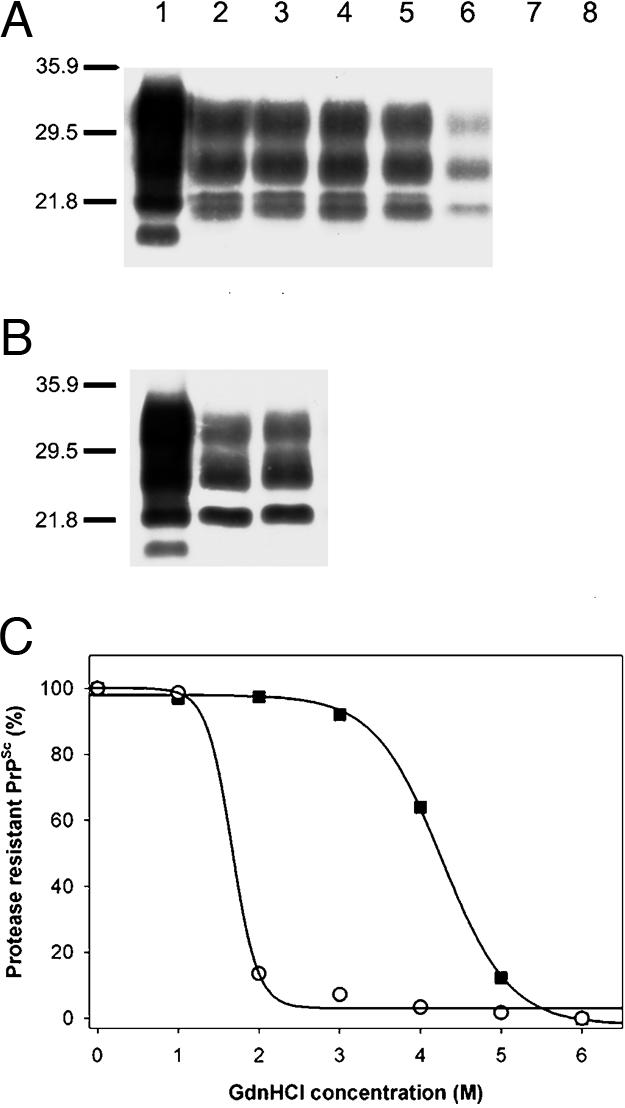
Conformational stability studies of MoSP1 and RML prions passaged in Tg9949 mice. Western blots of the denaturation transition of PrPSc in a brain extract of a Tg9949 mouse inoculated with MoSP1 (A) or RML prions (B) are shown. Ten microliters of undigested (lane 1) or PK-digested samples (lanes 2-8) was immunoblotted with the HuM-D18 Fab. In lanes 3-8, GdnHCl was added at concentrations of 1, 2, 3, 4, 5, and 6 M, respectively, followed by limited PK digestion. In lanes 1 and 2, no GdnHCl was added. Apparent molecular masses based on migration of protein standards are indicated in kDa. (C) Denaturation curves obtained by scanning of the PK-resistant PrPSc signals in Western blots are shown. ▪, MoSP1; ○, RML prions.
Similar experiments were performed by using aliquots of brain extracts prepared from Tg9949 mice inoculated with RML prions. In contrast to the samples from Tg9949 mice inoculated with MoSP1, PrPSc in these extracts was readily denatured by exposure to 2 M GdnHCl (Fig. 5 B and C). The Gdn1/2 value for RML prions passaged in Tg9949 mice is ≈1.8 M (Table 1).
We next examined the conformational stability of MoSP1 passaged in FVB mice to determine whether its conformational stability changed upon passage into FVB mice (Fig. 1B). The passage of MoSP1 to FVB mice resulted in an altered neuropathology that is indistinguishable from the lesions found in FVB mice inoculated with RML prions (Fig. 3). Aliquots of FVB mouse brain extract were exposed to increasing concentrations of GdnHCl, followed by PK digestion and Western immunoblotting of PrP 27-30 using the HuM-D18 Fab. PrPSc in brain homogenates of FVB mice inoculated with MoSP1 resisted denaturation at concentrations of GdnHCl up to 2.2 M (Fig. 6A). Densitometric scans of Western blots showed that the Gdn1/2 for MoSP1 passaged in FVB mice is ≈1.7 M (Fig. 6C).
Fig. 6.

Conformational stability studies of MoSP1 and RML prions passaged in FVB mice. Western blot of the denaturation transition of PrPSc in a brain extract of a FVB mouse inoculated with MoSP1 (A) or RML prions (B) is shown. Ten-microliter samples were immunoblotted with the HuM-D18 Fab. Lane 1, undigested, nondenatured sample; lane 2, sample not exposed to GdnHCl but subjected to limited PK digestion; lanes 3-11, GdnHCl was added at concentrations of 1.0, 1.2, 1.5, 1.7, 2.0, 2.2, 2.5, 2.7, and 3.0 M, respectively, followed by limited PK digestion. Apparent molecular masses based on migration of protein standards are indicated in kDa. (C) Denaturation curves obtained by scanning of the PK-resistant PrPSc signals in Western blots are shown. ▪, MoSP1; ○, RML prions.
Similar experiments were performed by using aliquots of brain extracts prepared from FVB mice inoculated with RML prions. Like MoSP1, PrPSc in these extracts was readily denatured by exposure to 2.2 M GdnHCl (Fig. 6 B and C). The Gdn1/2 value for RML prions passaged in FVB mice is ≈1.8 M, almost identical to that recorded for MoSP1 passaged in FVB mice.
Discussion
In addition to establishing beyond a reasonable doubt that prions are infectious proteins, the production of synthetic prions opens many approaches to the investigation of prion structure and biology. Before the production of WT synthetic prions using the 142-residue protein recMoPrP(89-230) (5), we used a 55-mer synthetic peptide carrying the analogous Gerstmann-Sträussler-Scheinker mutation at mouse residue 101 to create synthetic prions (4, 19). The strains of mutant synthetic prions were protease-sensitive and required Tg196 mice expressing low levels of MoPrP(P101L) to demonstrate prion infectivity. Moreover, the mutant prions in Tg196 mice could not be passaged into WT mice. Development of a system using recMoPrP(89-230) to produce synthetic prions has facilitated investigations because these prions are protease-resistant and can be passaged in WT mice.
Evidence that MoSP1 Is a Distinct Strain. The data presented here and in an earlier study (5) argue that MoSP1 is a distinct strain. The neuropathologic changes produced by MoSP1 in Tg9949 mice differ from all other mouse prion strains that we have previously investigated. Most noticeable are focal aggregates of large vacuoles in the cerebellar gray matter and brainstem (Figs. 2 and 3). This finding contrasts with Tg9949 mice inoculated with RML prions, in which vacuolation is widespread, including in the neocortex, hippocampus, thalamus, brainstem, and cerebellar white matter.
In our studies of the conformational stability of more than a dozen prion strains, we have not found any strain that is as stable as MoSP1 (Table 1). MoSP1 is more resistant to denaturation by GdnHCl (Fig. 5) than bovine spongiform encephalopathy prions in cattle (20) as well as the mouse 301V strain that originated in cattle (Table 1). In heat denaturation studies, 301V was more stable than four other mouse prion strains, all of which were derived from sheep with scrapie (21).
The incubation times, neuropathologic lesion profiles, and resistance to GdnHCl denaturation contends that MoSP1 is unlike any that we have previously studied. This finding is fortuitous because it provides yet another line of evidence supporting our earlier assertion that MoSP1 was produced from recMoPrP(89-230) polymerized into amyloid fibrils and not a contaminant derived from a naturally occurring prion (5).
Correlating Incubation Times, Neuropathology, and Stability. The mean incubation time for MoSP1 was reduced from 516 days to 258 days on second passage, presumably reflecting the higher titer of prions in the inoculum on second passage (Fig. 1 A). On first passage, the titer might well be low because the inoculum was composed of amyloid fibrils assembled from recMoPrP(89-230). When MoSP1 was passaged into FVB mice, the incubation period on first passage was 154 days and decreased to 130 days on second passage (Fig. 1B). This difference presumably reflects a transmission barrier caused by the difference in PrP sequences between FVB and Tg9949 mice: FVB mice express full-length MoPrP, and Tg9949 mice express only the C-terminal 65% of the protein, MoPrP(Δ22-88).
The neuropathology in Tg9949 mice induced by MoSP1 is distinct from that produced by RML prions. Most important, the neuropathology in Tg9949 mice produced by MoSP1 remained constant on second passage in the Tg9949 line. Passage of MoSP1 into FVB mice reduced the incubation time (Fig. 1), changed the distribution of neuropil vacuolation (Figs. 2 and 3), and dramatically reduced the conformational stability of the prions (Fig. 6). These changes in incubation times, neuropatho-logic lesions, and conformational stability are reminiscent of the alterations seen when Sc237 prions from Syrian hamsters were passaged in Tg mice expressing chimeric mouse-hamster (MH2M) PrP (22).
Whether MoSP1 passaged in FVB mice can regain its former properties by subsequent passage in Tg9949 mice remains to be determined. Although some investigators have reported that strain alterations are reversible (23), this idea is not well studied. Passage of MoSP1 through FVB mice changed the properties of this strain to resemble those of RML prions. It will be of considerable interest if passaging MoSP1 through FVB mice and back into Tg9949 mice results in the recovery of the original MoSP1 strain properties.
In contrast to the passage of MoSP1 into FVB mice, the converse passage of RML prions into Tg9949 mice was not accompanied by alterations in the incubation time and conformational stability even though there was a substantial change in the neuropathologic phenotype (compare Fig. 3 B and D). In Tg9949 mice inoculated with RML prions, vacuolation scores were diminished in all brain regions, and large vacuoles lined with PrPSc were seen. In this instance, it seems likely that the strain of prion did not change appreciably even though the PrP molecule is N-terminally truncated. Before the development of the conformational stability assay, the profound change in neuropathology might have been ascribed to a change in strain, but in fact, it seems to be dictated by the host. Teasing out strain and host PrP effects can at times be difficult (20).
Spectrum of Prion Strains. The discovery of MoSP1 dramatically widens the spectrum of prion strains. It will be important to determine the structure of MoSP1 passaged in Tg9949 mice and FVB mice; extreme differences in stability may be evident in how the protein is folded or assembled into oligomers. Unfortunately, such structural studies currently are limited by the insolubility of PrPSc. Whether electron crystallography can give sufficient detail to distinguish between two strains with such different conformational stabilities is unknown (24, 25).
As new synthetic prions are created, it will be important to build a catalog of strains. Identifying an array of strains with very different, readily measurable properties is critical to advancing our understanding of prion diversity. By correlating structural and biological characteristics of prion strains, it should be possible to begin to decipher how strain-specified properties are encrypted in the conformation of PrPSc. Clearly, learning the language of prion strains is an important avenue of investigation.
Supplementary Material
Acknowledgments
We thank the staff at the Hunters Point Animal Facility, San Francisco. This work was supported by National Institutes of Health Grants AG02132, AG10770, and AG021601 and a gift from the G. Harold and Leila Y. Mathers Charitable Foundation.
Author contributions: G.L., I.V.B., F.E.C., and S.B.P. designed research; H.-O.B.N. and S.J.D. performed research; G.L., S.J.D., and S.B.P. analyzed data; S.B.P. contributed new reagents/analytic tools; and G.L., S.J.D., and S.B.P. wrote the paper.
Abbreviations: PrP, prion protein; MoPrP, mouse PrP; PrPSc, disease-causing isoform of PrP; MoSP1, mouse synthetic prion1; rec, recombinant; Tg, transgenic; RML, Rocky Mountain Laboratory; Gdn1/2, half-maximal GdnHCl; HuM, human-mouse; PK, proteinase K.
References
- 1.Prusiner, S. B. (2004) in Prion Biology and Diseases, ed. Prusiner, S. B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 1-87.
- 2.Prusiner, S. B., McKinley, M. P., Bowman, K. A., Bolton, D. C., Bendheim, P. E., Groth, D. F. & Glenner, G. G. (1983) Cell 35, 349-358. [DOI] [PubMed] [Google Scholar]
- 3.McKinley, M. P., Meyer, R. K., Kenaga, L., Rahbar, F., Cotter, R., Serban, A. & Prusiner, S. B. (1991) J. Virol. 65, 1340-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tremblay, P., Ball, H. L., Kaneko, K., Groth, D., Hegde, R. S., Cohen, F. E., DeArmond, S. J., Prusiner, S. B. & Safar, J. G. (2004) J. Virol. 78, 2088-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Legname, G., Baskakov, I. V., Nguyen, H.-O. B., Riesner, D., Cohen, F. E., DeArmond, S. J. & Prusiner, S. B. (2004) Science 305, 673-676. [DOI] [PubMed] [Google Scholar]
- 6.Baskakov, I. V. (2004) J. Biol. Chem. 279, 7671-7677. [DOI] [PubMed] [Google Scholar]
- 7.Supattapone, S., Muramoto, T., Legname, G., Mehlhorn, I., Cohen, F. E., DeArmond, S. J., Prusiner, S. B. & Scott, M. R. (2001) J. Virol. 75, 1408-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandler, R. L. (1961) Lancet 1, 1378-1379. [DOI] [PubMed] [Google Scholar]
- 9.Carlson, G. A., Kingsbury, D. T., Goodman, P. A., Coleman, S., Marshall, S. T., DeArmond, S., Westaway, D. & Prusiner, S. B. (1986) Cell 46, 503-511. [DOI] [PubMed] [Google Scholar]
- 10.Prusiner, S. B., Cochran, S. P., Groth, D. F., Downey, D. E., Bowman, K. A. & Martinez, H. M. (1982) Ann. Neurol. 11, 353-358. [DOI] [PubMed] [Google Scholar]
- 11.Scott, M., Foster, D., Mirenda, C., Serban, D., Coufal, F., Waälchli, M., Torchia, M., Groth, D., Carlson, G., DeArmond, S. J., et al. (1989) Cell 59, 847-857. [DOI] [PubMed] [Google Scholar]
- 12.Muramoto, T., Kitamoto, T., Tateishi, J. & Goto, I. (1992) Am. J. Pathol. 140, 1411-1420. [PMC free article] [PubMed] [Google Scholar]
- 13.Carlson, G. A., Ebeling, C., Yang, S.-L., Telling, G., Torchia, M., Groth, D., Westaway, D., DeArmond, S. J. & Prusiner, S. B. (1994) Proc. Natl. Acad. Sci. USA 91, 5690-5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williamson, R. A., Peretz, D., Pinilla, C., Ball, H., Bastidas, R. B., Rozenshteyn, R., Houghten, R. A., Prusiner, S. B. & Burton, D. R. (1998) J. Virol. 72, 9413-9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peretz, D., Williamson, R. A., Kaneko, K., Vergara, J., Leclerc, E., SchmittUlms, G., Mehlhorn, I. R., Legname, G., Wormald, M. R., Rudd, P. M., et al. (2001) Nature 412, 739-743. [DOI] [PubMed] [Google Scholar]
- 16.Supattapone, S., Nguyen, H.-O. B., Cohen, F. E., Prusiner, S. B. & Scott, M. R. (1999) Proc. Natl. Acad. Sci. USA 96, 14529-14534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laemmli, U. K. (1970) Nature 227, 680-685. [DOI] [PubMed] [Google Scholar]
- 18.DeArmond, S. J., Kretzschmar, H. A. & Prusiner, S. B. (2002) in Greenfield's Neuropathology, eds. Graham, D. I. & Lantos, P. L. (Hodder Arnold, London), 7th Ed., pp. 273-323.
- 19.Kaneko, K., Ball, H. L., Wille, H., Zhang, H., Groth, D., Torchia, M., Tremblay, P., Safar, J., Prusiner, S. B., DeArmond, S. J., et al. (2000) J. Mol. Biol. 295, 997-1007. [DOI] [PubMed] [Google Scholar]
- 20.Scott, M. R., Peretz, D., Nguyen, H.-O. B., DeArmond, S. J. & Prusiner, S. B. (2005) J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 21.Taylor, D. M., Fernie, K., Steele, P. J., McConnell, I. & Somerville, R. A. (2002) J. Gen. Virol. 83, 3199-3204. [DOI] [PubMed] [Google Scholar]
- 22.Peretz, D., Williamson, R. A., Legname, G., Matsunaga, Y., Vergara, J., Burton, D., DeArmond, S. J., Prusiner, S. B. & Scott, M. R. (2002) Neuron 34, 921-932. [DOI] [PubMed] [Google Scholar]
- 23.Kimberlin, R. H., Cole, S. & Walker, C. A. (1987) J. Gen. Virol. 68, 1875-1881. [DOI] [PubMed] [Google Scholar]
- 24.Wille, H., Michelitsch, M. D., Guénebaut, V., Supattapone, S., Serban, A., Cohen, F. E., Agard, D. A. & Prusiner, S. B. (2002) Proc. Natl. Acad. Sci. USA 99, 3563-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Govaerts, C., Wille, H., Prusiner, S. B. & Cohen, F. E. (2004) Proc. Natl. Acad. Sci. USA 101, 8342-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.