

Abstract
Activation-induced cytidine deaminase (AID) is required for the DNA cleavage step of Ig somatic hypermutation (SHM). However, its molecular mechanism is controversial. The RNA editing hypothesis postulates that AID deaminates cytosine in an unknown mRNA to generate a new mRNA encoding SHM endonuclease. On the other hand, the DNA deamination hypothesis explains DNA cleavage by cytosine deamination in DNA, followed by uracil removal by uracil DNA glycosylase (UNG). By using the protein synthesis inhibitor cycloheximide, we showed that SHM requires de novo protein synthesis in accord with predictions by the RNA editing hypothesis. In addition, we found that cycloheximide but not Ugi (the specific inhibitor of UNG) inhibited AID-dependent DNA cleavage in the Ig gene during SHM, by using histone H2AX focus formation as a marker of DNA cleavage. The results indicate the following order of events: AID expression, protein synthesis, DNA cleavage, and SHM. The requirement of protein synthesis but not of UNG for the DNA cleavage step of SHM forces us to reconsider the DNA deamination hypothesis and strengthens the RNA editing hypothesis.
Antigen stimulation induces three types of genetic alterations, namely somatic hypermutation (SHM), gene conversion (GC), and class switch recombination (CSR) in activated B cells, giving rise to antigen-specific Ig with high affinity and appropriate effector functions. SHM introduces point mutations in variable-region (V) genes without template, whereas GC does so with pseudo-V genes as template. When SHM and GC are coupled with selection by limited amounts of antigen, highaffinity antibody-producing B cells are enriched. CSR is a genetic process that switches Ig isotypes from IgM to other isotypes such as IgG and IgE, adding diverse effector functions to Ig with a given antigen specificity (1, 2).
Activation-induced cytidine deaminase (AID), which is strictly expressed in activated B cells, especially in the germinal centers of lymphoid follicles, is essential for all three types of DNA alteration in B cells activated by antigen stimulation (3–6). AID has the strongest homology with apolipoprotein B mRNA editing catalytic subunit 1 (APOBEC-1) (reviewed in ref. 7). The genetic loci for AID and APOBEC-1 are tightly linked on mouse and human chromosomes (2). In addition to these evolutionary conservations, AID has functional similarities with APOBEC-1, including dimer formation, requirement of cofactors, and shuttling between cytoplasm and nucleus by N-terminal nuclear localization and C-terminal nuclear export domains (8–10).
Evolutionary conservation and biochemical similarities between AID and apolipoprotein B mRNA editing catalytic subunit 1 (APOBEC-1) led us to propose the RNA editing hypothesis that AID converts unknown mRNA precursors to novel mRNAs encoding putative endonucleases to cleave target DNA (1). The alternative hypothesis (DNA deamination) explains DNA cleavage by direct deamination of cytosine (C) to uracil (U) in target DNA by AID. To generate strand breakage, U should be removed by the base excision repair pathway, including uracil DNA glycosylase or uracil N-glycosylase (UNG) and apurinic/apyrimidinic-endonuclease (11). This model also proposes that uncorrected U/G mismatches introduce C/G→T/A mutations by DNA replication. Enhanced mutagenesis by overexpression of AID in a number of genes in Escherichia coli (12) and in vitro DNA deamination activity of AID is consistent with this hypothesis (13–16). The DNA deamination hypothesis is further supported by the fact that UNG deficiency causes marked reduction in class-switching efficiency (17) and target bias toward G and C of SHM, albeit no change of frequency (17, 18).
AID is required for DNA cleavage in CSR and SHM (19, 20). Both CSR and SHM require DNA breakage. Double-strand breakage (DSB) is essential for initiating CSR but not necessarily SHM. However, DSB is also associated with SHM, because frequent single-stranded cleavage can cause DSB. DSB in interphase DNA is under surveillance by a set of protein kinases, including ATM/ATR and DNA-dependent protein kinase catalytic subunit (21). Such kinases phosphorylate histone H2AX (γ-H2AX) in the DNA domain next to DSB soon after DNA lesion (22, 23) and recruit many proteins involved in DNA repair. Because γ-H2AX covers hundreds of kilobases of DNA adjacent to DSB, this accumulation serves as a good marker of DNA cleavage sites (19).
The mechanisms of AID-dependent DNA cleavage by the RNA editing and DNA deamination hypotheses assume involvement of de novo protein synthesis and UNG, respectively. We showed that DSB in S regions depend on de novo protein synthesis but not on UNG (24, 25). Furthermore, UNG mutants that abolished U removal activity still rescued class switching in UNG-deficient B cells (24). These results are consistent with the RNA editing hypothesis but not with the DNA deamination hypothesis.
Although both CSR and SHM depend on AID and transcription of the target locus, their detailed mechanisms could be distinct, because studies on AID mutants showed that the domains essential to CSR and SHM are separate (10, 26). It is therefore important to test whether either protein synthesis or UNG activity is required for DSB associated with SHM. We report here that AID-dependent DSB associated with SHM depends on de novo protein synthesis but not on UNG activity. These results further strengthen the RNA editing hypothesis.
Materials and Methods
Cells and Culture Conditions. NIR cells were established by transfection of NIH 3T3 cells with an AID fusion protein with the estrogen receptor hormine-binding domain (A IDER)-expressing construct, pAID-ER-BOSbsr (27), carrying an artificial SHM target gene (28, 29). JP8BdelER (pFB-ΔC-AIDERpuro) was generated by ligation of JP8Bdel, a C-terminal deletion mutant of human AID, and Flag-estrogen receptor hormone binding domain (ER) in the same reading frame. JP8BdelER was cloned into the pFB retrovirus vector (Stratagene) that has an internal ribosome entry site (IRES) and puromycin-resistant gene. BL2-ΔC-AIDER cells were established by drug selection and cloning after retrovirus infection of BTZ cells, which is a BL2 clone harboring pTet-tTAk-zeo (27). To obtain Ugi(+)BL2-ΔC-AIDER cells, BL2-ΔC-AIDER cells were infected with Ugi-expressing retrovirus carrying IRESGFP. GFP-positive cells were sorted by FACSVantage (BD Bioscience).
NIR cells were maintained as described (9), by the addition of 2.5 μg/ml Blastcidine S and 0.25 μg/ml tetracycline (TET). BTZ and BL2-ΔC-AIDER cells were maintained in RPMI medium 1640 containing 10% FCS, 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, 0.25 μg/ml TET, 0.5 μg/ml puromycin, and 100 μg/ml zeocine. Puromycin and Zeocine were removed at least 2 days before experiments. For activation of AIDER proteins, 4-hydroxytamoxifen (OHT) was added with 1 μM of concentration with or without 10 μg/ml cycloheximide (CHX).
Chromatin Immunoprecipitation (ChIP) Assay. ChIP was done as described (24, 25). Nonspecific γ-H2AX focus formation was induced by treatment of cells with 0.5 μM staurosporine for 2 h as a positive control (30). After immunoprecipitation and DNA purification, each DNA for ChIP and input samples (1/15 amount used for ChIP) was subjected to real-time PCR by using IQ SYBR green supermix and iCycler iQ (Bio-Rad) with cycles of 95°C for 30 seconds and 61°C for 30 seconds. The melting curves test was always done at the final step of reactions to confirm that appropriate amplification products were obtained. Arbitrary values were determined essentially as described (31); briefly, a relative intensity (X) was calculated by the formula: X = 2(Ct (input) – Ct (ChIP)); here, the threshold cycle (Ct) values of ChIP samples [Ct(ChIP)] were compared with that of the input [Ct (input)].
Primers used for detection are: Cμ5′, 5′-GCATCCGCCCCAACCCTTTTC-3′; Cμ3′, 5′-CCAACGGCCACGCTGCTC-3′; BL2V5′, 5′-TCACCTACTACAACCCATCCCTCGAGAGTC-3′; BL2V3′, 5′-CGCACAGTAATACACAGCCGTGTCTG-3′; RAG2-5′, 5′-CCCTGTGTTTTCCTGGTGGA-3′; RAG2-3′, 5′-CCAGGGGAAGATCAACCCTT-3′; huAID5′, 5′-GCTTCGTAGCACCATTACTGC-3′; huAID3′, 5′-GAAATGGAGTCTCAAAGCTTCA-3′; Cα2-5′, 5′-TGAGCTGAGGCCTAAGTTGA-3′; Cα2-3′, 5′-CAGCCCACATGGACGTGT-3′; Jh6-5′, 5′-CTACTGCCTGTGGGGTTTCCTGAGCATTG-3′; Jh6-3′, 5′-CCCCAGGCTCAGTTACTCCATCAGACGCAC-3′; V5-78P5′, 5′-GGCTGTTCTCCAAGGTC-3′; V5-78P3′, 5′-GGAGACACAAAATTTGCATC-3′; β2MG5′, 5′-CGGCTCTGCTTCCCTTAGAC-3′; β2MG3′, 5′-CGAAACCGCTTTGTATCACA-3′.
Western Blot and RT-PCR. Cells were directly lysed in SDS sample buffer (25 mM Tris/5% glycerol/1% SDS/1% 2-mercaptoethanol) after harvest. Lysates were separated by SDS/PAGE and blotted to Hybond-P membrane (Amersham Pharmacia). Antibodies used for Western blot are: anti-ER HC-20 (Santa Cruz Biotechnology); anti-tubulin Ab-1 (Oncogene Science); horseradish peroxidase (HRP)-conjugated goat anti-rabbit polyclonal (BioSource International, Camarillo, CA); and HRP-goat antimouse IgG (H+L) (Kirkegaard & Perry Laboratories). Signals were developed by the ECL Plus kit (Amersham Pharmacia). RNA was extracted by TRIzol (Invitrogen). First-strand cDNA was synthesized by the first-strand cDNA synthesis kit (Life Science, Arlington Heights, IL). Primers used for postswitch transcript of human are as described (32).
In Vitro UNG Assay and DNA Sequencing. The in vitro UNG assay was done as described (18, 24). The DNA sequence of Ig gene of BL2 cells was determined as described (33).
Results and Discussion
CHX Inhibits AID-Dependent SHM in Fibroblasts. We first examined whether overall SHM is sensitive to a protein synthesis inhibitor (CHX). To quantitate SHM, we used the fibroblast (NIH 3T3) system, in which AID expression can induce mutations on an artificial substrate gene as efficiently as SHM of Ig genes in germinal center B cells (29). The GFP gene with a premature stop codon serves as the artificial substrate gene to detect reversion mutations by monitoring GFP expression by flow cytometer (FACS) (28, 29). To avoid inhibition of AID expression by CHX, we took advantage of an AID fusion protein with the ER hormone-binding domain (AIDER), which can accumulate as an inactive form and then be activated by OHT (27). A transfectant of NIH 3T3 cells (NIR cell) carrying the artificial target under the TET repressible promoter and AIDER was treated with OHT for 2 h with or without CHX, as indicated in Fig. 1. To quickly cancel the effect of OHT treatment on SHM, TET was added to shut off target gene transcription, because SHM depends on the transcription of target DNA (28, 29). To ensure the complete decay of OHT, edited mRNA, and its translation products, cells were cultivated 6 days after withdrawal of OHT and/or CHX. After the withdrawal of TET at day 7, GFP-positive cells were counted by FACS at day 8. We observed a significant increase of GFP reversion in NIR cells incubated with OHT treatment in the absence of TET as compared with NIR cells incubated with OHT in the presence of TET (negative control) (Fig. 1, B and C). Significant GFP reversions in the negative control as compared with those without OHT (Fig. 1, A) could be due to a combination of leaky transcription in the presence of TET, remnant OHT, and slow decay of AID activation products (edited mRNA or putative endonuclease), which had some residual activity even 4 days after the washoff of OHT (data not shown). The addition of CHX before or at the time of OHT treatment clearly inhibited GFP reversion (Fig. 1, D and E), whereas the inhibition was not observed if the CHX treatment was done after OHT treatment (Fig. 1, F), suggesting that inhibition by CHX is specific to the time window before activation of AID. It is of note that 6-h CHX treatment did not suppress AIDER expression and target transcription (ref. 27 and results not shown). Taken together, we conclude that protein synthesis is required for AID-induced SHM in NIH 3T3 cells.
Fig. 1.
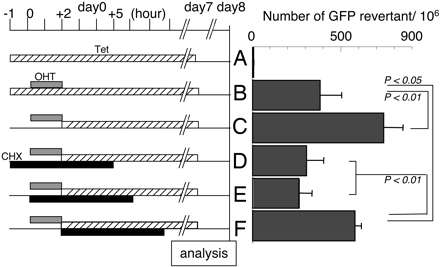
GFP reversion assay by NIR cells. NIR cells were plated with (A and B) or without TET (C–F) 1 day before day 0. (Left) Cells were treated on day 0 with OHT (gray bar) for 2 h. After washoff of OHT, TET (hatched bar) was added and maintained until day 7. CHX treatment (black bar) was done for D–F as indicated. Numbers of GFP revertants were shown (Right). The data are the average of triplicate culture with standard deviation (SD). Significant difference is indicated by the P value.
AID-Dependent γ-H2AX Focus Formation on Ig Genes in SHM-Inducible Cells. We next tried to examine whether protein synthesis is required for DNA breakage in SHM. BL2 is a Burkitt's lymphoma cell line, which mutates its Ig genes and forms γ-H2AX foci in the Ig locus by AID overexpression (33, 34). To eliminate DNA cleavage associated with CSR, we used a mutant AID, JP8Bdel, that lacks C-terminal 16-aa residues and fused it with ER (JP8BdelER). The C-terminal amino acid residues of AID have been shown to be critical for S region cleavage in CSR but not to SHM (8–10, 26). A BL2 transfectant, BL2-ΔC-AIDER, that expresses JP8BdelER was established (Fig. 2A). In this cell line, the V of the Ig heavy-chain (IgH) gene was hypermutated upon OHT treatment with the frequency ≈10–3 bp/3 days (Table 1). Without OHT, the BL2-ΔC-AIDER cells showed a mutation frequency similar to spontaneous ones in parental BL2. We found no obvious SHM of the constant region gene, in agreement with the in vivo feature of the Ig SHM in B cells (Table 1). We further confirmed that OHT treatment of BL2-ΔC-AIDER does not induce CSR, because postswitch transcripts for IgGs were undetectable (Fig. 2B). The results demonstrate that OHT treatment of BL2-ΔC-AIDER cells induces frequent SHM of the IgH gene but not CSR.
Fig. 2.
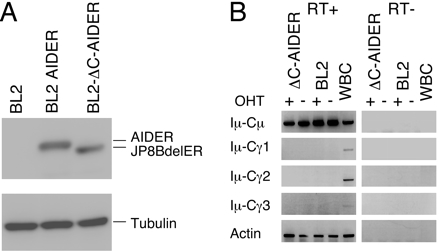
Establishment of JP8BdelER-expressing BL2 cells. (A) Lysate from 1.5 × 105 BL2-ΔC-AIDER cells was used for Western blotting. AIDER transfectant and parental BL2 cells are shown as controls. The ER fusion proteins were detected by anti-ER. Tubulin is shown as a protein loading control. (B) BL2-ΔC-AIDER and parental cells were cultured with OHT for 3 days and analyzed by RT-PCR to check the expression of postswitch transcripts. White blood cells (WBC) isolated from human peripheral blood were used as a positive control for postswitch transcripts. RT–samples were negative control that did not obtain reverse transcriptase treatment.
Table 1. Frequency of SHM on the IgH gene in OHT-treated BL2-ΔC-AIDER cells.
| Mutation frequency after 3 d
|
|||
|---|---|---|---|
| Cells | OHT(-) | OHT(+) | Day 0 |
| Mutation at the V region | |||
| BL2-ΔC-AIDER | 4.17 × 10-4* | 1.41 × 10-3* | 1.7 × 10-4 |
| 5/11,984 (4/27) | 17/11,977 (14/27) | 2/6,659 (2/15) | |
| BL2 | 2.5 × 10-4 | <3.8 × 10-4 | n.d. |
| 1/3,990 (1/9) | 0/2,664 (0/6) | ||
| Mutation at the Cμ region | |||
| BL2-ΔC-AIDER | 1.28 × 10-4 | 1.5 × 10-4 | n.d. |
| 1/7,812 (1/12) | 1/6,510 (1/10) | ||
Mutation frequency is expressed as mutations per base pair. Numbers below the frequency are as follows: numbers of mutation/total base pair (numbers of mutated sequence/total sequence). n.d., not determined; Cμ, IgH μ chain constant region. *, P = 0.0084.
Although it is still unclear whether DSB is an intermediate or a byproduct of SHM, DSB of target DNA associates with SHM reaction (34–37). To determine whether γ-H2AX focus formation on the IgH gene is detectable in OHT-treated BL2-ΔC-AIDER, we performed the ChIP assay with anti γ-H2AX antibodies. As shown in Figs. 3 and 4, OHT treatment significantly increased anti-γ-H2AX ChIP signals of Ig V and Cμ genes in BL2-ΔC-AIDER cells. However, no increase of ChIP signals was observed in the Ig locus in parental cells that have no AIDER. The present results are in accord with a previous report in which AID overexpression in BL2 causes γ-H2AX focus formation in V and Cμ loci (34). The signals were detectable as early as 30 min after OHT administration and were maintained thereafter for at least 6 h (Fig. 3). As previously observed in irradiated cells, the γ-H2AX focus formation seems to spread to a wide range of the IgH locus but not to other loci, such as the β2-microglobulin, Rag2, and AID genes (Figs. 3 and 4), indicating that DSB is specific to the Ig gene. We conclude that AID induces DSB of the Ig gene in OHT-treated BL2-ΔC-AIDER cells in association with SHM.
Fig. 3.

Rapid γH2AX association on the entire IgH locus in OHT-treated BL2-ΔC-AIDER cells. BL2-ΔC-AIDER cells were treated with OHT and used for ChIP experiment at indicated time points. Results of ChIP with anti-γH2AX (black) and nonspecific IgG (gray) are presented as intensity relative to input as described in Materials and Methods. Primer positions in the IgH gene locus are shown (Right). β2MG, β-2-microglobulin; VDJh5, rearranged VDJ; Cen., centromere; Tel., telomere; Chr., chromosome.
Fig. 4.
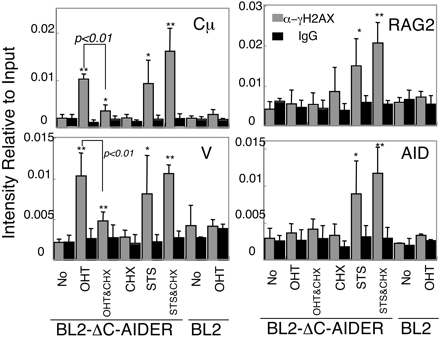
Specific induction of γH2AX focus formation on IgH genes in OHT-treated BL2-ΔC-AIDER cells and its inhibition by CHX. The cells indicated were treated by OHT for 6 h with or without addition of CHX1hin advance. The anti-γH2AX ChIP assay was carried out, and real-time PCR data relative to input signal were calculated as described in Materials and Methods. The column graphs show averages of four independent experiments of BZP cells with SD and those of two experiments of parental cell (BL2) used as negative controls. Data for Cμ, V, RAG2, and AID genes are shown. α-γH2AX (gray) and IgG (black) indicate the ChIP sample with anti-γH2AX and nonspecific mouse IgG, respectively. A significant increase in α-γH2AX over IgG is marked with single (P < 0.05) or double (P < 0.01) asterisks. A significant difference between CHX-treated and nontreated samples is indicated by P values. STS, staurosporin treated.
De Novo Protein Synthesis Is Required for DNA Breakage Associated with SHM. To determine whether de novo protein synthesis is required for DSB associated with SHM, we examined the effect of CHX addition on γ-H2AX focus formation in BL2-ΔC-AIDER cells. CHX addition 1 h in advance of the OHT treatment significantly inhibited γ-H2AX focus formation on the IgH gene (Fig. 4). The effect was not due to general inhibition of the γ-H2AX reaction, because γ-H2AX accumulation associated with apoptosis by staurosporine on nonspecific loci (30) was not inhibited by CHX (Fig. 4). The protein amount of JP8BdelER was not largely affected by CHX treatment, as shown by Western blot (Fig. 5A). Besides, the transcription efficiency of the IgH locus was checked by measuring the mRNA level as well as RNA polymerase II loading by RT-PCR and ChIP assay, respectively. No difference was observed between CHX-treated and nontreated cells (Fig. 5 B and C). Because neither the AIDER protein amount nor the target transcription level had been changed by CHX, the inhibition of DSB by CHX likely occurred at the step downstream of the AID activation. Taken together with SHM inhibition in fibroblasts by CHX, de novo protein synthesis is required for the AID-dependent DNA strand break associated with SHM.
Fig. 5.

CHX treatment showed negligible effects on JP8BdelER expression and target transcription. Lysates of BL2-ΔC-AIDER cells treated by OHT for 6 h with or without CHX addition 1 h in advance were analyzed by Western blot (A). The intensity of JP8BdelER was measured and normalized according to that of tubulin. Intensity relative to the OHT sample after normalization is shown by percentile. IgH gene transcription was checked by RT-PCR (B) and ChIP with anti-RNA polymerase II (C). The numbers on the RT-PCR gel image indicate the average intensity of each band that is obtained by photoshop (Adobe Systems, San Jose, CA) after image scanning. The column graph shows the average value of three independent experiments with standard deviation. RT, reverse transcriptase treatment.
Dispensability of UNG for DNA Breaks in SHM. If U excision on DNA by UNG would be the major pathway to introduce DNA breakage, Ugi, a specific inhibitor of UNG, should inhibit γ-H2AX focus formation in OHT-treated BL2-ΔC-AIDER cells. We therefore introduce the Ugi-expressing retrovirus with IRES GFP into BL2-ΔC-AIDER cells. Ugi-positive cells were enriched up to 98% by cell sorting. Inhibition of UNG was confirmed by in vitro U excision assay with cellular extracts (Fig. 6A). Ugi(+) BL2-ΔC-AIDER cells were treated with OHT and analyzed by the anti-γ-H2AX ChIP assay. As shown in Fig. 6B, the γ-H2AX focus formation at the V region gene in Ugi-expressing cells was similar to that of Ugi(–) BL2-ΔC-AIDER cells. We thus conclude that UNG is dispensable in the DSB formation associated with SHM.
Fig. 6.

Ugi is dispensable for γH2AX focusing. (A) FITC-labeled substrate oligo-DNA with single U was incubated with cellular extracts that contain 100 or 10 ng of protein. Protein extract from NIH 3T3 (3T3) is a positive control. The number indicates the percentage of cleaved substrate of each lane. (B) γH2AX focusing on the V region of IgH was detected by ChIP assay in BL2-ΔC-AIDER and Ugi(+) BL2-ΔC-AIDER cells treated with or without OHT for 2 h. The significant increase of α-γH2AX over IgG is marked with single (P < 0.05) or double (P < 0.01) asterisks. ΔC, BL2-ΔC-AIDER; ΔC Ugi, Ugi(+)BL2-ΔC-AIDER; No, without OHT.
CSR and SHM Have Similar Requirements for DNA Breakage. We have shown that DSB associated with SHM depends on de novo protein synthesis but not on UNG. These features are the same as reported for CSR (24, 25) and consistent with the RNA editing hypothesis but not with the DNA deamination hypothesis. Because AID has N- and C-terminal domains that are specifically required for SHM and CSR, respectively (10, 26), we proposed that AID interacts with SHM- and CSR-specific cofactors at the N and C termini, respectively. Taken together, the most likely scenario is that AID recognizes and edits two separate mRNA precursors for endonucleases in collaboration with SHM- and CSR-specific cofactors.
Since a causal role of DNA cleavage in SHM had first been proposed by Brenner and Milstein (38), a large number of investigators have reported the coincidence of DNA cleavage with SHM (34–37, 39). However, the direct requirement of DNA cleavage for SHM was not clearly demonstrated. We showed here that both DNA cleavage and SHM depend on de novo protein synthesis as well as AID expression, indicating that all these events are functionally related. Because there is no question about the beginning (AID expression) and the end (SHM), our results show the order of these events as follows: AID expression, protein synthesis, DNA cleavage, and SHM.
Recently, double-knockout mice of UNG and Msh2 have been shown to enhance phenotypes of UNG deficiency, more severe reduction in CSR efficiency, and stronger bias toward GC base mutations in SHM (40). This result was interpreted to indicate that both UNG and Msh2/6 mismatch repair enzymes recognize U/G mismatches generated by AID-dependent C deamination and complement each other. This interpretation may explain the dispensability of UNG in CSR and SHM. However, Msh2/6 is thought to serve for AT targeted mutation by short-patch synthesis repair initiated at least a few base pairs away from U/G mismatches (41). This repair pathway should be as efficient as UNG, because normally almost half of the SHM target is AT (42). If so, SHM by Msh2-mediated repair in the absence of UNG should shift toward the AT bases. However, UNG deficiency result in GC-biased SHM without changing the frequency (17, 18). In view of common requirements for DNA cleavage in CSR and SHM, dispensability of U removal activity in CSR (24) strongly suggests that SHM also does not depend on the U removal activity of UNG.
Acknowledgments
We thank Ms. Y. Sasaki for excellent technical assistance; Drs. S. Fagarasan, K. Kinoshita, and R. Shinkura for critical reading of the manuscript; Drs. J. C. Weill and C. A. Reynaud for the BL2 cells; and Ms. Y. Shiraki and T. Nishikawa for preparation of the manuscript. This work is supported by a Center of Excellence Grant from the Ministry of Education, Science, Sports, and Culture of Japan.
Author contributions: T.H. designed research; H.N., S.I., M.M., and M.N. performed research; ; M.M. contributed new reagents/analytic tools; H.N. analyzed data; and H.N. and T.H. wrote the paper.
Abbreviations: AID, activation-induced cytidine deaminase; SHM, somatic hypermutation; CSR, class switch recombination; DSB, double-strand breakage; UNG, uracil N-glycosylase; V, variable region; TET, tetracycline; OHT, 4-hydroxytamoxifen; CHX, cycloheximide; ChIP, chromatin immunoprecipitation; GC, gene conversion; ER, estrogen receptor hormone-binding domain; IgH, Ig heavy chain.
References
- 1.Honjo, T., Kinoshita, K. & Muramatsu, M. (2002) Annu. Rev. Immunol. 20, 165–196. [DOI] [PubMed] [Google Scholar]
- 2.Stavnezer, J., Kinoshita, K., Muramatsu, M. & Honjo, T. (2004) in Molecular Biology of B Cells, eds. Honjo, T., Alt, F. W. & Neuberger, M. S. (Elsevier Academic, London), pp. 307–319.
- 3.Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y. & Honjo, T. (2000) Cell 102, 553–563. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu, M., Sankaranand, V. S., Anant, S., Sugai, M., Kinoshita, K., Davidson, N. O. & Honjo, T. (1999) J. Biol. Chem. 274, 18470–18476. [DOI] [PubMed] [Google Scholar]
- 5.Arakawa, H., Hauschild, J. & Buerstedde, J. M. (2002) Science 295, 1301–1306. [DOI] [PubMed] [Google Scholar]
- 6.Revy, P., Muto, T., Levy, Y., Geissmann, F., Plebani, A., Sanal, O., Catalan, N., Forveille, M., Dufourcq-Labelouse, R., Gennery, A., et al. (2000) Cell 102, 565–575. [DOI] [PubMed] [Google Scholar]
- 7.Anant, S. & Davidson, N. O. (2001) Curr. Opin. Lipidol. 12, 159–165. [DOI] [PubMed] [Google Scholar]
- 8.Barreto, V., Reina-San-Martin, B., Ramiro, A. R., McBride, K. M. & Nussenzweig, M. C. (2003) Mol. Cell 12, 501–508. [DOI] [PubMed] [Google Scholar]
- 9.Ito, S., Nagaoka, H., Shinkura, R., Begum, N., Muramatsu, M., Nakata, M. & Honjo, T. (2004) Proc. Natl. Acad. Sci. USA 101, 1975–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ta, V. T., Nagaoka, H., Catalan, N., Durandy, A., Fischer, A., Imai, K., Nonoyama, S., Tashiro, J., Ikegawa, M., Ito, S., et al. (2003) Nat. Immunol. 4, 843–848. [DOI] [PubMed] [Google Scholar]
- 11.Petersen-Mahrt, S. K., Harris, R. S. & Neuberger, M. S. (2002) Nature 418, 99–103. [DOI] [PubMed] [Google Scholar]
- 12.Ramiro, A. R., Stavropoulos, P., Jankovic, M. & Nussenzweig, M. C. (2003) Nat. Immunol. 4, 452–456. [DOI] [PubMed] [Google Scholar]
- 13.Chaudhuri, J., Tian, M., Khuong, C., Chua, K., Pinaud, E. & Alt, F. W. (2003) Nature 422, 726–730. [DOI] [PubMed] [Google Scholar]
- 14.Dickerson, S. K., Market, E., Besmer, E. & Papavasiliou, F. N. (2003) J. Exp. Med. 197, 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pham, P., Bransteitter, R., Petruska, J. & Goodman, M. F. (2003) Nature 424, 103–107. [DOI] [PubMed] [Google Scholar]
- 16.Sohail, A., Klapacz, J., Samaranayake, M., Ullah, A. & Bhagwat, A. S. (2003) Nucleic Acids Res. 31, 2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rada, C., Williams, G. T., Nilsen, H., Barnes, D. E., Lindahl, T. & Neuberger, M. S. (2002) Curr. Biol. 12, 1748–1755. [DOI] [PubMed] [Google Scholar]
- 18.Di Noia, J. & Neuberger, M. S. (2002) Nature 419, 43–48. [DOI] [PubMed] [Google Scholar]
- 19.Petersen, S., Casellas, R., Reina-San-Martin, B., Chen, H. T., Difilippantonio, M. J., Wilson, P. C., Hanitsch, L., Celeste, A., Muramatsu, M., Pilch, D. R., et al. (2001) Nature 414, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagaoka, H., Muramatsu, M., Yamamura, N., Kinoshita, K. & Honjo, T. (2002) J. Exp. Med. 195, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhne, C., Tjornhammar, M. L., Pongor, S., Banks, L. & Simoncsits, A. (2003) Nucleic Acids Res. 31, 7227–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogakou, E. P., Boon, C., Redon, C. & Bonner, W. M. (1999) J. Cell Biol. 146, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S. & Bonner, W. M. (1998) J. Biol. Chem. 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- 24.Begum, N. A., Kinoshita, K., Kakazu, N., Muramatsu, M., Nagaoka, H., Shinkura, R., Biniszkiewicz, D., Boyer, L. A., Jaenisch, R. & Honjo, T. (2004) Science 305, 1160–1163. [DOI] [PubMed] [Google Scholar]
- 25.Begum, N. A., Kinoshita, K., Muramatsu, M., Nagaoka, H., Shinkura, R. & Honjo, T. (2004) Proc. Natl. Acad. Sci. USA 101, 13003–13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinkura, R., Ito, S., Begum, N. A., Nagaoka, H., Muramatsu, M., Kinoshita, K., Sakakibara, Y., Hijikata, H. & Honjo, T. (2004) Nat. Immunol. 5, 707–712. [DOI] [PubMed] [Google Scholar]
- 27.Doi, T., Kinoshita, K., Ikegawa, M., Muramatsu, M. & Honjo, T. (2003) Proc. Natl. Acad. Sci. USA 100, 2634–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachl, J., Carlson, C., Gray-Schopfer, V., Dessing, M. & Olsson, C. (2001) J. Immunol. 166, 5051–5057. [DOI] [PubMed] [Google Scholar]
- 29.Yoshikawa, K., Okazaki, I. M., Eto, T., Kinoshita, K., Muramatsu, M., Nagaoka, H. & Honjo, T. (2002) Science 296, 2033–2036. [DOI] [PubMed] [Google Scholar]
- 30.Rogakou, E. P., Nieves-Neira, W., Boon, C., Pommier, Y. & Bonner, W. M. (2000) J. Biol. Chem. 275, 9390–9395. [DOI] [PubMed] [Google Scholar]
- 31.Chakrabarti, S. K., James, J. C. & Mirmira, R. G. (2002) J. Biol. Chem. 277, 13286–13293. [DOI] [PubMed] [Google Scholar]
- 32.Cerutti, A., Zan, H., Schaffer, A., Bergsagel, L., Harindranath, N., Max, E. E. & Casali, P. (1998) J. Immunol. 160, 2145–2157. [PMC free article] [PubMed] [Google Scholar]
- 33.Faili, A., Aoufouchi, S., Gueranger, Q., Zober, C., Leon, A., Bertocci, B., Weill, J. C. & Reynaud, C. A. (2002) Nat. Immunol. 3, 815–821. [DOI] [PubMed] [Google Scholar]
- 34.Woo, C. J., Martin, A. & Scharff, M. D. (2003) Immunity 19, 479–489. [DOI] [PubMed] [Google Scholar]
- 35.Papavasiliou, F. N. & Schatz, D. G. (2000) Nature 408, 216–221. [DOI] [PubMed] [Google Scholar]
- 36.Bross, L., Fukita, Y., McBlane, F., Demolliere, C., Rajewsky, K. & Jacobs, H. (2000) Immunity 13, 589–597. [DOI] [PubMed] [Google Scholar]
- 37.Zan, H., Wu, X., Komori, A., Holloman, W. K. & Casali, P. (2003) Immunity 18, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brenner, S. & Milstein, C. (1966) Nature 211, 242–243.5965537 [Google Scholar]
- 39.Kong, Q. & Maizels, N. (2001) Genetics 158, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rada, C., Di Noia, J. M. & Neuberger, M. S. (2004) Mol. Cell 16, 163–171. [DOI] [PubMed] [Google Scholar]
- 41.Reynaud, C. A., Aoufouchi, S., Faili, A. & Weill, J. C. (2003) Nat. Immunol. 4, 631–638. [DOI] [PubMed] [Google Scholar]
- 42.Rada, C., Ehrenstein, M. R., Neuberger, M. S. & Milstein, C. (1998) Immunity 9, 135–141. [DOI] [PubMed] [Google Scholar]