

Abstract
Promoter DNA methylation is a major epigenetic mechanism for silencing genes and establishing commitment in cells differentiating from their precursors. The transcription factor T-bet is a key determinant of IFN-γ gene expression in helper T cells, but the mechanisms by which it achieves this effect are not clear. It is shown here that T-bet binds to a highly conserved T-box half-site in the IFN-γ promoter, is recruited to the endogenous IFN-γ promoter in T lymphoid cells, and transactivates gene expression through this sequence in a manner dependent on consensus T-box residues. This conserved promoter site is methylated in a model T cell line, and enforced T-bet expression did not alter its complete methylation. T-bet transactivated the conserved core promoter in transfection assays and collaborated functionally with C/EBPβ despite methylation of the conserved element. Importantly, enforced T-bet expression led to dissociation of the mSin3a corepressor from the endogenous, chromatinized IFN-γ promoter without decreasing loading of the methyl-CpG binding protein MeCP2. These data indicate that T-bet can override repressive epigenetic modification by a mechanism in which this master regulator acts through a T-box half-site to enforce the activation of IFN-γ gene expression in part by decreased loading of a corepressor on methylated DNA.
Keywords: corepressor, DNA methylation, transcription
Methylation of the DNA in regulatory sequences of eukaryotic promoters is a key mechanism of silencing gene expression (1–3). Two analogous mechanisms have been discerned by which methyl-CpG dinucleotides impose a repressive imprint on cis-acting regulatory sequences in genes, each of which involves the specific recognition of the methylated sequence by a protein sharing the methyl binding domain, and the interaction of these binding proteins with corepressor complexes. Methylated CpG residues are specifically recognized by MeCP2, which interacts with the pleiotropic corepressor mSin3A (4, 5), leading to recruitment of histone deacetylases that prevent the accumulation of acetylated histones H3 and H4 (3, 6). Alternatively, one of several related modified DNA-binding proteins (MBD2 or MBD3) binds to Me-CpG and interacts with NuRD/Mi-2, a different corepressor complex, instead of mSin3a (7–11). Gene-targeting studies in mice indicate that the various methyl-DNA-binding proteins play fundamentally different roles in development and function (12, 13), perhaps because of gene-specific differences in their targets. Cellular differentiation leads to derepression of target genes, but the mechanisms that mediate this reversal of silencing remain to be defined fully.
Differentiation of lymphocytes into T helper (Th) 1 or Th2 cells represents a key fate decision of CD4+ Th cells after activation (14). In the Th1 subset, the IFN-γ gene acquires the capacity for high rates of gene transcription, whereas differentiation into the Th2 subset suppresses the capacity to express this cytokine gene while selectively activating a program of other cytokine-encoding genes (15, 16). As a potent macrophageactivating factor, IFN-γ produced by Th1 cells is crucial for adaptive immunity to a broad range of microbial pathogens, so that a defect either in this cytokine or in the ability to develop Th1 cells leads to susceptibility to infectious diseases, particularly those of parasites, bacteria, and fungi for which phagocytosis and intracellular killing are important (14). IFN-γ also contributes to antiviral immunity and surveillance against cancer (17). One layer of mechanisms leading to selective transcriptional regulation at the IFN-γ locus involves networks of transcription factors whose induction or inhibition after T cell activation influences the induction of this cytokine gene (15, 16). Initially, proteins in latent cytoplasmic pools, such as NFAT, NF-κB, and the Stat transcription factors, are activated by T cell receptor signaling or cytokine receptor engagement (18–20). These transcriptional regulators, in turn, govern the induction or repression of subset-specific proteins such as the Brachyury-related T-box transcription factor T-bet in developing Th1 cells, and GATA-3 or c-maf in their Th2 counterparts (15, 21–23).
For these and other developmentally related genes, epigenetic modifications are a second layer of regulating expression (24–26). Thus, increases or de novo induction of posttranslational histone modifications favorable to transcription (e.g., histone acetylation, and methylation of certain residues in histone H3) have been correlated with activation of IFN-γ and the Th2 cytokine genes in Th1 and Th2 cells, respectively (27, 28). There is little direct evidence about subsequent events at cytokine gene loci, these epigenetic modifications enhance the ability of proteins mediating gene transcription to access the promoter and increase rates of initiation at other genes (25, 29, 30). DNA methylation may serve as one means of repressing IFN-γ expression in fully differentiated Th2 cells and, conversely, silencing the type 2 cytokine genes in Th1 cells (31–33). Experimentally induced decreases in methylation of the IFN-γ gene in a population of Th2 cells also suggested that CpG methylation of the IFN-γ locus inhibits expression of this cytokine (31–33). Also, a highly conserved regulatory element in the IFN-γ promoter was methylated in Th2 clones, and this DNA modification inhibited the binding of certain nuclear proteins (31, 34). However, enforced expression of T-bet can induce IFN-γ gene expression in populations of CD4 T cells which have undergone multiple cycles of effector development under Th2 conditions, and in Th2 clones which ordinarily are terminally differentiated (23). Furthermore, the IFN-γ promoter is methylated in a significant fraction of IFN-γ-expressing T cells (35, 36). Because Th2 cells ordinarily cannot override their fate, the mechanisms by which T-bet can reverse IFN-γ silencing are of significant interest. However, nothing is known about the mechanisms by which T-bet achieves the effect. In the present study, we have used a human T cell line to uncover evidence that T-bet acts through a conserved regulatory element in the IFN-γ promoter, which contains a T-box half-site, and that T-bet transactivates this sequence despite complete CpG methylation. This capacity to override promoter methylation involves a novel mechanism in which T-bet leads to decreased mSin3a at a methylated, MeCP2-bound IFN-γ promoter.
Materials and Methods
Plasmids, Cell Lines, and Transfections. The human IFN-γ promoter (–565, +64)-luciferase (P1P2) and proximal element (–70, +44)-luciferase (pIFN-γ) plasmids have been described (37). T-bet-GFP-R, and C/EBPβ (wild-type and transactivation-deficient mutant) expression constructs were obtained from L. Glimcher (Harvard University, Boston) and L. Sealy (Vanderbilt University, Nashville, TN), respectively. A SalI–XhoI T-bet cDNA fragment purified from T-bet-GFP-RV (23) was subcloned into pET-28, pcDNA3, and the MiT retrovirus vector (38). T-box consensus/mutant trimer and T-box IL2/mutant trimer oligonucleotides were subcloned into the luciferase-encoding plasmid pGT81 (Promega). The oligos of pIFN-γ mutant monomer and dimer were subcloned into the SnaBI site of the pIFN-γ plasmid vector. All constructs were reconfirmed by restriction analyses and DNA sequencing. Jurkat T cells were cultured exactly as described (37, 39). A T cell variant stably expressing T-bet was created by infection of Jurkat with retrovirus-containing supernatant collected 48 h after transfection of φNX amphitropic packaging cells with T-bet-MiT retrovector and centrifugation in the presence of polybrene (8 μg/ml) as described (39). Thy1.1+ cells were purified by positive selection using anti-CD90.1 (Thy1.1)-phycoerythrin (PE) antibody and anti-PE magnetic beads (Miltenyi Biotec). The purity of the selected cells was confirmed by FACS and Western blotting.
Antibodies and Cytokine Reagents. For chromatin immunoprecipitations (ChIPs), antibodies specific to MeCP2, acetylated histone H4, dimethyl-histone H3(lys4) (all from Upstate Biotechnology), T-bet (clone 4B10), mSin3A-K20, or equivalent amount of control IgG (all from Santa Cruz Biotechnology), were used. All fluorochrome-conjugated mAbs were from BD-Pharmingen.
Transient Transfections and Promoter Assays. The indicated expression and reporter plasmids, along with a constitutive β-galactosidase reporter, were transfected into Jurkat T cells by electroporation exactly as described (37). Cells were divided equally 24 h after transfection and then incubated 18 h, without stimulation or with PMA (50 ng/ml) and ionomycin (1 μg/ml). Promoter activity was measured by luciferase assays as described (40), normalizing to transfection efficiency according to the level of β-galactosidase. Mean values of three or more independent transfection experiments are shown. Complete methylation of CpG residues of the P1P2 and pIFN-γ plasmids before transfection was generated in vitro by using Sss1 methylase (41) (New England Biolabs, Beverly, MA) at 37°C for 1 h according to manufacturer's instructions, and verified by determination of cleavage efficiency using methylation-sensitive and -insensitive restriction enzymes.
EMSAs. To generate recombinant T-bet protein for mobility shift assays, coupled cell-free in vitro transcription and translation were performed by using the plasmid pET28b-Tbet and TNT Quick-Coupled Transcription/Translation Systems (Promega) according to manufacturer's instructions, and validated by Western blotting. Oligonucleotides for EMSAs were end-labeled with [γ-32P]ATP (>6,000 Ci/mmol; NEN Life Sciences; 1 Ci = 37 GBq) by using T4 polynucleotide kinase. Binding reactions (15 μl) for EMSA contained 2 μg poly(dI-dC) (Amersham Pharmacia), >25,000 cpm of probe, and incubation buffer (10 mM Hepes, pH 7.9/100 mM NaCl/10% glycerol/0.5 mM MgCl2/1 mM DTT). After the products of coupled in vitro transcription/translation reactions (5 μl) were preincubated with binding buffer for 15 min, 50-ng unlabeled competitor DNAs were added for 15 min, and radiolabeled DNA probe was added to the reaction for an additional 15 min, with all steps performed on ice. Complexes were then separated on native 4% polyacrylamide gels, followed by autoradiography.
RT-PCR and Western Blotting. RNAs were extracted from Jurkat cells by using TRIzol reagent (Invitrogen). cDNA synthesis was performed by the SuperScript First-Strand Synthesis System for RT-PCR (Life Technologies). The primers used for IFN-γ amplification were 5′-GCATCGTTTTGGGTTCTCTTGGCTGTTACTGC-3′ (forward) and 5′-CTCCTTTTTCGCTTCCCTGTTTTAGCTGCTGG-3′ (reverse). These primers were used in amplifications of 35 cycles of 1 min at 94°C; 1 min at 58°C; and 2 min at 72°C. For GAPDH amplification, the primers used were 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse), and the PCR consisted of 23 cycles of 1 min at 94°C; 1 min at 48°C; and 1 min at 72°C. For Western blotting, whole cell, cytoplasmic, and nuclear extracts were prepared as described (39, 40). Proteins (25 μg per lane) were separated by SDS/PAGE, transferred to poly(vinylidene difluoride) membranes, and probed with antibodies against T-bet, C/EBPβ, mSin3a, or MeCP2 according to the manufacturer's recommendations.
Southern Blots and Analyses of IFN-γ Promoter Methylation. DNAs were isolated with lysis buffer containing 100 mM Tris·Cl (pH 8.5), 5 mM EDTA (pH 8.0), 0.2% SDS, 200 mM NaCl, and 200 μg/ml Proteinase K, incubated overnight at 55°C, precipitated with isopropanol, and washed with ethanol. DNA samples (10–20 μg) were digested by restriction enzymes (5 units per μg of DNA) and then separated on 0.8% agarose gel; the enzyme activities were verified by parallel digestions of plasmid DNA (unmethylated) containing the relevant sites. After electrophoresis, DNA was transferred and UV cross-linked to nylon membranes that were then probed with a 32P-labeled 345-bp IFN-γ promoter fragment followed by autoradiography.
ChIP Assays. Jurkat T cells were incubated (10 min at room temperature) in 1% formaldehyde added to the tissue culture medium, which was then quenched with glycine. Nuclei were then incubated with micrococcal nuclease (Boehringer) (25 units/ml) to generate chromatin fragments with average lengths of 100–500 bp. Sheared chromatin was immunoprecipitated by using specific antibodies or control IgG at 4°C overnight followed by immobilized protein A presaturated with sheared salmon sperm DNA (2 h at 4°C). These protein A agarose bead-bound complexes were pelleted and washed extensively by using low-salt buffer, high-salt buffer, LiCl buffer, and twice with 1× 0.1 M Tris·Cl, pH 7.5/10 mM EDTA buffer. DNA–protein complexes were eluted from the immunoprecipitating antibody with 0.1 M NaHCO3 containing 1% SDS, followed by incubation at 65°C overnight in 200 mM NaCl to reverse cross-links. DNAs purified by proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation were assayed by PCR amplification using IFN-γ promoter primers (forward, 5′-CCAGTCCTTGAATGGTGTGAAG-3′; reverse, 5′-GCTGATCAGGTCCAAAGGAC-3′).
Results
Sequence-Specific Binding and Function of T-bet. The T-box transcription factors known to bind to DNA have a consensus nucleotide sequence for their binding sites (23, 42), a motif present in the 5′-flanking sequence of the IL-2 gene used to clone T-bet (Fig. 6A, which is published as supporting information on the PNAS web site) (23). To test whether T-bet functions as a sequence-specific transcription factor, we determined whether T-bet transactivates through the consensus T-box half-site in a manner dependent on the consensus residues and tested if this property is concordant with the effect of T-bet on the IL-2-derived T-box. In transfection assays using oligomerized T-box consensus and T-box IL-2 linked to a minimal promoter and reporter gene (pGT81-Tbox consensus.trimer-luc., pGT81-Tbox IL2.trimer-luc), T-bet transactivated the consensus T-box element >30-fold. When key residues of the T-box motif were mutated in this construct (pGT81-Tbox consensus mutant.trimer-luc, in which the CAC was mutated to CGT and the GTGT was mutated to GTAC), the T-box consensus mutant completely lost the ability to be activated in trans by T-bet (Fig. 6B). In sharp contrast to this finding with the T-box consensus sequence, T-bet did not transactivate the IL-2-derived T-box (Fig. 6B). Moreover, T-bet remained unable to transactivate when the IL-2 promoter T-box motif was mutated to make it more similar to the consensus T-box site (AGTGT to GGTGT) (Fig. 6B). These data show that T-bet transactivation is highly sequence-specific and depends on the precise T-box sequence, so that apparent T-box sites may not confer activity in cis.
The specificity suggested that T-bet interacts directly with T-box DNA, but there is scant biochemical evidence of such binding, which could require interaction with adjacent proteins. In mobility shift assays (EMSA) performed by using the consensus T-box site, no difference in pattern could be detected when comparing extracts of resting or activated Th1 and Th2 cells, T-bet overexpressing Jurkat T cells vs. wild-type cells, or 293 cells transfected with T-bet expression vector vs. empty vector controls (data not shown). However, a very weak but sequence-specific complex was detected when we programmed cell-free in vitro transcription/translation reactions with plasmid encoding T-bet and used these reticulocyte lysates for EMSA in comparison to controls (Fig. 6C). These findings indicate that T-bet binds to the consensus site directly albeit weakly.
T-bet Transactivates a Highly Conserved T-Box Half-Site in the IFN-γ Promoter. We observed previously that T-bet can transactivate a limited portion of the IFN-γ promoter (–110, +64) to an extent quantitatively similar to more extensive promoter constructs [(–565, +64), (–445, +64)], albeit at lower absolute levels of reporter gene expression (37). Moreover, a promoter fragment lacking the 5′-flanking (–1.2 and –1.3 kb) T boxes was sufficient to direct Th1-specific expression of a reporter gene in transgenic mice (37). A highly conserved sequence in the proximal IFN-γ promoter region (34, 43), previously termed “prox IFN-γ” (pIFN-γ), includes a potential T-box half-site (Fig. 1A). This proximal promoter element as a dimer (pIFN-γ.-luc, –70, –44) was strongly transactivated by T-bet (Fig. 1B). Mutants of the pIFN-γ.dimer-luciferase reporter constructs in which the site of “TGTG” in one (mutant 1, M1) or both (mutant 2, M2) of the monomers (sequence of –70 to –44) was mutated (Fig. 1 A) were then analyzed in transfection assays. pIFN-γ M1 exhibited decreased but partial ability to be transactivated by T-bet in comparison to the wild-type dimeric pIFN-γ construct, whereas pIFN-γ M2 could not be transactivated (Fig. 1B). Together with a mutational analysis showing that the same element mediated T-bet transactivation of the mouse IFN-γ promoter (44), also independent from full T-box consensus sequences further upstream, these findings indicate that this highly conserved half-site is a cis-acting element in T-bet transactivation.
Fig. 1.
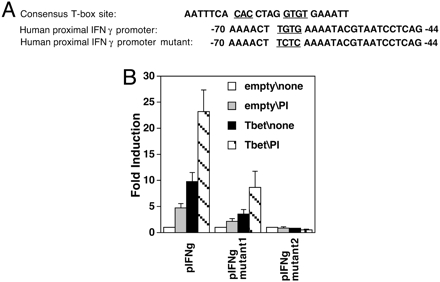
T-bet transactivates a highly conserved T-box half site in the IFN-γ promoter and induces IFN-γ expression. (A) Sequences of consensus T-box site and the conserved proximal IFN-γ promoter element or its mutant. (B) Reporter assays of T-bet or empty vector cotransfection with IFN-γ promoter element, or its mutants M1 and M2, were performed as in Fig. 6. After transfection, the cells were unstimulated or treated with PMA and ionomycin as indicated. Shown are the mean (± SEM) data from three independent experiments, each sample normalized to transfection efficiency by using β-galactosidase.
IFN-γ Promoter Recruitment of T-bet and Histone Modification. When levels of IFN-γ mRNA were assayed in Jurkat T cells transfected with T-bet expression vector or a control, short-term T-bet expression induced IFN-γ gene expression even when cells were unstimulated, and enhanced this expression in cells activated with PMA and ionomycin (Fig. 2A). Western blots probed with anti-T-bet antibodies showed that there is no endogenous T-bet expression when the cells were unstimulated, but PMA plus inonomycin induced T-bet expression in the line of parental Jurkat cells used in this work (data not shown). ChIP experiments with cells transduced with empty vector or T-bet, and left unstimulated or activated with PMA and ionomycin, were performed to test whether T-bet is recruited to the promoter region. The results showed that transfected T-bet associated with the endogenous IFN-γ promoter in the absence of T cell activation (Fig. 2B); this recruitment was further enhanced by the T cell activation. Acetylation of histone H4 and methylation of lysine 4 residue of histone H3 (K4) have been correlated with transcriptional activation. To test the effect of T-bet on these modifications, independent of other differentiation events, we measured the histone acetylation and methylation status of the IFN-γ promoter region as a function of T cell activation and levels of T-bet expression. ChIP assays revealed that T-bet induced increases in both H4 acetylation and, more strikingly, H3(K4) methylation in IFN-γ promoter chromatin (Fig. 2C). However, maximal increases were achieved when increased levels of T-bet were coupled with T cell activation.
Fig. 2.

T-bet recruitment to the endogenous IFN-γ promoter and enhancement of histone modifications. (A) T-bet transactivation of the endogenous IFN-γ gene. Shown are representative results of RT-PCR analyses of IFN-γ and GAPDH mRNAs in Jurkat cells. Cells were nontransfected (first two lanes) or subjected to short-term transfection with T-bet expression vector, followed by stimulation with nothing or PMA and ionomycin overnight, as indicated. RT-PCR of IFN-γ and GAPDH mRNAs were performed within the linear range of amplification, and products were detected by Southern blot hybridizations using internal oligonucleotide probes. (B and C) Jurkat T cells, stably infected with either an empty retrovector or one directing constitutive T-bet expression, were divided equally and stimulated with nothing or the combination of PMA and ionomycin as indicated. These cell populations were then used for ChIPs with antibodies specific for T-bet or IgG negative control or PCR from preimmunoprecipitation chromatin (input) (B) or anti-acetyl-histone H4, antidimethyl-histone H3(K4), or nonspecific IgG (C), as indicated. Western blots documented activation-induced expression of low levels of T-bet in the empty vector-infected cells and constitutive expression of T-bet in the transduced population (data not shown). In each case, similar results were obtained in short-term transfections with IRES-Thy1.1 expression vector plasmids followed by MACS selection of positive cells and ChIPs (data not shown).
T-bet Transactivates a Methylated IFN-γ Promoter. Previous investigations have shown that the highly conserved pIFN-γ regulatory element is completely methylated in Th2 clones and suggested that this methylation suppressed IFN-γ gene expression (31). However, T-bet activated the IFN-γ gene when transduced into either Th2 clones or primary Th2 populations (23), suggesting that T-bet can activate high-level IFN-γ gene expression despite the repressive epigenetic effect of CpG methylation at promoters. Because cells with complete methylation of the pIFN-γ CpG in the endogenous IFN-γ locus were needed for further mechanistic analyses and tumor cell lines exhibit increased methylation of the DNA in many genes, we evaluated the extent of methylation at the proximal conserved element of the IFN-γ promoter in Jurkat T cells. Sequencing of the IFN-γ promoter DNA in Jurkat showed that no mutation was present at the evolutionarily conserved site for the methylation-sensitive endonuclease SnaBI, previously reported as methylated in Th2 clones (Fig. 3A). No detectable cleavage at this CpG sequence (SnaBI site) of the IFN-γ gene occurred, indicating complete methylation, and stable overexpression of T-bet did not alter this methylated state (Fig. 3B). Thus, T-bet transactivated the endogenous IFN-γ promoter and increased the extent of its histone H4 acetylation in the Jurkat T cells despite complete CpG methylation. Further to test whether T-bet can activate via fully methylated CpGs in the pIFN-γ regulatory element, IFN-γ promoter reporters were methylated to completion and transfected into Jurkat T cells. After Sss1 methylase treatment and confirmation of complete loss of SnaBI cleavage, both the P1P2-luc (–565, +64) and pIFN-γ-luc (–70, –44) constructs were still activated in trans by T-bet (Fig. 3C), albeit with a modest decrease in efficiency.
Fig. 3.

T-bet transactivates through a methylated IFN-γ promoter. (A) Schematic of the conserved IFN-γ promoter element (100% identity mouse–human). (B) Complete methylation of the IFN-γ promoter SnaB1 site in the proximal conserved element. Shown are Southern blot analyses in which DNAs from control (empty vector-transduced) and T-bet overexpressing Jurkat T cells, stimulated with PMA and ionomycin overnight where indicated, were digested with either with HindIII alone or HindIII plus SnaBI. (C) T-bet activation of unmethylated and methylated IFN-γ promoters in trans. pcDNA3-T-bet, or the empty vector, and a constitutive β-galactosidase reporter were transfected into T cells along with luciferase reporter plasmids as indicated [IFN-γ promoter reporter (P1P2-luc, –565, +64), unmethylated or Sss1-methylated; or proximal IFN-γ promoter element (pIFN-γ-luc, –70, +44), unmethylated or methylated]. Samples were processed as in Fig. 1. Shown are the mean (± SEM) β-galactosidase-normalized luciferase activities from three independent experiments.
T-bet Collaborates Functionally with C/EBP. The T-box half site in the proximal element is located immediately upstream of a composite C/EBP-AP-1-ATF motif, which contains the CpG subject to DNA methylation (34, 43). Activated T cells, especially those of the Th2 subset, express an array of b-Zip transcription factors, including C/EBPβ (45). To test how this protein would influence T-bet, C/EBPβ, or a C/EBPβ mutant lacking a transactivation domain was transfected into Jurkat cells along with T-bet and reporter constructs. A larger IFN-γ promoter [(–565, +64)] and the conserved T-bet binding region [(–70, –44)] were activated by transfection of C/EBPβ, but not the mutated C/EBPβ (Fig. 4A). More than 30-fold induction was observed when C/EBPβ and T-bet were cotransfected into the cells, whereas the C/EBPβ mutant was unable to collaborate with T-bet and appeared to inhibit T-bet transactivation of the IFN-γ promoter instead (Fig. 4B). When the IFN-γ promoter and pIFN-γ-luc reporters were methylated in vitro and transfected along with combinations of C/EBPβ, C/EBPβ mutant, and T-bet, T-bet synergized with C/EBPβ despite complete methylation of the IFN-γ promoter. Note that the decrease in transactivation (fold induction) noted when T-bet was tested on its own was reversed when the T-box protein could cooperate with a b-Zip transcription factor (C/EBP) at the highly conserved composite element. Collaboration of T-bet with C/EBPβ was also observed at the fully chromatinized promoter methylated at the proximal conserved element, inasmuch as short-term overexpression of T-bet and C/EBPβ increased expression of the endogenous IFN-γ gene in the Jurkat T cells (Fig. 4C). C/EBPβ alone did not induce IFN-γ gene expression, but C/EBPβ collaborated with T-bet to increase IFN-γ mRNA to a level greater than that observed with T-bet alone (Fig. 4C). The specific role of C/EBPβ in developing Th1 or Th2 cells may be complex, and overexpression of this protein in cells lacking T-bet may decrease expression of an IFN-γ gene of unknown methylation status (46). These data show that a b-Zip transcription factor can enhance T-bet function at the methylated IFN-γ promoter.
Fig. 4.

Collaboration of T-bet with C/EBPβ in IFN-γ promoter transactivation. Jurkat T cells were cotransfected with pcDNA3-Tbet, pcDNA3-C/EBPβ, or pcDNA3-C/EBPβ mutant along with a constitutive β-galactosidase reporter. Shown are the results (mean ± SEM from three independent experiments) of transactivation assays using the indicated fusions of promoter sequences with a luciferase reporter, either without (A) or with (B) complete methylation of reporter plasmid DNA in vitro. The transactivation of promoter activity is presented as “fold induction” (normalized activity in transcription factor-transfected cells/empty vector controls, after normalizing to β-galactosidase activity programmed by a constitutive lacZ reporter construct). (C) Collaborative induction of the endogenous IFN-γ gene. T cells were transfected with the indicated combinations of T-bet (or its empty vector) along with empty vector, C/EBPβ, or the transactivation-defective C/EBPβ mutant as indicated. RT-PCR analyses of IFN-γ and GAPDH mRNAs were then performed within the linear range of amplification as in Fig. 1C. Shown is the result of one experiment representative of three with the same result.
T-bet Decreases mSin3a but Not MeCP2 Density at the IFN-γ Promoter. To investigate the ability of T-bet to trans-activate efficient IFN-γ gene expression despite methylation of the conserved element or the entire IFN-γ promoter, ChIPs were performed by using antibodies against MeCP2 and mSin3a (5–11) (Fig. 5A). MeCP2 displacement has been identified as one mechanism by which gene silencing can be bypassed without changing the DNA methylation status of a locus (47–49), but MeCP2 loading of the IFN-γ promoter did not decrease overexpression of T-bet (Fig. 5A, third and fourth lanes vs. first and second). Surprisingly, T cell activation led to MeCP2 recruitment to the IFN-γ promoter (first vs. second lanes) even though MeCP2 protein levels in the nucleus were little affected by T cell activation or T-bet (Fig. 5B). In sharp contrast to this finding, T cell activation decreased mSin3a at the promoter (Fig. 5A). Because T cell activation induced T-bet but also a variety of other transcriptional pathways, similar ChIPs were performed at the same time by using short-term transduction to achieve constitutive T-bet expression in the absence of T cell activation (Fig. 5). The results showed decreased mSin3a at the promoter as a consequence of T-bet expression. Thus, the ability of T-bet to counter the repressive influences of CpG methylation at the IFN-γ promoter and activate expression of this cytokine gene is associated with corepressor dissociation from this site.
Fig. 5.
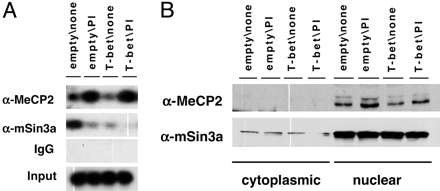
Dissociation of mSin3a but not MeCP2 from the methylated IFN-γ promoter. Shown are T cells transduced with empty retrovirus or T-bet-IRES-Thy1.1. virus, selected for Thy1.1-positive cells, and treated with PMA plus ionomycin (PI) or not as indicated. Cells (the same polyclonal populations as in Fig. 2) were then used for ChIP (A) or fractionated into cytoplasmic and nuclear extracts to use in Western blots (B), in each case using antibodies against MeCP2 or mSin3a as indicated. Separate controls documented activation-induced expression of T-bet in the empty vector-infected cells and constitutive expression of T-bet in the transduced population (data not shown).
Discussion
The data presented here show that a master regulator of IFN-γ gene expression, T-bet, is recruited to the promoter of the IFN-γ gene, activating the promoter in trans and collaborating with C/EBPβ when the conserved DNA element with which it specifically interacts is fully methylated. ChIPs using T cells, either activated or engineered to express elevated levels of T-bet without T cell activation, provide evidence of a previously undescribed mechanism leading to this ability to overcome the repressive effect of methylation. Instead of leading to loss of CpG methylation or dissociation of a methyl-CpG-binding protein loaded at this promoter, MeCP2, T-bet caused decreased density of mSin3a, a corepressor identified as the link between MeCP2 and later steps in gene silencing (3, 6).
Gene silencing by DNA methylation, and relief of this repressive effect, are fundamental processes in cellular differentiation (1–3). The present findings represent an example of selective dissociation of the corepressor while retaining promoter interaction with the methyl-DNA-binding protein. Other mechanisms leading to derepression of a methylated gene have been reported, but each differs from the mechanism reported here (49). An association of a DNA methyltransferase, DNMT1, with MeCP2 reinforces the epigenetic imprint during the cell cycle (50, 51). However, the methylcytosine is sometimes lost during DNA replication, thereby terminating the repressive effect in a daughter cell (49, 52). A substantial fraction of activated, IFN-γ-producing CD8 T cells have extensive methylation of the promoter and its conserved element (35). This finding indicates that, although some loss of cytosine methylation does occur during growth and differentiation of T cell populations, this mechanism is only one of several that are used to achieve high-level IFN-γ gene expression.
Loss of interaction between the modified DNA and a Me-CpG-binding protein is an alternative mechanism that contributes to relieving the silencing imposed by promoter methylation (47, 48). At least two fundamentally different complexes used to mediate silencing have been identified, MeCP1/MBD2 and MeCP2/mSin3a (5–11). Functional distinctions between MeCP2 and MBD2/MeCP1 can be inferred from differences in their patterns of MeCpG binding and the phenotypes of the respective MBD protein-deficient mice (12, 13). The first example of regulated relief of MeCP2-mediated repression involves the gene encoding brain-derived neurotrophic factor (BDNF), which is subject to silencing by methylation and activation by neuronal depolarization. Although this stimulus led to decreased promoter methylation, ChIP analyses showed evidence of MeCP2 dissociation from the BDNF promoter as an additional mechanism contributing to gene activation (47, 48). MeCP1, formed by the association of the methyl-DNA-binding protein MBD2 with NuRD/Mi-2 and HDACs (1 or 2), represents a second silencing complex (8, 10). Evidence of an analogous dissociation mechanism has been presented for MBD2 at the gene encoding a Th2 cytokine IL-4 (53). MBD2-deficient T cells were found to exhibit derepression of the IL-4 gene so that 8% of cells became IL-4 producers when grown under conditions that promote Th1 differentiation and ordinarily block Th2 development. Enforced expression of a master regulator of Th2 differentiation, GATA3, followed by ChIP assays indicated that forcing IL-4 gene activation in the Th1-promoting environment decreased the association of MBD2 with sequences in intron 2 and a conserved 5′-flanking regulatory element (53). In a manner analogous to the findings presented here, this process was effected despite complete methylation of these regulatory elements and was accompanied by increased histone acetylation at the sequences assayed. However, the status of events at the IL-4 promoter is not known, and the relationship of these findings to regulatory events impacting IFN-γ gene activation was not investigated.
The IFN-γ promoter region studied here is sufficient to direct Th1-specific gene expression (37), and promoter regions are the sites at which DNA methylation effects gene silencing most consistently and potently. However, two other conserved regulatory elements have recently been reported, and maximal levels of IFN-γ gene expression may depend on promoter interactions with these sites (54). A 5′-flanking site, (IFNg)CNS-1, was activated by T-bet in trans but was demethylated to an equivalent degree in Th1 and Th2 clones, so the functional role of methylation was not analyzed further (55). An IFNgCNS-2 site ≈17.4 kb downstream from the IFN-γ promoter contained an elevated density of histone modifications favorable to increased gene transcription (acetylated H3 and H4 as well as K4-methylated H3) (56), but its function, methylation status, transactivation by T-bet, and interactions with the promoter are not yet known.
Although naive and activated, uncommitted CD4 T cells can turn on high levels of IFN-γ transcription, the potential to activate this gene is lost during successive cycles of T cell receptor stimulation and growth under conditions promoting Th2 differentiation (15, 24, 57). Early in Th2 differentiation, the major mechanism by which the potential to turn on the IFN-γ gene appears to be the inhibition of expression of several cytokine receptor chains that signal T-bet induction (57). However, prior work showed that, in long-term Th1 and Th2 clones, this restriction of potential gene expression correlated with the establishment of CpG methylation at a conserved regulatory element in the IFN-γ promoter (31). More recently, an unexplained surprise was posed by the discovery that enforced expression of T-bet overcame these barriers and drove high-level IFN-γ production in a substantial fraction of the transduced Th2 cells (23). Furthermore, a significant portion of IFN-γ-expressing Th1 or CD8 T cells exhibit methylation of the promoter, and T-bet drives IFN-γ activation under nonpermissive Tc2 conditions in CD8 T cells (23, 35). Such findings suggest that T-bet, or a related T-box protein, eomesodermin (58), may also override promoter methylation in CD8 T cells. As such, the mechanism revealed herein may be central to the role of T-bet as a master regulator.
Supplementary Material
Acknowledgments
We thank S. Brandt for helpful discussions and L. Glimcher for generous gifts of reagents. This work was supported by National Institutes of Health Grant AI49460; core support was provided through Center Grants CA68485 and DK20593.
Author contributions: Y.T. and M.B. designed research; Y.T. performed research; T.A. contributed new reagents/analytic tools; Y.T. and M.B. analyzed data; Y.T., T.A., and M.B. wrote the paper; and T.A. provided editorial input.
Abbreviations: Th, T helper; ChIP, chromatin immunoprecipitation.
L.G.H. has equity in and is on the Corporate Board of the Bristol-Myers Squibb Company and has equity in and is a paid consultant for Healthcare Ventures SAB and Mannkind Corporation, a biopharmaceutical company focused on the development and commercialization of treatments for diseases including cancer and autoimmune diseases.
References
- 1.Razin, A. (1998) EMBO J. 17, 4905–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bird, A. P. & Wolffe, A. P. (1999) Cell 99, 451–454. [DOI] [PubMed] [Google Scholar]
- 3.Lorincz, M., Schubeler, D. & Groudine, M. (2001) Mol. Cell. Biol. 21, 7913–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis, J. D., Meehan, R., Henzel, W., Maurer-Fogy, I., Jeppesen, P., Klein, F. & Bird, A. (1992) Cell 69, 905–914. [DOI] [PubMed] [Google Scholar]
- 5.Nan, X., Campoy, F. & Bird, A. (1997) Cell 88, 471–481. [DOI] [PubMed] [Google Scholar]
- 6.Nan, X., Ng, H., Johnson, C., Laherty, C., Turner, B., Eisenman, R. N. & Bird, A. (1998) Nature 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 7.Hendrich, B. & Bird, A. (1998) Mol. Cell. Biol. 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng, H., Zhang, Y., Hendrich, B., Johnson, C., Turner, B., Erdjument-Bromage, H., Tempst, R., Reinberg, D. & Bird, A. (1999) Nat. Genet. 23, 58–61. [DOI] [PubMed] [Google Scholar]
- 9.Ng, H., Jeppesen, P. & Bird, A. (2000) Mol. Cell. Biol. 20, 1394–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang, Y., Ng, H., Erdjument-Bromage, H., Tempst, P., Bird, A. & Reinberg, D. (1999) Genes Dev. 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saito, M. & Ishikawa, F. (2002) J. Biol. Chem. 277, 35434–35439. [DOI] [PubMed] [Google Scholar]
- 12.Hendrich, B., Guy, J., Ramsahoye, B., Wilson, V. A. & Bird, A. (2001) Genes Dev. 15, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guy, J., Hendrich, B., Holmes, M., Martin, J. & Bird, A. (2001) Nat. Genet. 27, 322–326. [DOI] [PubMed] [Google Scholar]
- 14.Abbas, A. K., Murphy, K. M. & Sher, A. (1996) Nature 383, 787–793. [DOI] [PubMed] [Google Scholar]
- 15.Murphy, K. M., Ouyang, W., Farrar, J. D., Yang, J., Ranganath, S., Asnagli, H., Afkarian, M. & Murphy, T. L. (2000) Annu. Rev. Immunol. 18, 451–494. [DOI] [PubMed] [Google Scholar]
- 16.Ho, I. C. & Glimcher, L. H. (2002) Cell 109, S109–S120. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan, D. H., Shankaran, V., Dighe, A., Stockert, E., Aguet, M., Old, L. J. & Schreiber, R. D. (1998) Proc. Natl. Acad. Sci. USA 95, 7556–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao, A., Luo, C. & Hogan, P. G. (1997) Annu. Rev. Immunol. 15, 707–747. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh, S., May, M. & Kopp, E. (1998) Annu. Rev. Immunol. 16, 225–260. [DOI] [PubMed] [Google Scholar]
- 20.O'Shea, J. J. (1997) Immunity 7, 1–10. [DOI] [PubMed] [Google Scholar]
- 21.Zheng, W. & Flavell, R. A. (1997) Cell 89, 587–596. [DOI] [PubMed] [Google Scholar]
- 22.Ouyang, W., Lohning, M., Gao, Z., Assenmacher, M., Ranganath, S., Radbruch, A. & Murphy, K. M. (2000) Immunity 12, 27–37. [DOI] [PubMed] [Google Scholar]
- 23.Szabo, S. J., Kim, S. T., Costa, G., Zhang, X., Fathman, C. G. & Glimcher, L. H. (2000) Cell 10, 655–669. [DOI] [PubMed] [Google Scholar]
- 24.Murphy, K. M. & Reiner, S. L. (2002) Nat. Rev. Immunol. 2, 933–944. [DOI] [PubMed] [Google Scholar]
- 25.Jenuwein, T. & Allis, C. D. (2001) Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 26.Ansel, K. M., Lee, D. U. & Rao, A. (2003) Nat. Immunol. 4, 616–623. [DOI] [PubMed] [Google Scholar]
- 27.Avni, O., Lee, D., Macian, F., Szabo, S. J., Glimcher, L. H. & Rao, A. (2002) Nat. Immunol. 3, 643–651. [DOI] [PubMed] [Google Scholar]
- 28.Fields, P. E., Kim, S. T. & Flavell, R. A. (2002) J. Immunol. 169, 647–650. [DOI] [PubMed] [Google Scholar]
- 29.Urnov, F. D. & Wolffe, A. P. (2001) Oncogene 20, 2991–3006. [DOI] [PubMed] [Google Scholar]
- 30.Narlikar, G., Fan, H. Y. & Kingston, R. E. (2002) Cell 108, 475–487. [DOI] [PubMed] [Google Scholar]
- 31.Young, H. A., Ghosh, P., Ye, J., Lederer, J., Lichtman, A., Gerard, J., Penix, L., Wilson, C., Melvin, A., McGurn, M., et al. (1994) J. Immunol. 153, 3603–3610. [PubMed] [Google Scholar]
- 32.Bird, J. J., Brown, D. R., Mullen, A. C., Moskowitz, N. H., Mahowald, M. A., Sider, J. R., Gajewski, T. F., Wang, C. R. & Reiner, S. L. (1998) Immunity 9, 229–237. [DOI] [PubMed] [Google Scholar]
- 33.Makar, K. W., Perez-Melgosa, M., Shnyreva, M., Weaver, W. M., Fitzpatrick, D. R. & Wilson, C. B. (2003) Nat. Immunol. 4, 1183–1190. [DOI] [PubMed] [Google Scholar]
- 34.Penix, L., Weaver, W. M., Pang, Y., Young, H. A. & Wilson, C. B. (1993) J. Exp. Med. 178, 1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitzpatrick, D. R., Shirley, K. M., McDonald, L. E., Bielefeldt-Ohmann, H., Kay, G. F. & Kelso, A. (1998) J. Exp. Med. 188, 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mullen, A. C., Hutchins, A. S., High, F. A., Lee, H. W., Sykes, K. J., Chodosh, L. A. & Reiner, S. L. (2002) Nat. Immunol. 3, 652–658. [DOI] [PubMed] [Google Scholar]
- 37.Soutto, M., Zhang, F., Enerson, B., Tong, Y., Boothby, M. & Aune, T. M. (2002) J. Immunol. 169, 4205–4212. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell, T., Hildeman, D., Kedl, R., Teague, T., Schaefer, B., White, J., Zhu, Y., Kappler, J. & Marrack, P. (2001) Nat. Immunol. 2, 397–402. [DOI] [PubMed] [Google Scholar]
- 39.Mora, A., Stephenson, L., Enerson, B., Youn, J. H., Keegan, A. & Boothby, M. (2003) J. Immunol. 171, 1891–1900. [DOI] [PubMed] [Google Scholar]
- 40.Goenka, S., Marlar, C., Schindler, U. & Boothby, M. (2003) J. Biol. Chem. 278, 50362–50370. [DOI] [PubMed] [Google Scholar]
- 41.Chuvpilo, S., Jankevics, E., Tyrsin, D., Akimzhanov, A., Moroz, D., Jha, M., Schulze-Luehrmann, J., Santner-Nanan, B., Feoktistova, E., Konig, T., et al. (2002) Immunity 16, 881–895. [DOI] [PubMed] [Google Scholar]
- 42.Muller, C. W. & Herrmann, B. G. (1997) Nature 389, 884–888. [DOI] [PubMed] [Google Scholar]
- 43.Penix, L., Sweetser, M., Weaver, W., Hoeffler, J., Kerppola, T. & Wilson, C. B. (1996) J. Biol. Chem. 271, 31964–31972. [DOI] [PubMed] [Google Scholar]
- 44.Cho, J., Grigura, V., Murphy, T. L. & Murphy, K. (2003) Int. Immunol. 15, 1149–1160. [DOI] [PubMed] [Google Scholar]
- 45.Davydov, I., Krammer, P. & Li-Weber, M. (1995) J. Immunol. 155, 5273–5279. [PubMed] [Google Scholar]
- 46.Berberich-Siebelt, F., Klein-Hessling, S., Hepping, N., Santner-Nanan, B., Lindemann, D., Schimpl, A., Berberich, I. & Serfling, E. (2000) Eur. J. Immunol. 30, 2576–2585. [DOI] [PubMed] [Google Scholar]
- 47.Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., Fan, G. & Sun, Y. E. (2003) Science 302, 890–893. [DOI] [PubMed] [Google Scholar]
- 48.Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A., Griffith, E., Jaenisch, R. & Greenberg, M. E. (2003) Science 302, 885–889. [DOI] [PubMed] [Google Scholar]
- 49.Klose, R. & Bird, A. (2003) Science 302, 793–795. [DOI] [PubMed] [Google Scholar]
- 50.Fuks, F., Burgers, W. A., Brehm, A., Hughes-Davies, L. & Kouzarides, T. (2000) Nat. Genet. 24, 88–91. [DOI] [PubMed] [Google Scholar]
- 51.Kimura, H. & Shiota, K. (2003) J. Biol. Chem. 278, 4806–4812. [DOI] [PubMed] [Google Scholar]
- 52.Brunk, B., Goldhammer, D. & Emerson, C. P., Jr. (1996) Dev. Biol. 177, 490–503. [DOI] [PubMed] [Google Scholar]
- 53.Hutchins, A. S., Mullen, A. C., Lee, H. W., Sykes, K. J., High, F., Hendrich, B., Bird, A. P. & Reiner, S. L. (2002) Mol. Cell 10, 81–91. [DOI] [PubMed] [Google Scholar]
- 54.Soutto, M., Zhou, W. & Aune, T. M. (2002) J. Immunol. 169, 6664–6667. [DOI] [PubMed] [Google Scholar]
- 55.Lee, D., Avni, O., Chen, L. & Rao, A. (2004) J. Biol. Chem. 279, 4802–4810. [DOI] [PubMed] [Google Scholar]
- 56.Shnyreva, M., Weaver, W., Blanchette, M., Taylor, S. L., Tompa, M., Fitzpatrick, D. & Wilson, C. B. (2004) Proc. Natl. Acad. Sci. USA 101, 12622–12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szabo, S., Jacobson, N., Dighe, A., Gubler, U. & Murphy, K. M. (1995) Immunity 2, 665–675. [DOI] [PubMed] [Google Scholar]
- 58.Pearce, E., Mullen, A. C., Martins, G., Krawczyk, C., Hutchins, A., Zediak, V., Banica, M., DiCioccio, C., Gross, D., Mao, C. A., et al. (2003) Science 302, 1041–1043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.