

Abstract
New therapeutic strategies are urgently needed to improve clinical outcomes in patients with multiple myeloma (MM). Daratumumab is a first‐in‐class, CD38 human immunoglobulin G1κ monoclonal antibody approved for treatment of relapsed or refractory MM. Identification of an appropriate dose regimen for daratumumab is challenging due to its target‐mediated drug disposition, leading to time‐ and concentration‐dependent pharmacokinetics. We describe a thorough evaluation of the recommended dose regimen for daratumumab in patients with relapsed or refractory MM.
Daratumumab is a first‐in‐class, CD38 human immunoglobulin G1κ (IgG1κ) monoclonal antibody (mAb) that recently received accelerated approval from the US Food and Drug Administration (FDA) for use in patients with multiple myeloma (MM) who have received ≥3 lines of treatment including a proteasome inhibitor and an immunomodulatory drug, or who are double refractory to these agents, and received conditional marketing authorization from the European Medicines Agency for the treatment of adults with relapsed and refractory MM.1 Daratumumab binds with high affinity to CD38, which is ubiquitously expressed on myeloma cells. The antimyeloma activity of daratumumab is mediated through a number of pathways.1
The phase I/II first‐in‐human study of daratumumab, GEN501, evaluated doses ranging from the minimal anticipated biological effect level (0.005 mg/kg) to 24 mg/kg, administered intravenously (i.v.). The maximum tolerated dose was not reached.2 Over the range of evaluated doses, increases in area under the curve were more than dose proportional; increasing dose and repeated dosing led to decreased clearance, suggesting target‐mediated clearance.3 Therefore, a tapered dosing schedule was established. Results from GEN501 showed encouraging efficacy and a favorable safety profile with daratumumab monotherapy in patients with heavily pretreated and refractory MM.2 However, pharmacokinetic analyses from GEN501 suggested that the 8 mg/kg dose was lower than the trough threshold for target saturation.4 At the same time, efficacy analyses of the 8 mg/kg dose in the phase II SIRIUS study demonstrated a low overall response rate (ORR).4 In contrast, daratumumab 16 mg/kg demonstrated deep and durable responses while maintaining a favorable safety profile; therefore, daratumumab 16 mg/kg once weekly (QW) for 8 weeks, every 2 weeks (Q2W) for 16 weeks, and every 4 weeks (Q4W) thereafter was established as the recommended dosing schedule.4
Identification of an appropriate dose and dosing schedule is complex for daratumumab due to its target‐mediated drug disposition (TMDD), which leads to time‐ and concentration‐dependent pharmacokinetics (PK).3 Additionally, concentrations decrease over time as daratumumab is tapered from QW to Q4W dosing. Therefore, it is important to determine whether most patients can achieve efficacious concentrations after the QW 16 mg/kg dosing of daratumumab, and whether sufficient target saturation can be maintained during Q2W or Q4W dosing to reduce the risk of disease progression.
We evaluated PK and efficacy/safety data collected from GEN501 (N = 104; ClinicalTrials.gov Identifier: NCT00574288)2 and SIRIUS (N = 124; NCT01985126)4 to understand the clinical implications of the complex PK on the daratumumab dose regimen. Details on patient eligibility, study designs for GEN501 and SIRIUS, and population PK modeling are described in the Supplemental Methods, Supplemental Table 1, and Supplemental Figures 1 and 2. We first investigated the relationship between maximal trough concentration (Ctrough,max) and the primary efficacy endpoint, ORR, to identify the effective induction concentration during the intensive QW dosing. Among the tested exposure metrics, Ctrough,max had the strongest correlation with ORR (Supplemental Table 2). Second, analyses of time to progression (TTP) and duration of response (DOR) were performed to determine whether there was any association between decreases in daratumumab trough concentrations (Ctrough,delta; the reduction in trough concentration at the last dose from Ctrough,max) and the likelihood of disease progression. A PK model incorporating the drug‐target binding was developed5 and utilized to infer the pharmacodynamics of the daratumumab‐CD38 complex (i.e., target saturation over time). The exposure‐safety relationship between the peak concentration after multiple doses (Cmax) and thrombocytopenia, anemia, neutropenia, lymphopenia, or infections was assessed. Because the majority of infusion‐related reactions (IRRs) occurred during the first dose, the peak concentration after the first dose (Cmax,1st) was used to evaluate the relationship between exposure and IRRs.
ORR significantly increased with Ctrough,max (P < 0.0001) via a maximal effect (Emax) relationship (Figure 1 a), where 90% Emax on ORR ( ) was achieved at 274 μg/mL. Limited additional benefit to ORR could be obtained when Ctrough,max was above the . The Ctrough,max‐ORR relationship was virtually unchanged after adjusting for baseline disease‐state variables (Supplemental Table 3). Although the observed ORR appeared to be higher for the 8th quantile (Ctrough,max = ∼900 μg/mL), the increase was not statistically significant, as the confidence interval overlapped with the predicted ORR. Also, a hyperbolic Emax relationship was identified between daratumumab concentration and target saturation (Figure 1 b). The concentration estimated to provide 99% target saturation ( ) was 236 μg/mL, suggesting that a >99% target saturation at the end of weekly dosing may be required to induce the clinical effect. Simulations demonstrated that the 16‐mg/kg dose was the lowest tested i.v. dose at which the majority of patients (∼80%) achieved Ctrough,max above and after QW dosing (QW for 8 weeks). The 8‐mg/kg QW dose produced Ctrough,max above the and in only 40% to 50% of patients. Although the 24‐mg/kg dose increased the number of patients above the and to ∼90%, the higher i.v. dose will also produce much higher peak concentrations than the 16‐mg/kg dose and potentially compromise the safety profile.
Figure 1.
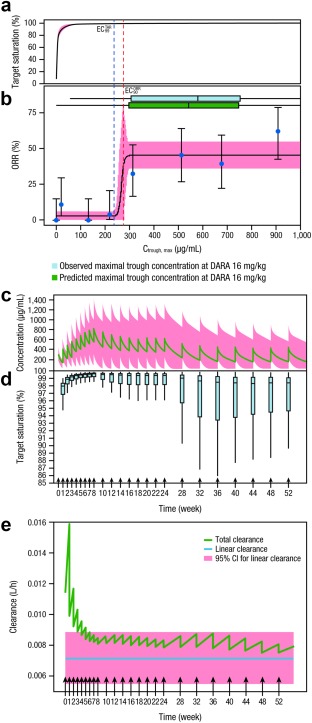
Exposure‐response relationship between daratumumab concentration and target saturation (a), and between ORR and predicted Ctrough,max (b). Representative PK profile of daratumumab (c), including boxplots for the target saturation profile of daratumumab at pre‐infusion time points for the patient population at the recommended dose and schedule (d). Total and linear clearance vs. time for the daratumumab 16‐mg/kg dose regimen (e). In Panel a, the centered curves and shaded areas represent predicted target saturation and 95% CI, respectively. In Panel b, the solid blue dots represent the proportion of responders grouped by eight quantiles of Ctrough,max and plotted at the geometric mean for each group. The bars represent the 95% CI for the proportion in each group. The centered curves and shaded areas represent predicted ORR values and 95% CIs of model‐predicted response rate, respectively. The horizontal boxplots represent the predicted (blue) and observed (green) maximal trough concentration at daratumumab 16 mg/kg. In Panel c, the green line indicates simulated population mean values and the pink shaded area indicates the 95% prediction intervals. In Panels c–e, arrows indicate the daratumumab 16‐mg/kg dose regimen: QW for 8 weeks, Q2W for 16 weeks, and then Q4W thereafter. ORR, overall response rate; Ctrough,max, maximal trough concentration; PK, pharmacokinetic; CI, confidence interval; QW, once weekly; Q2W, every 2 weeks; Q4W, every 4 weeks; , concentration estimated to provide 99% target saturation; , concentration estimated to provide 90% maximal effect on ORR; DARA, daratumumab; CI, confidence interval.
There was no significant relationship between Ctrough,delta and either TTP (P = 0.16) or DOR (P = 0.44; Supplemental Figure 3), suggesting that the decrease in concentration during less frequent dosing intervals (Figure 1 c) was unlikely to result in shorter DOR or higher risk of disease progression. This is not surprising, as the decrease in target saturation over time was minimal in the majority of patients, with a median above 98% for trough concentrations at 52 weeks (Figure 1 d), despite the decrease in daratumumab concentration over time. Further analysis demonstrated that the total clearance of daratumumab decreased over time (Figure 1 e) and approached the nonspecific clearance for IgGs by the end of the initial QW dosing period, indicating that the saturation and depletion of CD38 is achieved by the end of the QW dosing period (8 doses) and maintained over time during the Q2W and Q4W dosing intervals.
Large‐molecule mAbs for cancer treatment often exhibit TMDD, resulting in higher clearance in the presence of the target.6, 7 Following effective treatment for cancer, the amount of target or receptors will most likely decrease rapidly due to killing and depletion of tumor cells that carry the target receptors. This may explain the time‐dependent PK (i.e., decreasing clearance over time) for daratumumab and some other recently developed mAbs. The decrease in clearance over time for these mAbs may require a more frequent dosing at the beginning of the treatment to overcome the initially higher clearance of the drug, but a less frequent dosing at later times to maintain the target saturation. Therefore, QW dosing for daratumumab at the beginning of treatment helped overcome the high initial, target‐mediated clearance and rapidly established efficacious concentrations. Thereafter, although the concentration of daratumumab tended to decrease following the Q2W and Q4W dosing until reaching steady state, the concentration levels during less frequent dosing intervals were sufficient to maintain target saturation, thus reducing the risk of disease progression.
There was no apparent relationship within the investigated concentration range between Cmax,1st and IRRs, or between Cmax and thrombocytopenia, anemia, neutropenia, and lymphopenia (Figure 2 a,b). Although the overall event rate of infection appeared to increase numerically with drug exposure, this trend was not observed for grade ≥3 infections. Incidence of grade ≥3 adverse events was slightly numerically lower in patients with higher daratumumab exposure quartiles than in patients with lower exposure. These observations are consistent with the clinical data, in which no dose‐related safety signal was observed.2, 4 The safety profile of daratumumab doses >16 mg/kg has not been investigated beyond three patients treated with 24 mg/kg in the GEN501 study.
Figure 2.
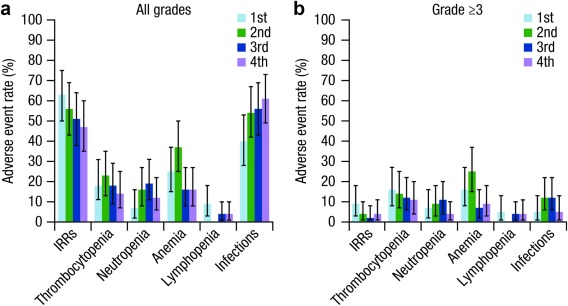
Exposure‐response relationship between adverse events and predicted Cmax (a,b). Panels a,b show the rate of adverse events of interest by exposure quartile. Cmax,1st was used as the exposure measure for analyses of IRRs. Cmax was used as the exposure measure for analyses of other adverse events. The quartiles for Cmax,1st were: 1st quartile (≤134 μg/mL), 2nd quartile (134–245 μg/mL), 3rd quartile (245–310 μg/mL), and 4th quartile (310–470 μg/mL). The quartiles for Cmax are: 1st quartile (≤270 μg/mL), 2nd quartile (270–511 μg/mL), 3rd quartile (511–907 μg/mL), and 4th quartile (907–1,840 μg/mL). IRR, infusion‐related reaction; Cmax, overall maximum concentration; Cmax,1st, peak concentration after the first infusion.
Taken together, daratumumab exhibits complex time‐ and concentration‐dependent PK. The drug exposure was strongly correlated with efficacy, but not with the safety endpoints analyzed. The target effective trough concentration ( ) and the required target saturation were identified to be 274 μg/mL and >99%, respectively, at the end of QW dosing. The currently recommended dose regimen of daratumumab (16 mg/kg; QW for 8 weeks, Q2W for 16 weeks, and Q4W thereafter) provides a balanced benefit–risk profile for i.v. administration in the relapsed or refractory MM patient population (i.e., ∼80% of patients could achieve the identified effective concentration with an acceptable safety profile). Further optimization of the PK and dose through development of new formulations and routes of delivery (e.g., subcutaneous administration) may allow more patients to attain the identified effective trough concentration without a significant increase in peak concentrations, therefore further improving the clinical benefit‐risk profile for daratumumab. Lastly, due to the depletion of the target on tumor cells over time following treatments, mAb therapies with TMDD may display decreasing clearance over time. Therefore, after an intensive induction period to overcome the initially higher clearance, less frequent dosing may be sufficient to maintain target saturation, thus reducing the risk of disease progression.
AUTHOR CONTRIBUTIONS
X.S.X. contributed to the scientific literature search, data acquisition, and interpretation and analysis. X.Y. contributed to data acquisition and interpretation and analysis. T. Puchalski contributed to data acquisition and interpretation and analysis. S.L. contributed to the accrual and treatment of patients and data acquisition. T. Plesner and H.M.L. contributed to the accrual and treatment of patients and data acquisition. P.M.V. contributed to the accrual and treatment of patients and data acquisition. K.L. contributed to data interpretation and analysis. I.K. contributed to data acquisition and interpretation and analysis. R.J. contributed to data interpretation and analysis. T.A. contributed to data acquisition and interpretation and analysis. J.J.P.R. contributed to interpretation and analysis. H.Z. contributed to interpretation and analysis. P.L.C. contributed to data acquisition and interpretation and analysis. All authors drafted and reviewed the article, approved the final version, and decided to publish this report; all authors vouch for data accuracy and completeness.
CONFLICT OF INTEREST/DISCLOSURE
X.S.X., X.Y., T. Puchalski, K.L., I.K., R.J., T.A., J.J.P.R., H.Z., and P.L.C. are employees of Janssen Research & Development, LLC. X.S.X., X.Y., T. Puchalski, R.J., J.J.P.R., H.Z., and P.L.C. own stock in Johnson & Johnson, and J.J.P.R. owns stock in Amgen. S.L. reports consultancy and research funding from Millennium, Novartis, Bristol‐Myers Squibb, Onyx, Celgene, and Janssen. H.M.L. reports honoraria from Amgen and honoraria and research funding from Genmab and Janssen. P.M.V. reports consultancy from Janssen, Millennium Pharmaceuticals: A Takeda Oncology Company, Celgene, Novartis, Array BioPharma, and Oncopeptides; and research funding from Janssen, Celgene, GlaxoSmithKline, Onyx Pharmaceuticals, and Oncopeptides. T. Plesner reports research funding from Janssen and has served on advisory boards for Janssen.
Supporting information
Supporting Information S1
Supporting Information S2
Supporting Information S3
Supporting Information S4
ACKNOWLEDGMENTS
This study was sponsored by Janssen Research & Development, LLC. The authors wish to thank the patients participating in this study and their families, as well as the global network of investigators, research nurses, study coordinators, and operations staff. Medical writing and editorial support were provided by Erica S. Chevalier‐Larsen of MedErgy (Yardley, PA, USA), and were funded by Janssen Global Services, LLC.
References
- 1. McKeage, K. & Lyseng‐Williamson, K.A. Daratumumab in multiple myeloma: a guide to its use as monotherapy in the EU. Drugs Ther. Perspect. 32, 463–469 (2016). [Google Scholar]
- 2. Lokhorst, H.M. et al Targeting CD38 with daratumumab monotherapy in multiple myeloma. N. Engl. J. Med. 373, 1207–1219 (2015). [DOI] [PubMed] [Google Scholar]
- 3. Clemens, P.L. et al Pharmacokinetics of daratumumab following intravenous infusion in relapsed or refractory multiple myeloma (MM) after prior proteasome inhibitor (PI) and immunomodulatory drug (IMiD) treatment. Clin. Lymphoma Myeloma Leuk. 15, e269–e270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lonial, S. et al Daratumumab monotherapy in patients with treatment‐refractory multiple myeloma (SIRIUS): an open‐label, randomised, phase 2 trial. Lancet 387, 1551–1560 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Yan, X. et al Target‐mediated drug disposition of daratumumab following intravenous infusion in relapsed or refractory multiple myeloma after prior proteasome inhibitors and immunomodulatory drugs: a population pharmacokinetic analysis. Blood 126, 4222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alinari, L. et al Alemtuzumab (Campath‐1H) in the treatment of chronic lymphocytic leukemia. Oncogene 26, 3644–3653 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Gibiansky, E. et al Population pharmacokinetics of obinutuzumab (GA101) in chronic lymphocytic leukemia (CLL) and non‐Hodgkin's lymphoma and exposure‐response in CLL. CPT Pharmacometrics Syst. Pharmacol. 3, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Supporting Information S2
Supporting Information S3
Supporting Information S4