

Abstract
The voltage-dependent calcium channel γ4 subunit protein, CACNG4, is closely related to the γ2 subunit, CACNG2. Both are expressed primarily in the brain and share 53% amino acid identity. The Cacng2 gene is disrupted in the stargazer mouse, with its distinctive phenotype including ataxia, frequent absence seizure episodes, and head elevation. A disruption within Cacng4 was engineered to assess its particular function. The homozygous Cacng4-targeted mutant mouse appeared normal with no ataxic gait or absence seizures, suggesting that other members of the γ subunit family might functionally compensate for the absence of CACNG4. To test this hypothesis, the targeted Cacng4 mutation was combined with alleles of Cacng2. Absence seizures were observed in combination with the stargazer 3J mutation, which itself does not have seizures, and increased seizure activity was observed in combination with the waggler allele. Furthermore, within the corticothalamic loop, where absence seizures arise, CACNG4 expression is restricted to the thalamus. Our studies show that the CACNG4 protein has seizure suppressing activity, but this effect is revealed only when CACNG2 expression is also compromised, suggesting that CACNG subunits have in vivo overlapping functions.
Keywords: stargazer mutants, absence epilepsy, γ4 expression
The voltage-dependent calcium channel is regulated by three accessory proteins: CACNB (β), CACNA2D (α2δ), and CACNG (γ). Each protein has multiple isoforms, but little is known about the individual or overlapping functions of the γ and α2δ proteins (1). The γs are an intriguing family of proteins as they appear to have the least profound effect on in vitro calcium channel function of the three molecules (1). The CACNG2 or stargazin subunit has also been shown to be a member of the transmembrane α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate (AMPA) receptor regulatory proteins and participates in AMPA receptor synaptic localization (2).
The CACNG1 molecule was first identified from skeletal muscle. Mice with targeted mutations in the skeletal muscle Cacng1 gene show a significant increase in the amplitude of peak dihydropyridine-sensitive voltage-dependent calcium channel in isolated myotubes (3). The protein mutated in stargazer mice, CACNG2, was identified based on its similarity to the CACNG1 protein (4). The Cacng gene family now includes eight members, seven of which are expressed in brain. CACNG2 is very closely related to CACNG3 and CACNG4, sharing 66% and 53% amino acid identity with each, respectively, in mouse (5–7).
From the analysis of the stargazer mouse, it is clear that a mutation in the Cacng2 gene has severe consequences. This mutant has an ataxic gait, distinctive head-tossing, and frequent absence seizures (8). There are many biochemical changes in the cerebellum of stargazer (2, 9–12) that, along with inner ear vestibular defects (13), may cause the ataxic and neck extensor movements of stargazer.
Histological and biochemical changes are also detected in the stargazer midbrain, including increased mossy fiber sprouting in the hippocampus (14), increased neuropeptide Y expression (15), altered hyperpolarization-activated channel activity in the cortical pyramidal neurons (16), and increased calcium channel activity (17). This region is most closely associated with the generation of absence seizures; in particular the corticothalamic loop encompassing the cortex, thalamus, and reticular thalamic nuclei and their interconnecting neurons (18). When the balance of excitatory and inhibitory discharges within this loop is disrupted, recurrent spike-wave discharges (SWDs) are evoked that ultimately spread throughout the cortex. These discharges are measured by electroencephalographic (EEG) recordings and occur spontaneously and frequently in the stargazer mouse.
There are three spontaneous allelic mutations in the Cacng2 gene; namely stargazer, Cacng2stg; waggler, Cacng2stg-wag; and stargazer 3J, Cacng2stg-3J (5). Each mutant shows differing degrees of severity, both at the level of Cacng2 expression and overall phenotype (5). The stargazer (stg/stg) mouse is the most severely affected, and the stargazer 3J (stg3J/stg3J) has the mildest phenotype. Stargazer and waggler have very little Cacng2 mRNA by RT-PCR real-time analysis and Northern blots, and no detectable protein by Western blot analysis (5). In contrast, stargazer 3J retains 28% of normal Cacng2 message level compared to the B6 strain, and CACNG2 protein is also present (5). Furthermore, in contrast to stargazer and waggler, stargazer 3J mutants do not have absence seizures but do exhibit ataxia, which is common to all three allelic mutants.
Cacng2 appears to be the most highly expressed of the Cacng genes in the brain (7, 19, 20). Relatively little is known about the in vivo function of the other brain-specific Cacng gene family members, and we describe here the results from targeting a mutation of the Cacng4 gene.
Materials and Methods
Mice. The stargazer, waggler, and stargazer 3J mice arose as spontaneous mutations at The Jackson Laboratory and, along with mice of the parental and control strains (A/J, MRL/MpJ-Tnfrsf6lpr/J, BALB/cJ, and C57BL/6J), continue to be maintained at The Jackson Laboratory. All animal procedures were approved by the Animal Care and Use Committee.
The stargazer mutation arose on the A/J inbred background, and has since been crossed onto a C3FeLe.B6-a (B6C3) hybrid background, retaining only a small region of A/J encompassing the stargazer mutation. The waggler mutation arose on the MRL/MpJ background and, through repeated backcrossing (>N10) is now on a primarily C57BL/6J (B6) background. The stargazer 3J mutant allele arose on the BALB/cJ inbred line and is now maintained on the B6 and B6C3 backgrounds.
RT-PCR. Brain RNA was prepared by using TRIzol (Invitrogen) and treated with DNase1 (Promega) using the manufacturers' suggested conditions. Two micrograms of RNA was transcribed with avian myeloblastosis virus reverse transciptase (Promega). PCR was performed under the following conditions: 1 min at 94°C, 2 min at 55°C, and 2 min at 72°C, for a total of 25–35 cycles. The primers were as follows: B2mF, 5′-CACGCCACCCACCGGAGAATG-3′; B2mR, 5′-GATGCTGATCACATGTCTCG-3′; Cacng2F, 5′-TCCGGAAGACGCGGACTAC-3′; Cacng2R, 5′-ATGATGTTGTGGCGTGTCTTG-3′; Cacng3F, 5′-TTGTGGAGGACCTGCTGCTT-3′; Cacng3R, 5′-TGACGCTGAGGATGGGAAAG-3′; Cacng4F, 5′-CCCATCCTCAGCACCATTCT-3′; Cacng4R, 5′-CCCGTGTTGCTGGAAATGTA-3′. The Cacng primers were selected to amplify the Cacng product spanning adjacent exons. To confirm the specificity of each primer pair, Cacng2, Cacng3, and Cacng4 cDNA clones were amplified with each primer combination. PCR products were observed only with the appropriate primer pairs.
Production of Cacng4-Targeted Mutant Mice. The targeting vector was constructed as illustrated in Fig. 1 and electroporated into ES129-R1 cells, derived from a cross between 129× 1/SvJ and 129S1/SvF1-Kitl mice. Clones were initially selected for resistance to G418 (neomycin-resistant gene integration). Correctly targeted colonies were further selected by Southern analysis, and homologous recombinant clones were injected into C57BL/6J blastocysts for implantation into pseudopregnant mice. Chimeric mice were crossed to B6 females to generate heterozygous Cacng4 mutant mice.
Fig. 1.
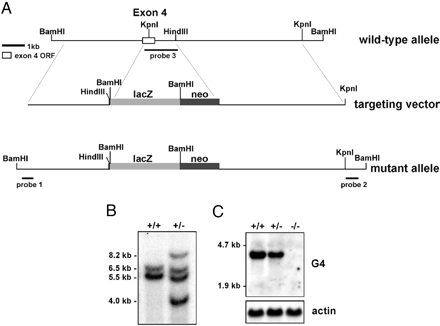
Targeted disruption of the fourth exon of Cacng4. (A) The targeting vector included a 3.5-kb genomic fragment upstream and 19 bp of exon 4 ligated in frame to lacZ, thus deleting the remaining 520 bp of the exon 4 ORF. (B) Southern blot showing DNA from ES cells digested with BamHI and HindIII, and probed with genomic DNA proximal and distal to the targeted insertion site. The genomic bands are 5.5 and 6.5 kb (+/+ lane). The targeted disruption had two additional bands of 4.0 and 8.2 kb (+/- lane). (C) Northern blot probed with Cacng4 cDNA. Lane 1, +/+ control; lane 2, heterozygous +/g4tm1; lane 3, g4tm1/g4tm1 homozygous mutant. (Lower) The β-actin control.
Southern and Northern Blot Analysis. DNA was prepared from ES cells and digested with BamHI and HindIII according to the manufacturers' protocols (Promega) and run on 0.7% agarose gels. After blotting onto Nytran Plus membrane in 0.4 M NaOH, the blot was probed in formamide/hybridization buffer at 42°C with 32P-labeled fragments from probes 1 and 2 (Fig. 1). Total RNA was prepared from adult mouse brains and run on a 1.2% agarose/formaldehyde gel (21). After blotting, the blot was hybridized with probe 3 (exon 4, Fig. 1) in formamide/hybridization buffer at 42°C. All final washes after hybridization were 0.1× SSC, 0.1% SDS at 65°C. A 1.8-kb mouse actin probe was labeled and used as a control for equal RNA loading.
β-Galactosidase Staining. Fresh embryos were fixed in 4% para-formaldehyde (PFA) in 1× PBS for 1 h at 22°C. After three washes in wash buffer (0.1 M phosphate buffer, pH 7.3/2 mM MgCl2/0.01% Na-deoxycholate/0.02% Nonidet P-40), they were incubated overnight at 37°C in a stain solution containing 1 mg/ml Bluo-Gal (GIBCO/BRL), 5 mM K-ferricyanide, and 5 mM K-ferrocyanide in wash buffer. A blue precipitate was only observed in embryos with the targeted allele, and not in +/+ mice. For adult sections, brains were fixed for 4–6 h in 4% PFA, and 100-μm sections were cut by using a vibratome. The sections were stained in 1 mg/ml X-Gal in the dye solution described above at 37°C overnight and washed in PBS. Sections were imaged on a Nikon eclipse microscope by using a Spot RT camera.
Electrode Implantation and EEG Measurements. Mice aged between 8 and 26 weeks were tested for spontaneous SWD activity. Mice were anesthetized with tribromoethanol (400 mg/kg i.p.) and placed in a stereotaxic holder fitted with a mouse incisor bar. Burr holes were drilled (posterior to bregma, 1 mm lateral to midline) on both sides of the skull. Two procedures were used to measure EEG activity. Two Teflon-coated bipolar electrodes were implanted at 0.1–0.5 mm below the dura. Screws were placed at the periphery of the skull to anchor the dental cap. Alternatively, four silver electrodes soldered onto a microconnector were slid between the skull and the dura, two on each side of the cortex, and a dental cap was applied. After the mice recovered from surgery, EEG recordings were taken over a 3-day period, for a maximum of 3 h each day, by using a Grass EEG Model 12 Neurodata Acquisition System and polyviewpro software program. The parameters for detecting SWDs have been described (22).
Rotarod and Open Maze Testing. Five male and five female homozygous mutants along with four female and six male wild-type littermate controls from the F2 B6;129 generation were tested between 6 and 8 weeks of age by rotarod (23) and open maze (24) procedures.
Results
The Cacng4-Targeted Mouse Appears Normal. The targeting plasmid included the β-galactosidase cDNA (lacZ) and neomycin resistance gene on a 5-kb fragment. The lacZ expression cassette was ligated in-frame to the beginning of exon 4 of the Cacng4 gene (Fig. 1) allowing the expression of the chimeric Cacng4 gene to be followed by staining for β-galactosidase activity. We chose to target this exon because the surrounding genomic DNA had been fully sequenced, allowing us to prepare a targeting plasmid with left 4-kb and right 5.5-kb arms for homologous recombination in ES cells. The HindIII site beyond exon4 is not retained in the targeted construct. After selection of correctly targeted ES cells (7 of 384), three clones were selected for injection and two showed germ-line transmission. Chimeric mice were generated, bred to C57BL/6J (B6) mice, and intercrossed to generate mice homozygous for the targeted mutation (B6;129-Cacng4 tm1Frk, F2 generation). We also backcrossed the mutation onto the B6 background for 10 generations to construct the congenic strain, retaining the 129 lineage only in the region surrounding the Cacng4 locus (B6.129-Cacng4tm1Frk). To confirm the targeting event, expression of Cacng4 was shown to be reduced in the heterozygote and absent from the homozygous mutant by Northern blot analysis (Fig. 1).
Both the F2 and congenic mice were viable and fertile. The F2 mice showed no obvious ataxia, and were tested for more subtle movement disorders by using rotarod and open maze procedures. No movement differences were observed on comparing the homozygous and heterozygous mice. EEG recordings of the congenic and F2 mice revealed that none had absence seizures. In addition, the homozygous F2 mice had normal rod and cone recordings as determined by electroretinograms (25) and normal hearing as measured by auditory brainstem responses (26).
Expression of Cacng2, Cacng3, and Cacng4 Genes. RNA was prepared from the head and trunk of B6 embryos at embryonic day 10.5 (E10.5), E12.5, and E14.5. Cacng4 expression was observed in all regions at the three time points by RT-PCR analysis (Fig. 2). Cacng2 expression was also seen in E12.5 and E14.5 embryos, but no embryonic Cacng3 expression was observed, although Cacng3 expression was clearly present in the adult brain (Fig. 2 Center, lane 8). Expression of both Cacng2 and Cacng4 continued through embryogenesis to birth.
Fig. 2.
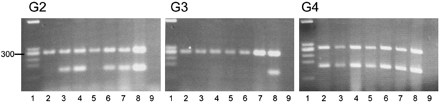
Embryonic RT-PCR analysis from B6 E10.5, E12.5, and E14.5 heads (lanes 2–4) and bodies (lanes 5–7). Lane 1 is the PhiX–HaeIII standard, lane 8 is adult brain, and lane 9 is a water control. Each gel shows the 279-bp amplification product of the internal control transcript, β2 microglobulin. The lower bands in each panel represent (Left), Cacng2 primer amplification product (156 bp) (Center), Cacng3 product (151 bp) and Cacng4 product (151 bp) (Right).
LacZ expression of the chimeric g4tm1 construct with was first observed faintly in heterozygous embryos at E11.5. By E12.5, strong lacZ staining was present. Characteristically, embryos showed blue staining around the rhombic lip of the brain, where the fourth ventricle and cerebellum later develop, and in dorsal stripes along the spinal cord as illustrated in Fig. 3.
Fig. 3.

Dorsal view of E12.5 heterozygous embryo showing lacZ staining in rhombic lip and spinal cord.
CACNG4 Is Expressed in Discrete Regions of the Adult Brain. In the adult, we confined our analysis to the brain region where mRNA analysis showed the strongest Cacng4 expression (7). The major lacZ expression within the brain included the olfactory bulb, caudate putamen, habenulae, inferior colliculus, amygdala, and the dorsal brainstem (Fig. 4). The CA3 region of the hippocampus also showed bright, punctate staining (Fig. 4 C and E). We consistently observed no staining in the cortex, but coronal sections revealed lacZ staining within the thalamus (Fig. 4B). The staining within the cerebellum was localized primarily to the Purkinje cell layer between the granule and molecular cell layers (Fig. 4D). There was also staining within the granule cell layer in the rostral lobes of the cerebellum.
Fig. 4.

LacZ-stained brain sections from homozygous B6.129-g4tm1/g4tm1 mice at 1 month of age. (A–D) Coronal sections showing caudate putamen (CP) (A), thalamus (TH) (B), habenulae (H), and CA3 region of the hippocampus (HC) (C), and Purkinje cell layer (PCL) of cerebellum (D), between the granule (G) and molecular (M) cell layers. (E) Sagittal section showing cortex (CO), olfactory bulb (OB), amygydala (AM), inferior colliculus (IC), cerebellum (CE), and dorsal brainstem (DBS).
Introducing the Cacng4 Mutation Onto a Compromised Cacng2 Background. The similarity in structure between CACNG2 and CACNG4 led us to think that CACNG2 expression could be compensating for the loss of functional CACNG4 expression, although we detected no obvious up-regulation of CACNG2 expression (results not shown). In contrast to CACNG4 expression, CACNG2 is expressed ubiquitously throughout the brain, with particularly high expression in the cortex, hippocampus, and cerebellum (7, 19, 20). We crossed the g4tm1/g4tm1 mice to stargazer (stg/stg), waggler (wag/wag), and stargazer 3J (stg3J/stg3J) to create double homozygotes. The formal and abbreviated forms of nomenclature for these single and double mutant combinations are described in Table 1.
Table 1. Formal and abbreviated mouse mutant nomenclature referred to in this study.
| Symbol (formal) | Symbol (abbreviated) | Mutant genotype |
|---|---|---|
| Single mutations | ||
| stargazer | Cacng2stg | stg/stg |
| waggler | Cacng2stg-wag | wag/wag |
| stargazer 3 Jackson | Cacng2stg-3J | stg3J/stg3J |
| Cacng4tm1Frk | Cacng4tm1Frk | g4tml/g4tml |
| Double mutation combinations | ||
| stargazer; Cacng4tm1Frk | Double homozygous | stg/stg;g4tml/g4tml |
| waggler; Cacng4tmlFrk | Double homozygous | wag/wag;g4tml/g4tml |
| stargazer 3J; Cacng4tmlFrk | Double homozygous | stg3J/stg3J;g4tml/g4tml |
| Homozygous;heterozygous | stg3J/stg3J;g4tml/+ | |
| Heterozygous;homozygous | stg3J/+;g4tml/g4tml | |
| Double heterozygous | stg3J/+;g4tml/+ |
It became evident that there was a problem in generating stg/stg;g4tm1/g4tm1 double homozygotes. It appeared that the double mutants were failing to thrive; indeed, very few double homozygous embryos were born, and even fewer mice survived beyond four weeks of age. From matings between double heterozygous stg/+;g4tml/+ animals, 141 pups were born. Onesixteenth (eight or nine) would be expected to be double homozygotes, but only one survived to 4 weeks of age.
The crosses between waggler, stargazer 3J, and Cacng4tm1 were more productive, and we were able to generate double homozygous mutant combinations. Combining the Cacng4-targeted mutation with these Cacng2 mutations appeared to confer no additional overt phenotype, although the double homozygotes were occasionally runted, most noticeably between 2 and 10 weeks of age.
Increased Seizure Activity in Cacng2;Cacng4 Double Mutants. We recorded EEGs from the doubly homozygous mice and the single-mutant controls. In the single mutants, waggler had variable seizures (reflected in the large standard deviation). On average, waggler had one SWD burst lasting 1.8 s every 3 min (Table 2). We never detected any SWDs from stargazer 3J mice, the mildest allelic member of the stargazer series.
Table 2. Incidence of SWDs from single and double mutant combinations.
| Mutant genotype | Average seizure duration, s ± 1 SD | Average seizures per h ± 1 SD | No. of mice with SWDs/no. of mice tested |
|---|---|---|---|
| wag/wag | 1.8 ± 1.5 | 18.7 ± 40.9 | 10/11 |
| stg3J/stg3J | 0 | 0 | 0/8 |
| g4tm1/g4tm1 | 0 | 0 | 0/8 |
| wag/wag;g4tm1/g4tm1 | 4.8 ± 4.6 | 37.4 ± 32.1 | 11/11 |
| stg3J/stg3J;g4tm1/g4tm1 | 2.3 ± 1.4 | 24.6 ± 24.9 | 7/9 |
| stg3J/stg3J;g4tm1/+ | 3.9 ± 4.5 | 48.6 ± 29.6 | 7/7 |
| stg3J/+;g4tm1/g4tm1 | 0 | 0 | 0/6 |
| stg3J/+;g4tm1/+ | 1.8 ± 0.5 | 0.3 ± 0.7 | 1/6 |
The double homozygotes showed a pronounced increase in seizure activity compared to the single homozygotes. Typical EEG recordings are shown in Fig. 5. Table 2 shows the average SWD period and the average number of seizures each hour. In the wag/wag;g4tm1/g4tm1 double homozygotes, there was a consistent increase in both the seizure duration compared to the wag/wag single homozygotes (t test, one degree of freedom; P < 7 × 10-20) and recurrence (t test, P < 0.02). Interestingly, the stg3J/stg3J;g4tm1/g4tm1 double homozygotes also had seizures, but no seizures were observed in either of the single mutants (Fisher exact test, P < 0.002). Included in these results are two double stg3J/stg3J;g4tm1/g4tm1 homozygotes that did not have SWD episodes during the entire recording period (Table 2), indicating that there is considerable heterogeneity in the recordings from these mice.
Fig. 5.

Typical EEG tracings recorded from wag/wag (A), wag/wag; g4tm1/g4tm1 (B), stg3J/stg3J (C), and stg3J/stg3J; g4tm1/g4tm1 (D) mice.
We further pursued homozygous;heterozygous combinations of stargazer 3J- and γ4-targeted mutants. All of the stg3J/stg3J;g4tm1/+ mutants showed increased numbers of SWDs compared to the single homozygotes (Table 2), but the incidence was not significantly different compared to the double homozygotes (t test, P < 0.06). No seizures were observed in mice with the stg3J/+;g4tm1/g4tm1 combination and only one of six stg3J/+;g4tm1/+ mutants had four brief SWD episodes lasting a total of 7 s during 150 min of recording.
Strain Background of Crosses. To reduce the possibility that genes segregating from different inbred backgrounds are influencing our results, each mutant combination was restricted to a combination of, at most, three inbred strain backgrounds, C57BL/6J (B6), 129 (129), and C3FeLe.B6 (C3). The congenic B6.129-g4tm1 was crossed to stargazer 3J with a mixed B6C3 background. The waggler × g4tm1 cross included both the B6 (waggler) and B6129 background (g4tm1). The stargazer mutation was maintained on a B6C3 mixed background, and was also crossed to the targeted Cacng4 mutation on a B6129 mixed background. Neither the F2 B6129 nor congenic B6.129-g4tm1-targeted mice had absence seizures, and the same was true for the B6C3-stg3J/stg3J and congenic B6-stg3J/stg3J mice.
Discussion
The Cacng4-targeted mutant appeared normal and showed none of the characteristic phenotypes displayed by the three Cacng2 mutants, stargazer, waggler, and stargazer 3J (4, 5). Because we could discern no EEG SWDs in the Cacng4-targeted homozygous mutants and the control parental strains, we combined this mutation with mutations in the Cacng2 gene. Viable double mutants were generated with the two milder alleles, waggler and stargazer 3J. These double mutants resembled the single waggler and stargazer3J mutants; the targeted Cacng4 mutation did not confer any change in the ataxia, but in large litters the double mutants were sometimes smaller than their littermates around weaning age. Noticeably, both the duration and recurrence of seizure episodes increased in the double homozygotes, exacerbating the seizures compared to the waggler mutant and introducing seizures into the previously seizure-free stargazer 3J. These results suggest that CACNG4 has a role in seizure susceptibility, but this can only be revealed when expression of CACNG2 is also compromised.
Our results confirmed that reducing the expression of both CACNG2 and CACNG4 proteins leads to an increase in seizure incidence, and by testing all stargazer 3J;g4tml mutant combinations, we sought to learn more about the interplay between these two proteins. Both the stg3J/stg3J; g4tm1/g4tm1 and stg3J/stg3J;g4tm1/+ double mutant combinations showed frequent seizure activity. In contrast, the lack of seizures in the stg3J/+;g4tm1/g4tm1 combination indicated that the loss of CACNG2 expression is more critical to seizure predilection than CACNG4. Our results suggest that the level of Cacng2 expression in stargazer 3J is sufficient to suppress seizures, but appears to be close to the threshold of seizure induction. Tipping the balance over the threshold can be accomplished by reducing CACNG4 expression as observed in the stg3J/stg3J;g4tm1/+ combination. However, we do not think that there is a sharp cutoff between the normal and seizure state. Rather, it seems that there is a continuum between the two, because we have examples of mice with the same mutations that show considerable variations in both the duration and recurrence of seizures, ranging from a high seizure profile to having no seizures at all [e.g., B6-wag/wag mice single mutants (all with the same genetic background; ref. 5) and B6C3-stg3J/stg3J;g4tm1/g4tm1 double mutant combinations (with segregating B6 and C3H backgrounds)]. Some of this variation in phenotype may be explained by the nature of the Cacng2 mutations. Two of the three stargazer alleles are associated with aberrant splicing into a novel early transposon element (ETn) in intron 2 of the Cacng2 gene (5), and recent data confirm that the waggler mutation is also caused by an ETn insertion in the first intron (V.A.L. and W.N.F., unpublished results). The amount of normal Cacng2 message is potentially variable from cell to cell depending on the level of normal versus aberrant splicing events, in contrast to the Cacng4-targeted mutation where every copy of the gene is disrupted and no normal message is present in the homozygote. We propose that the variability in Cacng2 expression may be a major factor in contributing to the seizure differences observed within the same mutant colony.
To disrupt the Cacng4 gene, we removed the fourth exon encoding the last two transmembrane domains and the C terminus. The chimeric protein retains the N terminus, the first two transmembrane regions and terminates within the construct containing the lacZ gene. We know that this chimeric protein is expressed from the lacZ-stained embryo and brain sections. It is possible that this protein is transported to the membrane and blocks calcium function, resulting in a dominant-negative effect. We predict that, if this were the case, we would expect to see some evidence reflected in the phenotype of the single g4tm1/g4tm1 homozygous mutant, but this mouse appeared to be normal. However we cannot rule out that the chimeric protein does not affect the phenotype when the Cacng2 expression is reduced. In these circumstances, Cacng4 expression may be required to compensate for the Cacng2 loss and the role of the chimeric protein would become more pronounced. Interestingly, when Cacng2 expression is reduced, there is no discernible compensation of Cacng3 or Cacng4 expression (20). If our targeting construct is obstructing membrane function, we might have expected to see seizures within the stg3J/+; g4tm1/g4tm1 heterozygous homozygous combination, but none were observed. We interpret these results to indicate that CACNG4 does have a role in seizure suppression, and we think that future studies to target a disruption of the first exon of the Cacng4 gene will confirm that the loss of CACNG4, rather than the presence of the chimeric protein, is the reason for our seizure results.
The chimeric construct allowed us to locate the sites of CACNG4 expression both in the mouse embryo and adult brain. In chicken embryo studies, CACNG4 expression was found in hind limb buds, cranial neural plate, spinal cord, hind limb buds, dorsal root ganglia, and myotomes (27), but our studies indicate a more reduced expression in mouse, confined to the posterior brain and spinal cord. The timing of CACNG4 expression coincides with the onset of neuronal differentiation, around E11 in neuronal cortical cells (28). In the adult brain, there is good correlation between our lacZ-stained areas and Cacng4 mRNA expression by in situ hybridization in mouse (19), and CACNG4 expression studies in rat brain (29). We observed CACNG4 expression in the thalamus and surrounding areas, but not the cortex. Absence seizures are believed to be caused by perturbations within the corticothalamic loop, including the cortical pyramidal cells, the reticular thalamic nuclei, and the thalamocortical neurons (18). The expression analysis suggests that the contribution of the Cacng4-targeted mutation to the seizures observed in the double mutants is not directly attributable to abnormal cortical neuron activity, but more likely to be associated with neuronal pathways within the thalamus.
We observe no obvious association between the sites of CACNG4 expression and the CACNA1, CACNA2D, or CACNB subunits of the high or low voltage-dependent calcium channels (30–34). Thus, we cannot deduce which of these subunits might preferentially associate with the CACNG4 protein based solely on their expression patterns. Furthermore, stargazin (CACNG2) and CACNG4 are required for the synaptic trafficking of α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate (AMPA) receptors (2, 29). Detailed studies in the rat and Macaque brains reveal that three AMPA subunit transcripts, GluR1, GuR2, and GluR3, are widely distributed, including within the cortex, and GluR4 has a more restricted expression profile (35, 36). Again, we can determine no clear association between these subunits with CACNG4 expression.
We have found that the Cacng4-targeted homozygous mutant is overtly normal. However, by combining this mutation with mutations in the Cacng2 gene, there is an overall increase in SWD activity. These results indicate that CACNG2 compensates for the loss of CACNG4 expression. Only if both proteins are reduced in the same mouse, the prevalence of spontaneous SWDs is increased revealing that both CACNG2 and CACNG4 are involved in suppressing absence seizure activity. Additionally, there is in vitro evidence that the γ subunits may be able to substitute for each other. In Xenopus oocytes, it was found that CACNG1 could functionally replace CACNG2 in voltage-dependent calcium channel activity, even though CACNG1 expression is normally confined to skeletal muscle (37). Thus, there is something intrinsic to the common structure of these CACNG subunits that allows them to compensate for each other, although the heterogeneity of the γ family suggests that each γ molecule also retains more specialized functions.
Acknowledgments
We gratefully appreciate the technical expertise of Kevin Seburn, Louise A. Dionne, Ron Hurd, and Qing Y. Zheng. We also thank The Jackson Laboratory histology, multimedia, and gene-targeting services (supported by Cancer Center Support Grant CA034196) and our colleagues, Ken Johnson, Susan Ackerman, Kevin Seburn, and Yan Yang, for critical advice. This work was supported by National Institutes of Health Grant NS32801 (to V.A.L.).
Author contributions: V.A.L. designed research; V.A.L., C.L.M., B.B., and W.N.F. performed research; V.A.L. analyzed data; and V.A.L. wrote the paper.
Abbreviations: SWD, spike-wave discharge; EEG, electroencephalogram; En, embryonic day n.
References
- 1.Black, J. L., III (2003) J. Bioenerg. Biomembr. 35, 649-660. [DOI] [PubMed] [Google Scholar]
- 2.Chen, L., Chetkovich, D. M., Petralia, R. S., Sweeney, N. T., Kawasaki, Y., Wenthold, R. J., Bredt, D. S. & Nicoll, R. A. (2000) Nature 408, 936-943. [DOI] [PubMed] [Google Scholar]
- 3.Freise, D., Held, B., Wissenbach, U., Pfeifer, A., Trost, C., Himmerkus, N., Schweig, U., Freichel, M., Biel, M., Hofmann, F., et al. (2000) J. Biol. Chem. 275, 14476-14481. [DOI] [PubMed] [Google Scholar]
- 4.Letts, V. A., Felix, R., Biddlecome, G. H., Arikkath, J., Mahaffey, C. L., Valenzuela, A., Bartlett, F. S., Jr., Mori, Y., Campbell, K. P. & Frankel, W. N. (1998) Nat. Genet. 19, 340-347. [DOI] [PubMed] [Google Scholar]
- 5.Letts, V. A., Kang, M. G., Mahaffey, C. L., Beyer, B., Tenbrink, H., Campbell, K. P. & Frankel, W. N. (2003) Mamm. Genome 14, 506-513. [DOI] [PubMed] [Google Scholar]
- 6.Chu, P. J., Robertson, H. M. & Best, P. M. (2001) Gene 280, 37-48. [DOI] [PubMed] [Google Scholar]
- 7.Burgess, D. L., Davis, C. F., Gefrides, L. A. & Noebels, J. L. (1999) Genome Res. 9, 1204-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noebels, J. L., Qiao, X. Bronson, R. T., Spencer, C. & Davisson, M. T. (1990) Epilepsy Res. 7, 129-135. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto, K., Fukaya, M., Qiao, X., Sakimura, K., Watanabe, M. & Kano, M. (1999) J. Neurosci. 19, 6027-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao, X., Chen, L., Gao, H., Bao, S., Hefti, F., Thompson, R. F. & Knusel, B. (1998) J. Neurosci. 18, 6990-6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson, C. A. & Leitch, B. (2002) J. Comp. Neurol. 453, 85-99. [DOI] [PubMed] [Google Scholar]
- 12.Thompson, C. L., Tehrani, M. H., Barnes, E. M., Jr., & Stephenson, F. A. (1998) Brain Res. Mol. Brain Res. 60, 282-290. [DOI] [PubMed] [Google Scholar]
- 13.Khan, Z., Carey, J., Park, H. J., Lehar, M., Lasker, D. & Jinnah, H. A. (2004) Neuroscience 127, 785-796. [DOI] [PubMed] [Google Scholar]
- 14.Qiao, X. & Noebels, J. L. (1993) J. Neurosci. 13, 4622-4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chafetz, R. S., Nahm, W. K. & Noebels, J. L. (1995) Brain Res. Mol. Brain Res. 31, 111-121. [DOI] [PubMed] [Google Scholar]
- 16.DiPasquale, E., Keegan, K. D. & Noebels, J. L. (1997) Neurophysiology 77, 621-631. [DOI] [PubMed] [Google Scholar]
- 17.Zhang, Y., Mori, M., Burgess, D. L. & Noebels, J. L. (2002) J. Neurosci. 22, 6362-6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crunelli, V. & Leresche, N. (2002) Nat. Rev. Neurosci. 3, 371-382. [DOI] [PubMed] [Google Scholar]
- 19.Klugbauer, N., Dai, S., Specht, V., Lacinova, L., Marais, E., Bohn, G. & Hofmann, F. (2000) FEBS. Lett. 470, 189-197. [DOI] [PubMed] [Google Scholar]
- 20.Sharp, A. H., Black, J. L., III, Dubel, S. J., Sundarraj, S., Shen, J. P., Yunker, A. M., Copeland, T. D. & McEnery, M. W. (2001) Neuroscience 105, 599-617. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY), 2nd Ed., Vol. 1.
- 22.Hosford, D. A., Lin, F. H., Kraemer, D. L., Cao, Z., Wang, Y. & Wilson, J. T. Jr. (1995) J. Neurosci. 15, 7367-7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein, J. A., Longo-Guess, C. M., Rossmann, M. P., Seburn, K. L., Hurd, R. E., Frankel, W. N., Bronson, R. T. & Ackerman, S. L. (2002) Nature 419, 367-374. [DOI] [PubMed] [Google Scholar]
- 24.Tarantino, L. M., Gould, T. J., Druhan, J. P. & Bucan, M. (2000) Mamm. Genome 11, 555-564. [DOI] [PubMed] [Google Scholar]
- 25.Hawes, N. L., Chang, B., Hageman, G. S., Nusinowitz, S., Nishina, P. M., Schneider, B. S., Smith, R. S., Roderick, T. H., Davisson, M. T. & Heckenlively, J. R. (2000) Invest. Ophthalmol. Vis. Sci. 41, 3149-3157. [PubMed] [Google Scholar]
- 26.Zheng, Q. Y., K. R. Johnson, K. R. & Erway, L. C. (1999) Hear. Res. 130, 94-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kious, B. M., Baker, C. V., Bronner-Fraser, M. & Knecht, A. K. (2002) Dev. Biol. 243, 249-259. [DOI] [PubMed] [Google Scholar]
- 28.Faure, A. V., Grunwald, D., Moutin, M. J., Hilly, M., Mauger, J. P., Marty, I., De Waard, M., Villaz, M. & Albrieux, M. (2001) Eur. J. Neurosci. 14, 1613-1622. [DOI] [PubMed] [Google Scholar]
- 29.Tomita, S., Chen, L., Kawasaki, Y., Petralia, R. S., Wenthold, R. J., Nicoll, R. A. & Bredt, D. S. (2003) J. Cell. Biol. 161, 805-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hobom, M., Dai, S., Marais, E., Lacinova, L., Hofmann, F. & Klugbauer, N. (2000) Eur. J. Neurosci. 12, 1217-1226. [DOI] [PubMed] [Google Scholar]
- 31.Klugbauer, N., Marais, E., Lacinova, L. & Hofmann, F. (1999) Pflugers Arch. 437, 710-715. [DOI] [PubMed] [Google Scholar]
- 32.Ludwig, A., Flockerzi, V. & Hofmann, F. (1997) J. Neurosci. 17, 1339-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moosmang, S., Biel, M., Hofmann, F. & Ludwig, A. (1999) Biol. Chem. 380, 975-980. [DOI] [PubMed] [Google Scholar]
- 34.Yunker, A. M., Sharp, A. H., Sundarraj, S., Ranganathan, V., Copeland, T. D. & McEnery, M. W. (2003) Neuroscience 117, 321-335. [DOI] [PubMed] [Google Scholar]
- 35.Beneyto, M. & Meador-Woodruff, J. H. (2004) J. Comp. Neurol. 468, 530-554. [DOI] [PubMed] [Google Scholar]
- 36.Petralia, R. S. & Wenthold, R. J. (1992) J. Comp. Neurol. 318, 329-354. [DOI] [PubMed] [Google Scholar]
- 37.Kang, M. G., Chen, C. C., Felix, R., Letts, V. A., Frankel, W. N., Mori, Y. & Campbell, K. P. (2001) J. Biol. Chem. 276, 32917-32924. [DOI] [PubMed] [Google Scholar]