

Abstract
Blood flow through healthy human vessels releases nitric oxide (NO) to produce vasodilation, whereas in patients with coronary artery disease (CAD), the mediator of dilation transitions to mitochondria-derived hydrogen peroxide (mtH2O2). Excessive mtH2O2 production contributes to a proatherosclerotic vascular milieu. Loss of PGC-1α is implicated in the pathogenesis of CAD. We hypothesized that PGC-1α suppresses mtH2O2 production to reestablish NO-mediated dilation in isolated vessels from patients with CAD. Isolated human adipose arterioles were cannulated, and changes in lumen diameter in response to graded increases in flow were recorded in the presence of PEG-catalase (H2O2 scavenger) or L-NAME (NOS inhibitor). In contrast to the exclusively NO- or H2O2-mediated dilation seen in either non-CAD or CAD conditions, respectively, flow mediated dilation in CAD vessels was sensitive to both L-NAME and PEG-catalase after PGC-1α upregulation using ZLN005 and alpha-lipoic acid. PGC-1α overexpression in CAD vessels protected against the vascular dysfunction induced by an acute increase in intraluminal pressure. In contrast, downregulation of PGC-1α in non-CAD vessels produces a CAD-like phenotype characterized by mtH2O2-mediated dilation (no contribution of NO). Loss of PGC-1α may contribute to the shift towards the mtH2O2-mediated dilation observed in vessels from subjects with CAD. Strategies to boost PGC-1α levels may provide a therapeutic option in patients with CAD by shifting away from mtH2O2-mediated dilation, increasing NO bioavailability, and reducing levels of mtH2O2. Furthermore, increased expression of PGC-1α allows for simultaneous contributions of both NO and H2O2 to flow-mediated dilation.
Keywords: pgc-1 alpha, coronary artery disease, microcirculation, flow-mediated dilation
Introduction
Cardiovascular disease remains a pressing global health issue. One unifying etiological factor in the development of cardiovascular disease is endothelial dysfunction, manifest as an impaired vasodilatory response to increased blood flow (i.e. shear stress) or pharmacological agonists, an abnormality typically associated with endothelial inflammation and oxidative stress.1, 2 Although most interventions are aimed at mitigating the influence of a single risk factor pathway associated with endothelial dysfunction, such as diabetes or hypercholesterolemia, targeting the participating mechanisms of endothelial dysfunction itself is an exciting approach to combat cardiovascular diseases such as atherosclerosis.3
Microvascular dysfunction is strongly prognostic for cardiovascular events, 4 suggesting that endothelial mechanisms in the microcirculation carry disease significance as either indicators of, or underlying contributors to, cardiovascular disease. However, microvascular dysfunction in humans has not been extensively examined. We have reported that the mediator of microcirculatory dilation to shear stress, the most physiologically important mechanism of endothelium-dependent dilation, is different between healthy and diseased vessels. Nitric oxide (NO) elicits flow-mediated dilation (FMD) in vessels from subjects without cardiovascular disease. In contrast, in vessels from subjects with coronary artery disease (CAD), NO bioavailability is reduced as NO reacts with rising endothelial superoxide levels, and microvascular dilation is maintained by compensatory release of mitochondria-derived hydrogen peroxide (mtH2O2).5-7 Excessive mtH2O2 production can contribute to an inflammatory vascular milieu8 and may be a key early pathogenic step in the progression of CAD. Pathologic stimuli associated with CAD, including telomerase inhibition,7 an acute increase in intraluminal pressure,9 and exogenous administration of the sphingolipid ceramide,6 can induce a switch to mtH2O2-mediated dilation, but the endogenous regulator of this shear-sensitive switch is not known.
In this study, we sought to elucidate a novel role for the transcriptional coactivator Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) in regulating the mechanism of dilation in the human microcirculation. Although largely studied in relation to obesity, skeletal muscle, and diabetes, interest in the vascular effects of PGC-1α and its protective role in atherogenesis has risen in the past several years.10-13 Given the shear-sensitive,14 redox-modulating15 vascular properties of PGC-1α, as well as its known interaction with NO in cultured endothelial cells and animal vessels,16, 17 we considered whether PGC-1α could be an endogenous switch determining the mechanism of FMD between health and disease. We hypothesized that loss of PGC-1α in non-CAD arterioles produces a CAD phenotype, characterized by a switch from NO-mediated to mtH2O2-mediated dilation. We further hypothesized that PGC-1α upregulation in microvessels from subjects with CAD will restore a non-CAD vascular phenotype characterized by a return to NO-mediated dilation. We discovered that PGC-1α upregulation uniquely recruits both NO and H2O2 during FMD, and protects against acute increases in intraluminal pressure, in vessels from subjects with CAD.
Materials and Methods
Materials
ZLN005 (Sigma), a known small-molecule activator of PGC-1α,18, 19 was prepared in DMSO. Bio-Enhanced® Na-Rala (GeroNova Research) and dissolved in distilled water. Endothelin-1 (Sigma) was prepared in 1% bovine serum albumin. Lentiviral constructs (GFP and PGC-1α siRNA) were produced by the Blood Center of Wisconsin Hybridoma Core Lab and dissolved in distilled water. mitoPBA/mitoB (Cayman) was prepared in ethanol. Rotenone (Sigma) and MitoPY1 (Cayman) were prepared in DMSO.
Statistical Analysis
Data are expressed as mean ± SEM. FMD is expressed as a percentage of maximal dilation to papaverine after endothelin-1 constriction. To compare flow–response relationships, a 2-way repeated measures ANOVA was used with pressure gradient and intervention as parameters. When a significant difference was observed between control and inhibitor curves, responses at individual concentrations were compared using a Holm–Sidak multiple comparison test. An unpaired Students t-test was used to compare baseline characteristics for patients with and without CAD. Differences in Western blot protein levels and H2O2 production in HUVECs were also assessed with an unpaired Student's t-test. Analyses were performed using SigmaPlot and GraphPad. Statistical significance was defined as P<0.05.
Results
Subject Demographics
Discarded adipose tissue was obtained from 49 patients. 28 of those patients had a clinical diagnosis of CAD. Detailed patient demographics are summarized in Table S1 (online-only Data Supplement).
PGC-1α protein levels are decreased in heart tissue from subjects with CAD
To determine whether a decline in PGC-1α levels occurs in the presence of CAD, we compared PGC-1α protein expression in non-CAD and CAD human left ventricular tissue. Western blotting revealed that PGC-1α protein content is lower in human CAD tissue (Figure S1 in the online-only Data Supplement). In human microvessels, we performed IHC, which, though not quantitative, also suggests a reduction in staining for PGC-1α in the microvasculature of subjects with CAD (Figure S2A vs Figure S2D in the online-only Data Supplement). Therefore, a diagnosis of CAD is associated with a relative decline of PGC-1α in both the human heart and microcirculation.
PGC-1α levels in the human microcirculation
48-hour incubation with lentiviral PGC-1α siRNA decreased PGC-1α levels in non-CAD vessels relative to untreated control (Figure S2B vs Figure S2A in the online-only Data Supplement). Overnight treatment (16-24 hr) with either alpha-lipoic acid (ALA) or ZLN005, both activators of PGC-1α, increased PGC-1α levels in vessels from subjects with CAD (Figure S2 E&F in the online-only Data Supplement).
Downregulation of PGC-1α in non-CAD vessels produces a CAD phenotype
Treatment of non-CAD vessels with lentiviral PGC-1α siRNA established a CAD phenotype characterized by normal magnitude H2O2-mediated dilation to shear (inhibited by PEG-catalase) [% max diameter at 100 cm H2O: vehicle 81.3±4.3, PEG-catalase 0.7±9.6]. In contrast, L-NAME, which abolishes dilation in untreated non-CAD vessels, had no effect on FMD following PGC-1α downregulation (Figure 1C) [% max diameter at 100 cm H2O: vehicle 81.3±4.3, L-NAME 71.9±6.3]. These data suggest that loss of PGC-1α in non-CAD vessels shifts away from NO-mediated dilation and exposes H2O2 as the compensatory vasodilator.
Figure 1.
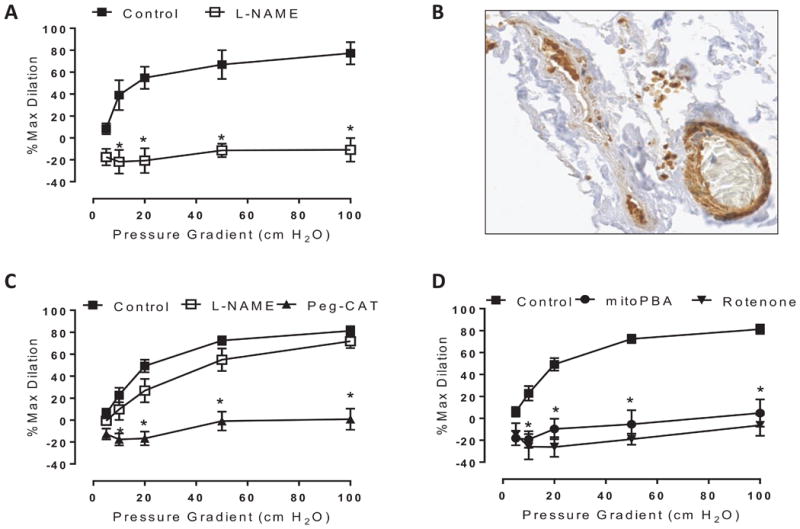
Effect of PGC-1α downregulation on flow-mediated dilation (FMD) in non-CAD vessels. A) The magnitude of FMD is preserved in human arterioles following 48-hour treatment with lentiviral GFP. L-NAME (eNOS inhibitor) acts to inhibit FMD. B) Confirmation of lentiviral GFP uptake in non-CAD vessel via immunohistochemistry. C) The magnitude of FMD is preserved in human arterioles following 48-hour treatment with PGC-1α siRNA. PEG-catalase (H2O2 scavenger) acts to inhibit FMD, whereas L-NAME has no effect. D) To determine the source of H2O2 following PGC-1α knockdown, vessels were incubated with mitochondria-targeted inhibitors rotenone (1 μM) and mitoPBA (5 μM) for 30 minutes prior to initiation of flow. n=4-7 per treatment condition. *P < 0.05 vs control curves at specific pressure gradients.
Since a relative reduction in PGC-1α levels is observed in CAD vessels, and CAD vessels dilate to mtH2O2, we anticipated that a forced downregulation of PGC-1α in non-CAD arterioles using siRNA would also result in the unmasking of compensatory H2O2 production from the mitochondria. Indeed, after PGC-1α knockdown, rotenone (inhibitor of electron transport chain complex 1) and mitoPBA (mitochondrial targeted H2O2 scavenger) both inhibited FMD (Figure 1D) [% max diameter at 100 cm H2O: vehicle 81.3±4.3, rotenone -6.3±9.6, mitoPBA 4.8±12.5].
To control for possible off-target effects of the lentivirus, we also treated non-CAD vessels with a lentivirus harboring a green fluorescent protein segment (GFP). The mechanism of dilation in these non-CAD vessels was unchanged (i.e., FMD was still inhibited by L-NAME; Figure 1A) [% max diameter at 100 cm H2O: vehicle 77.2±10.1, L-NAME -10.9±10.8], and successful transfection of GFP into arterioles was confirmed (Figure 1B). These findings indicate that non-CAD vessels dilate to NO, consistent with our previous reports.6, 7 Moreover, the switch from NO- to H2O2-mediated dilation that we observed in non-CAD vessels following PGC-1α downregulation originates from mitochondrial sources and is attributable to changes in PGC-1α levels, not the lentivirus itself.
Overexpression of PGC-1α confers vasodilatory plasticity in CAD vessels
After observing that loss of PGC-1α in non-CAD vessels produces a switch to mtH2O2-mediated dilation, we hypothesized that, conversely, upregulation of PGC-1α levels in CAD vessels, which normally dilate to increased shear via mtH2O2, might reverse the disease phenotype and restore NO-mediated dilation. To test this hypothesis, we treated vessels with two chemically distinct compounds to increase PGC-1α expression: alpha-lipoic acid (ALA), an over-the-counter supplement known to increase PGC-1α,11 and ZLN005, a novel small molecule transcriptional activator of PGC-1α.18, 19 Prior to treatment, PEG-catalase blocked dilation in CAD vessels (Figure 2A) [% max diameter at 100 cm H2O: vehicle 75.3±11.1, PEG-catalase 24.2±5.6], as we have previously observed.6 Unexpectedly, PGC-1α overexpression in CAD vessels exposed a novel phenotype characterized by contributions of both NO and H2O2 to FMD. Following treatment with either ALA or ZLN005, dilation was only partly inhibited by either L-NAME or PEG-catalase. Instead, dilation was abolished after combined co-incubation with both L-NAME and PEG-catalase following PGC-1α upregulation with these two distinct compounds (Figure 2 B&C) [ZLN treatment, % max diameter at 100 cm H2O: vehicle 80.7±4.1, L-NAME+PEG-catalase 19.0±6.5; ALA treatment: vehicle 79.4±4.4, L-NAME+PEG-catalase 14.0±7.7]. We also compared the responses between PGC-1α overexpressing and control CAD vessels (Figure 2D), which indicates that PGC-1α overexpression using ZLN005 or ALA renders dilation in CAD vessels less susceptible to inhibition by catalase (% maximal dilation at 100 cmH2O flow in vessels that are: untreated: 24.2+5.6; versus ALA-treated: 56.3+11.0, and ZLN-treated: 54.4+8.5). Thus, although a component of NO-mediated dilation was restored in CAD vessels, we observed a maintained contribution of H2O2 to dilation, highlighting that forced PGC-1α overexpression allows for more than one dilator mechanism to be present and that inhibition of dilation can only be achieved by simultaneously interfering with NO production and scavenging H2O2.
Figure 2.
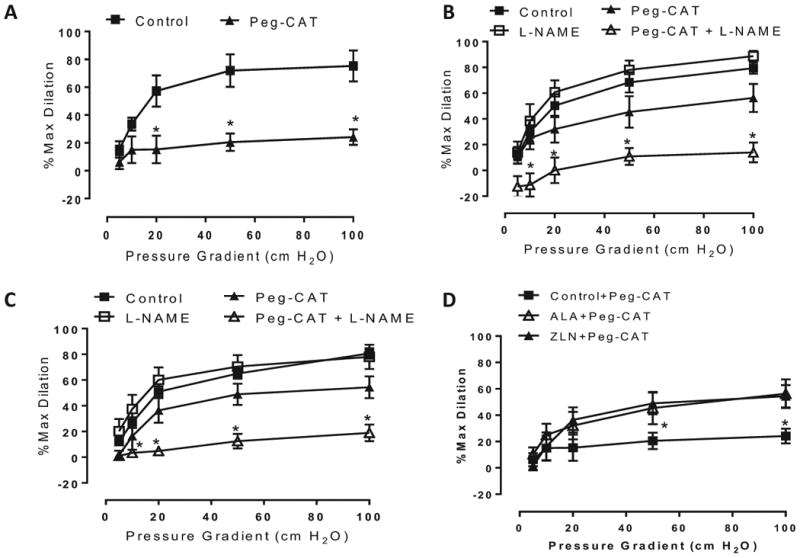
Effect of PGC-1α overexpression on flow-mediated dilation (FMD) in CAD vessels. Mechanism of FMD in CAD vessels following A) relies on H2O2 at baseline, n=4 (control) and 5 (PEG-catalase), *P < 0.05 Peg-CAT vs control curve at specific pressure gradients; both NO and H2O2 following treatment with B) 250 μM ALA (16-24 hr) and C) 15 μM ZLN005 (16-24 hr). n=5-8 per curve. *P < 0.05 Peg-CAT+L-NAME vs control curve at specific pressure gradients. D) Effect of Peg-CAT on FMD between different treatment groups. *P < 0.05 ALA/ZLN+Peg-CAT vs control+Peg-CAT curve at specific pressure gradients.
Source of H2O2 after overexpression of PGC-1α in CAD vessels
We next examined whether the mitochondria remain the subcellular source of H2O2 after PGC-1α upregulation in CAD vessels. FMD in CAD vessels is mediated entirely by mtH2O2 and can be inhibited by either PEG-catalase or specific mitochondrial H2O2 inhibitors or scavengers.5 In contrast, following PGC-1α overexpression in CAD vessels, these mitochondrial inhibitors (in combination with L-NAME) no longer influenced FMD (Figure 3B). These findings suggest that, despite the continued presence of H2O2 as a vasodilator, there is a transition away from the mtH2O2 production observed in untreated CAD vessels. To further investigate these findings using a complementary technique, we assessed mtH2O2 production using a mitochondria-targeted H2O2 probe (MitoPY1) in untreated and PGC-1α-overexpressing CAD vessels. Exposure of these vessels to shear stress revealed that PGC-1α overexpression suppresses mtH2O2 production in ALA-treated vessels from subjects with CAD relative to untreated CAD vessels (Figure 3A).
Figure 3.
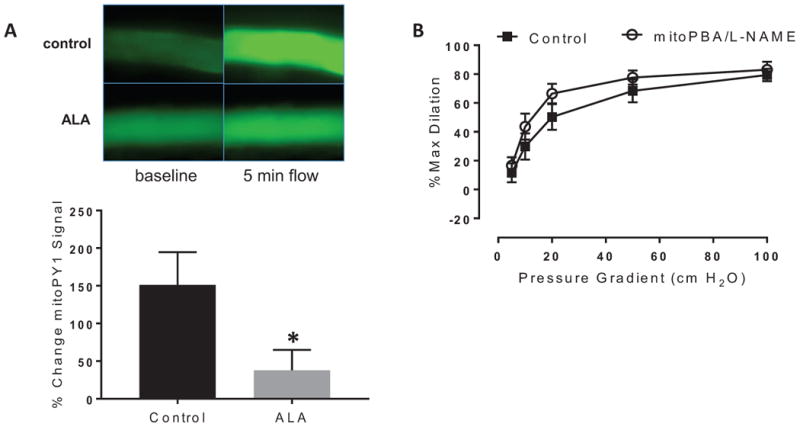
Source of H2O2 after PGC-1α overexpression on flow-mediated dilation (FMD) in CAD vessels. A) Reduction in MitoPY1 fluorescence following 24-hour ALA treatment (250 μM). Changes in fluorescence intensity in response to shear stress were evaluated in untreated or ALA-treated CAD vessels. n=6-7 per treatment group. *P < 0.05 ALA vs control; B) mitoPBA had no additional effect on dilation after incubation with the eNOS inhibitor L-NAME following 24-hour ALA treatment (250 μM) in CAD vessels. P=NS vs control between flow curves.
Overexpression of PGC-1α leads to simultaneous release of both NO and H2O2 in HUVECS in response to shear
To confirm the dual release of NO and H2O2, PGC-1α-overexpressing HUVECS were exposed to shear stress (15 dynes/cm2) for 1 hour, and levels of NO and H2O2 were determined relative to untreated, sheared HUVEC controls. DAF-2 fluorescence signal (NO) increased in ALA-treated versus untreated sheared cells (Figure 4A), illustrating the increase in NO production in response to shear stress as a result of increasing endothelial PGC-1α levels. Amplex Red fluorescence signal (H2O2) also increased after shear in ALA-treated versus untreated cells, indicating that H2O2 release is elevated in response to shear stress following PGC-1α overexpression (Figure 4B). To determine whether shear-induced H2O2 release occurs tonically (basal release) or only following inhibition of NO production (compensatory release), we repeated the Amplex Red measurements following 30-minute incubation with L-NAME to block NO production. No change in the Amplex Red fluorescence signal was observed when NO was inhibited (Figure 4B), suggesting that H2O2 coexists alongside NO during shear and is not acting as a compensatory vasodilator that only emerges when NO bioavailability is reduced.
Figure 4.
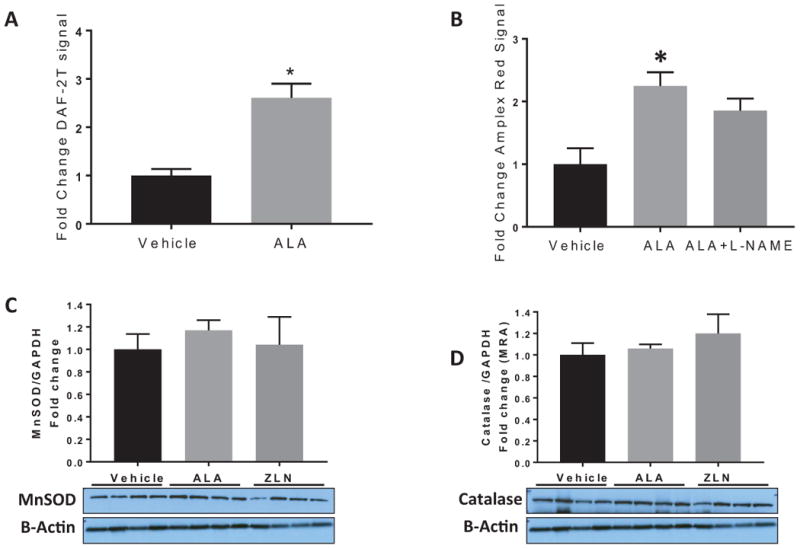
Assessment of NO and H2O2 production after shear and antioxidant levels following PGC-1α overexpression in HUVECS. A) PGC-1α overexpression using 250 μM ALA treatment increased NO production during 1 hour shear stress (15 dynes/cm2), as determined by DAF-2A/HPLC; B) PGC-1α overexpression using 250 μM ALA treatment increased global H2O2 production during 1 hour shear stress (15 dynes/cm2), as determined by Amplex Red fluorescence. Inhibition of NO production (L-NAME) did not further increase H2O2 release. N=4-6 per treatment group, *P < 0.05 ALA vs vehicle control, Data reported as mean + SEM. C&D) Western blot evaluation of the antioxidant levels of MnSOD and catalase following overnight treatment with ALA and ZLN in HUVECs. P=NS ALA/ZLN versus control.
Overexpression of PGC-1α does not reduce antioxidant levels
Since endogenous antioxidants modulate H2O2 levels in the vasculature, we evaluated whether the appearance of H2O2 as a vasodilator was associated with a loss of antioxidant defense mechanisms in HUVECs. Interestingly, PGC-1α upregulation did not lead to a decrease in either catalase or MnSOD levels in ALA-treated HUVECs (Figure 4 C&D), revealing maintained antioxidant defense mechanisms alongside the increase in H2O2 production.
Overexpression of PGC-1α protects against acute increases in intraluminal pressure
After observing this novel phenotype wherein more than one vasodilator contributes to dilation in the presence of chronic disease (CAD), we sought to determine the functional relevance of this discovery. We questioned whether this additional vasodilatory plasticity confers a broader increased protection against acute vascular insults. Adipose microvessels from CAD subjects experienced a severely impaired dilation to flow after 30-minute exposure to increased intraluminal pressure (IILP; 150 mmHg) [% max diameter at 100 cm H2O: vehicle 75.3±4.3, IILP 22.7±10.7]. In contrast, PGC-1α upregulation with ALA fully prevented IILP-induced vascular dysfunction, showing no reduction in maximal FMD following exposure to IILP (Figure 5A). Endothelium-independent dilation to papaverine was not different between the treatment groups (Figure 5B), highlighting that our results are related to endothelial-dependent mechanisms.
Figure 5.
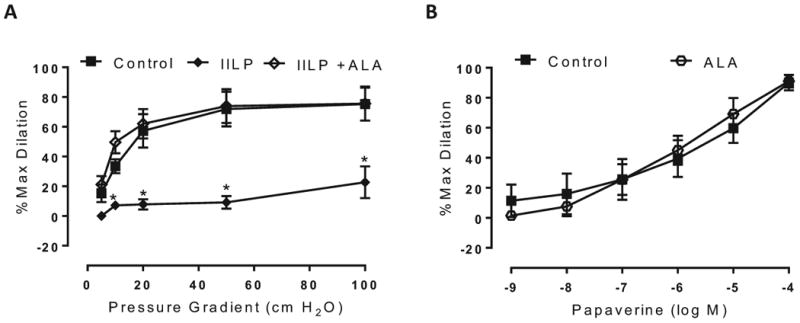
Effect of increased intraluminal pressure following PGC-1α overexpression in CAD vessels. A) PGC-1α overexpression using 250 μM ALA preserved the magnitude of FMD in response to acute increases in intraluminal pressure (IILP: 150 mmHg, 30 min), whereas acute hypertension severely impaired the overall magnitude of dilation in untreated CAD vessels; *P<0.05 IILP vs control at specific pressure gradients. B) Endothelium-independent, papaverine-induced dilation is not altered by overnight treatment with lipoic acid. N=4 per treatment group. P=NS vs control.
Discussion
There are several major findings of this study. First, we have identified a functionally-relevant effect of PGC-1α on flow-mediated dilation, adopting a reverse translational approach in human tissue to explore the reported link between loss of PGC-1α and development of CAD. As hypothesized, loss of PGC-1α in non-CAD arterioles produced a diseased (CAD) phenotype characterized by a shift from NO- to mtH2O2-mediated dilation to flow, positioning PGC-1α as a key factor in microvascular atherosclerotic disease development. Second and unexpectedly, restoring PGC-1α in vessels from subjects with CAD produced a novel phenotype wherein both NO and H2O2 contribute to dilation. Third, the source of H2O2 was no longer mitochondrial in CAD vessels following PGC-1α upregulation. Fourth, the maintained, non-mitochondrial H2O2 release was accompanied by conserved mitochondrial and cytosolic antioxidant expression. From a therapeutic perspective, we have identified two distinct compounds, ALA and ZLN005, that can produce upregulation of PGC-1α in the human microcirculation. We have uncovered a novel phenotype characterized by the simultaneous presence of NO and H2O2, lending plasticity to the mechanism of dilation to shear. We also report that microvascular FMD in CAD vessels is severely compromised following increased intraluminal pressure (IILP) (more so than non-CAD vessels9) and that PGC-1α provides protection against this IILP-induced vascular dysfunction. These findings position PGC-1α as a promising therapeutic target for the microvascular complications of CAD via attenuation of mtH2O2 release during shear, restoration of antiatherogenic NO as a vasodilator, and added resistance to acute barotrauma in CAD vessels.
That PGC-1α overexpression results in the coexistence of two vasodilators, NO and H2O2, which are canonically viewed as antagonistic, warrants additional discussion. H2O2 is traditionally viewed as a pro-thrombotic and pro-inflammatory vasoactive substance that contributes to atherosclerotic disease burden.8 However, recent evidence suggests that NO and H2O2 may act in a synergistic fashion in certain circumstances, such as H2O2-induced activation of endothelial NOS,20, 21 and that H2O2 may even function as a primary mediator of dilation in healthy animal models.20, 22-24 In addition, the detrimental effects of H2O2 in relation to cardiovascular disease are mainly attributable to the source (mitochondria-localized) and local concentrations of H2O2.25-27 For example, recent evidence suggests that NOX4-derived H2O2 is atheroprotective28, 29 and can improve vasodilation without harmful oxidative effects on the vascular wall.30 Data from other labs illustrate that excessive NO, and deficient H2O2, may increase, rather than decrease, endothelial dysfunction and that achieving moderate levels of these two vasodilators simultaneously is most cardioprotective.31, 32 Although recent studies provide evidence to support this stance, few provide therapeutic strategies or molecular mechanisms to achieve this homeostatic balance between these two vasodilators. Given our results, PGC-1α may serve as a fundamental “molecular switch” that unlocks this compensatory pathway wherein NO and H2O2 contribute to dilation, providing a window to advance our understanding surrounding this issue.
Our data support the protective role of H2O2 by demonstrating that the presence of >1 vasodilator (NO and H2O2) in CAD vessels fully preserves the overall magnitude of FMD following exposure to acute hypertension, a stimulus known to precipitate endothelial damage,33 impair microvascular FMD,9 and worsen development of CAD.34, 35 This endothelial resilience following PGC-1α upregulation is not observed following acute hypertension in vessels relying on a single mediator, including both CAD vessels (H2O2) and non-CAD vessels (NO).9 This observation underscores that PGC-1α upregulation not only restores NO bioavailability and reduces mtH2O2 (i.e., reversal of the CAD phenotype) but also confers additional protection against barotrauma within the context of CAD. This finding corroborates the anti-hypertensive effects of PGC-1α described in a recent mouse model of angiotensin II infusion.17
Given the known relationship between PGC-1α and mitochondrial dynamics,36-40 and the results from previous studies in our lab indicating a central role for mtH2O2, we chose to examine the contribution of mtH2O2 during FMD during PGC-1α knockdown in non-CAD vessels and PGC-1α upregulation in CAD vessels. We further demonstrate the continued presence of H2O2, but no longer of mitochondrial H2O2, in PGC-1α-overexpressing CAD vessels, emphasizing that one must look beyond the mere presence of H2O2 and focus on its more specific subcellular source. It is also possible that there is a concentration-dependent effect of H2O2, with low levels serving a physiological role while high levels are detrimental.41 Our data highlight that H2O2 production may exist alongside conserved antioxidant defense mechanisms. This is a particularly intriguing finding, considering the high-profile failure of several global exogenous antioxidant trials in the past,42, 43 believed to be the result of blocking pathological ROS in addition to the physiological ROS needed for proper cellular signaling. As a result, recent attempts to develop and clinically test more targeted antioxidants (MitoQ, Mito-VitE) that specifically limit mitochondrial – rather than total cellular – free radical production have been reported,44, 45 with preliminary preclinical results describing beneficial effects of these compounds within the context of cardiovascular disease.27, 46 Our results suggest that PGC-1α upregulation may be an effective strategy to specifically dampen mtROS production, in the same manner as MitoQ and MitoVitE, while also preserving endogenous antioxidant levels and promoting release of cellular, non-mitochondrial H2O2. The mechanism underlying this relationship between PGC-1α and mtROS production warrants continued investigation.
We do not identify the molecular event caused by PGC-1α overexpression that results in dual contributions of NO and H2O2 to FMD. We speculate that several possibilities exist based on past literature. Caveolin-1 may act as a the mechanosensor regulating this pathway, given previous reports from the Shimokawa laboratory indicating that caveolin-1 may be a key mechanism responsible for setting the balance between NO and H2O2 in resistance vessels.32 Regarding potential intracellular mediators of this pathway, PKG-1α is downstream of both NO and H2O2 and establishes balance between NO and H2O2 during dilation.24, 47, 48 One pathway responsible for the decreased mtROS production following PGC-1α overexpression may relate to increases in NO bioavailability. Since NO inhibits mtROS in vessels,49 restoring NO in CAD vessels may decrease mtROS during FMD without impairing ROS formation at other sites.
We offer both practical and mechanistic rationales for the use of ALA, a compound that can be administered orally, as an adjunct treatment in cardiovascular disease via upregulation of NO bioavailability and downregulation of mtROS. Several studies have noted broad associations between ALA supplementation and an improvement in cardiovascular parameters, such as brachial artery FMD in CAD patients,50 and our data both support and extend these previous findings. Of note, the concentration of lipoic acid that we used for incubation studies (250 μM) is consistent with therapeutically-achievable plasma concentrations of ALA found in human studies.51
Limitations
Please see Supplemental Material for a complete list of study limitations.
Perspectives
This study suggests that loss of PGC-1α is sufficient to shift the mechanism of flow-mediated dilation from NO to H2O2, thus establishing a link between decreased PGC-1α and coronary artery disease (CAD) pathogenesis in the microcirculation. Overexpression of PGC-1α has a beneficial effect in vessels from subjects with CAD by restoring a component of NO-mediated dilation and conferring protection against acute increases in intraluminal pressure. Lipoic acid supplementation has the potential to produce therapeutically-advantageous effects in patients with CAD, but in-human clinical trials are needed to advance this concept.
Supplementary Material
Figure 6.
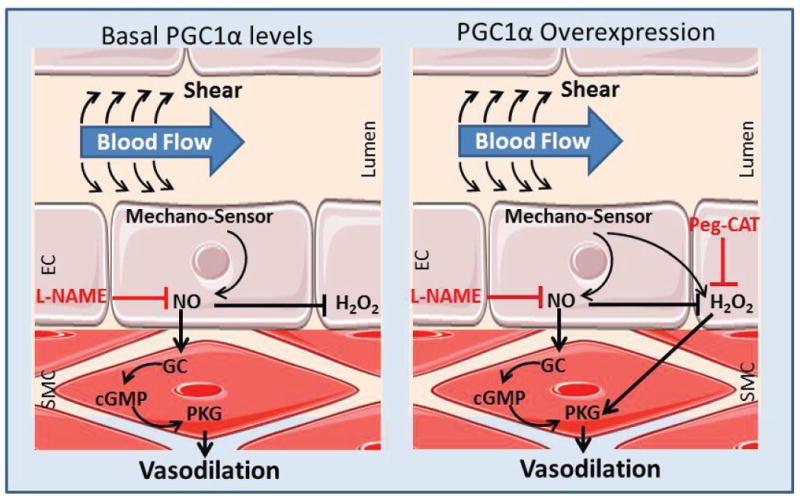
Proposed schematic of the regulation of FMD by PGC-1α. A) Basal PGC-1α in non-CAD vessels allows for NO-mediated dilation (inhibitable by L-NAME alone). B) PGC-1α overexpression provides plasticity in non-CAD and CAD vessels, resulting in contributions of both NO and H2O2 to dilation.
Novelty and Significance.
What is New?
Loss of PGC-1α contributes to the microvascular phenotype observed in subjects with coronary artery disease (CAD)
PGC-1α upregulation allows both nitric oxide and hydrogen peroxide to contribute to FMD and decreases mitochondrial reactive oxygen species production in CAD vessels
Microvessels from subjects with CAD experience severe dysfunction following acute increases in intraluminal pressure (IILP)
What is Relevant?
PGC-1α protects against acute IILP-induced vascular dysfunction and may provide a therapeutic target to combat CAD
Summary.
This study demonstrates the functional significance of targeting PGC-1α in the human microcirculation and that loss of PGC-1α can expose a CAD phenotype. For the first time, to our knowledge, we show that overexpressing an endogenous molecule can provide plasticity during flow-mediated dilation that is preserved in the presence of CAD. We also provide evidence for the therapeutic potential of lipoic acid and ZLN005 within the setting of CAD (chronic) and acute hypertension.
Acknowledgments
The authors thank Dr. Neil Hogg's Redox Biology program for assistance with HPLC. We also thank the following locations for their assistance in providing tissue for this study: Wisconsin Donor Network, St Joseph's Hospital, Froedtert Memorial Lutheran Hospital, Aurora St Luke's Medical Center, and Wheaton Franciscan Healthcare's Elmbrook Memorial Hospital.
Source of funding: This work was supported by the National Institutes of Health Grants R01-HL-135901-01 (to D.D. Gutterman) and T32-GM-080202 (to MCW Medical Scientist Training Program), and the American Heart Association Predoctoral Fellowship Grant 16PRE29130003 (to A.O. Kadlec).
Footnotes
Conflict of interest: The authors report no conflict of interest pertaining to this work.
References
- 1.Vanhoutte PM. Endothelial dysfunction and atherosclerosis. Eur Heart J. 1997;18(Suppl E):E19–29. doi: 10.1016/s0195-668x(97)90005-1. [DOI] [PubMed] [Google Scholar]
- 2.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III-27–III-32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 3.Gimbrone MA, Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murthy VL, Naya M, Taqueti VR, Foster CR, Gaber M, Hainer J, Dorbala S, Blankstein R, Rimoldi O, Camici PG, Di Carli MF. Effects of sex on coronary microvascular dysfunction and cardiac outcomes. Circulation. 2014;129:2518–2527. doi: 10.1161/CIRCULATIONAHA.113.008507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of h2o2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 6.Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res. 2014;115:525–532. doi: 10.1161/CIRCRESAHA.115.303881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beyer AM, Freed JK, Durand MJ, Riedel M, Ait-Aissa K, Green P, Hockenberry JC, Morgan RG, Donato AJ, Peleg R, Gasparri M, Rokkas CK, Santos JH, Priel E, Gutterman DD. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ Res. 2016;118:856–866. doi: 10.1161/CIRCRESAHA.115.307918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol. 2014;307:H1587–1593. doi: 10.1152/ajpheart.00557.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rowe GC, Jiang A, Arany Z. Pgc-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–838. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong S, Patrushev N, Forouzandeh F, Hilenski L, Alexander RW. Pgc-1alpha modulates telomere function and DNA damage in protecting against aging-related chronic diseases. Cell Rep. 2015;12:1391–1399. doi: 10.1016/j.celrep.2015.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue Y, Wei Z, Ding H, Wang Q, Zhou Z, Zheng S, Zhang Y, Hou D, Liu Y, Zen K, Zhang CY, Li J, Wang D, Jiang X. Microrna-19b/221/222 induces endothelial cell dysfunction via suppression of pgc-1alpha in the progression of atherosclerosis. Atherosclerosis. 2015;241:671–681. doi: 10.1016/j.atherosclerosis.2015.06.031. [DOI] [PubMed] [Google Scholar]
- 13.McCarthy C, Lieggi NT, Barry D, Mooney D, de Gaetano M, James WG, McClelland S, Barry MC, Escoubet-Lozach L, Li AC, Glass CK, Fitzgerald DJ, Belton O. Macrophage ppar gamma co-activator-1 alpha participates in repressing foam cell formation and atherosclerosis in response to conjugated linoleic acid. EMBO Mol Med. 2013;5:1443–1457. doi: 10.1002/emmm.201302587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY. Shear stress, sirt1, and vascular homeostasis. Proc Natl Acad Sci U S A. 2010;107:10268–10273. doi: 10.1073/pnas.1003833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. Pgc-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 16.Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator pgc-1alpha. FASEB J. 2006;20:1889–1891. doi: 10.1096/fj.05-5189fje. [DOI] [PubMed] [Google Scholar]
- 17.Craige SM, Kroller-Schon S, Li C, Kant S, Cai S, Chen K, Contractor MM, Pei Y, Schulz E, Keaney JF., Jr Pgc-1alpha dictates endothelial function through regulation of enos expression. Sci Rep. 2016;6:38210. doi: 10.1038/srep38210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang LN, Zhou HY, Fu YY, Li YY, Wu F, Gu M, Wu LY, Xia CM, Dong TC, Li JY, Shen JK, Li J. Novel small-molecule pgc-1alpha transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes. 2013;62:1297–1307. doi: 10.2337/db12-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Li X, Wang B, Chen Y, Xiao A, Zeng D, Ou D, Yan S, Li W, Zheng Q. Zln005 protects cardiomyocytes against high glucose-induced cytotoxicity by promoting sirt1 expression and autophagy. Experimental cell research. 2016;345:25–36. doi: 10.1016/j.yexcr.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Lauer N, Suvorava T, Ruther U, Jacob R, Meyer W, Harrison DG, Kojda G. Critical involvement of hydrogen peroxide in exercise-induced up-regulation of endothelial no synthase. Cardiovasc Res. 2005;65:254–262. doi: 10.1016/j.cardiores.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Thomas SR, Chen K, Keaney JF., Jr Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. 2002;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- 22.Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breton-Romero R, Gonzalez de Orduna C, Romero N, Sanchez-Gomez FJ, de Alvaro C, Porras A, Rodriguez-Pascual F, Laranjinha J, Radi R, Lamas S. Critical role of hydrogen peroxide signaling in the sequential activation of p38 mapk and enos in laminar shear stress. Free Radic Biol Med. 2012;52:1093–1100. doi: 10.1016/j.freeradbiomed.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 24.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase g eliminates oxidant sensing to cause hypertension. Nat Med. 2012;18:286–290. doi: 10.1038/nm.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR. The mitochondria-targeted antioxidant mitoq decreases features of the metabolic syndrome in atm+/-/apoe-/- mice. Free Radic Biol Med. 2012;52:841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 27.Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (mitoq) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592:2549–2561. doi: 10.1113/jphysiol.2013.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, Calkin AC, Biessen EA, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Reactive oxygen species can provide atheroprotection via nox4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol. 2016;36:295–307. doi: 10.1161/ATVBAHA.115.307012. [DOI] [PubMed] [Google Scholar]
- 29.Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, Sugamura K, Fitzgibbons T, Keaney JF., Jr Endothelial nadph oxidase 4 protects apoe-/- mice from atherosclerotic lesions. Free Radic Biol Med. 2015;89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial nox4 nadph oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–1376. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 31.Godo S, Shimokawa H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.12.019. http://doi.org/10.1016/j.freeradbiomed.2016.12.019. [DOI] [PubMed]
- 32.Godo S, Sawada A, Saito H, Ikeda S, Enkhjargal B, Suzuki K, Tanaka S, Shimokawa H. Disruption of physiological balance between nitric oxide and endothelium-dependent hyperpolarization impairs cardiovascular homeostasis in mice. Arterioscler Thromb Vasc Biol. 2016;36:97–107. doi: 10.1161/ATVBAHA.115.306499. [DOI] [PubMed] [Google Scholar]
- 33.Lamping KG, Dole WP. Acute hypertension selectively potentiates constrictor responses of large coronary arteries to serotonin by altering endothelial function in vivo. Circ Res. 1987;61:904–913. doi: 10.1161/01.res.61.6.904. [DOI] [PubMed] [Google Scholar]
- 34.Olafiranye O, Zizi F, Brimah P, Jean-Louis G, Makaryus AN, McFarlane S, Ogedegbe G. Management of hypertension among patients with coronary heart disease. Int J Hypertens. 2011;2011:653903. doi: 10.4061/2011/653903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrera VM, Didishvili T, Lopez LV, Zander K, Traverse S, Gantz D, Herscovitz H, Ruiz-Opazo N. Hypertension exacerbates coronary artery disease in transgenic hyperlipidemic dahl salt-sensitive hypertensive rats. Mol Med. 2001;7:831–844. [PMC free article] [PubMed] [Google Scholar]
- 36.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating sirt1 and pgc-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 37.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: The central role of pgc-1alpha. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 38.Anderson RM, Barger JL, Edwards MG, Braun KH, O'Connor CE, Prolla TA, Weindruch R. Dynamic regulation of pgc-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soriano FX, Liesa M, Bach D, Chan DC, Palacin M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783–1791. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- 40.Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the pgc-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Yun J, Rocic P, Pung YF, Belmadani S, Carrao AC, Ohanyan V, Chilian WM. Redox-dependent mechanisms in coronary collateral growth: The “redox window” hypothesis. Antioxid Redox Signal. 2009;11:1961–1974. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinhubl SR. Why have antioxidants failed in clinical trials? The American journal of cardiology. 2008;101:S14–S19. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Kris-Etherton PM, Lichtenstein AH, Howard BV, Steinberg D, Witztum JL, Nutrition Committee of the American Heart Association Council on Nutrition PA, Metabolism Antioxidant vitamin supplements and cardiovascular disease. Circulation. 2004;110:637–641. doi: 10.1161/01.CIR.0000137822.39831.F1. [DOI] [PubMed] [Google Scholar]
- 44.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase ii study of hepatitis c patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 45.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, Protect Study G A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant mitoq as a disease-modifying therapy in parkinson's disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 46.Dare AJ, Logan A, Prime TA, Rogatti S, Goddard M, Bolton EM, Bradley JA, Pettigrew GJ, Murphy MP, Saeb-Parsy K. The mitochondria-targeted anti-oxidant mitoq decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J Heart Lung Transplant. 2015;34:1471–1480. doi: 10.1016/j.healun.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friederich-Persson M, Nguyen Dinh Cat A, Persson P, Montezano AC, Touyz RM. Brown adipose tissue regulates small artery function through nadph oxidase 4-derived hydrogen peroxide and redox-sensitive protein kinase g-1alpha. Arterioscler Thromb Vasc Biol. 2017;37:455–465. doi: 10.1161/ATVBAHA.116.308659. [DOI] [PubMed] [Google Scholar]
- 48.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2o2-induced dilation in human coronary arterioles: Role of protein kinase g dimerization and large-conductance ca2+-activated k+ channel activation. Circ Res. 2012;110:471–480. doi: 10.1161/CIRCRESAHA.111.258871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hockenberry J, Zinkevitch N, Beyer A, Gutterman D. Acute and chronic inhibition of nos causes a switch in vasodilator mechanism from nitric oxide to hydrogen peroxide in the human microcirculation. The FASEB Journal. 2015;29:794–792. [Google Scholar]
- 50.McMackin CJ, Widlansky ME, Hamburg NM, Huang AL, Weller S, Holbrook M, Gokce N, Hagen TM, Keaney JF, Jr, Vita JA. Effect of combined treatment with alpha-lipoic acid and acetyl-l-carnitine on vascular function and blood pressure in patients with coronary artery disease. J Clin Hypertens (Greenwich) 2007;9:249–255. doi: 10.1111/j.1524-6175.2007.06052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carlson DA, Smith AR, Fischer SJ, Young KL, Packer L. The plasma pharmacokinetics of r-(+)-lipoic acid administered as sodium r-(+)-lipoate to healthy human subjects. Altern Med Rev. 2007;12:343–351. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.