

Abstract
The protein Triggering receptor expressed on myeloid cells-2 (TREM2) is an immunomodulatory receptor with a central role in myeloid cell activation and survival. In recent years, the importance of TREM2 has been highlighted by the identification of coding variants that increase risk for Alzheimer’s disease and other neurodegenerative diseases. Animal studies have further shown the importance of TREM2 in neurodegenerative and other inflammatory disease models including chronic obstructive pulmonary disease, multiple sclerosis, and stroke. A mechanistic understanding of TREM2 function remains elusive, however, due in part to the absence of conclusive information regarding the identity of endogenous TREM2 ligands. While many TREM2 ligands have been proposed, their physiological role and mechanism of engagement remain to be determined. In this review, we highlight the suggested roles of TREM2 in these diseases, recent advances in our understanding of TREM2, and discuss putative TREM2-ligand interactions and their potential roles in signaling during health and disease. We develop a model based on the TREM2 structure to explain how different TREM2 ligands might interact with the receptor and how disease-risk variants may alter ligand interactions. Finally, we propose future experimental directions to establish the role and importance of these different interactions on TREM2 function.
Keywords: Alzheimer’s disease, Inflammation, Immune Signaling, Neurodegeneration, Microglia
Graphical abstract
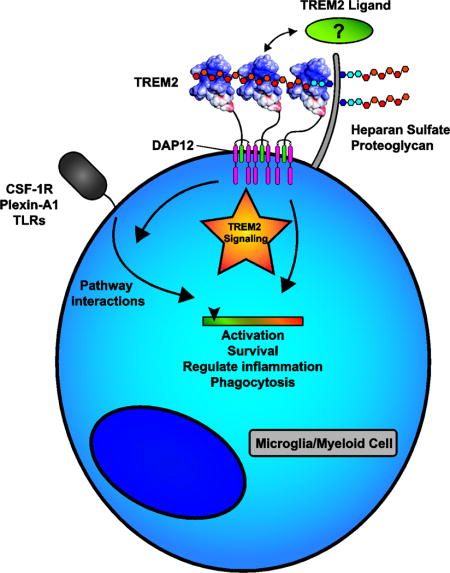
Introduction
Triggering receptor expression on myeloid cells-2 (TREM2) is an extracellular innate immune receptor expressed on myeloid lineage cells such as dendritic cells (DC) and resident tissue macrophages (including osteoclasts and microglia). The receptor consists of an extracellular V-type immunoglobulin (Ig) domain followed by a short stalk (ectodomain = 19–172 a.a.) leading to a single transmembrane helix which interacts with DNAX-activation protein 12 (DAP12, also known as TYROBP) to mediate downstream signaling (Fig 1 A, B). TREM2 terminates with a short cytosolic tail (195–230 a.a.) lacking known signaling or trafficking motifs [1]. In addition to the membrane-bound form, soluble TREM2 ectodomains (sTREM2) can be generated by proteolytic processing that occurs within the protein stalk [2] or by alternative splicing [3] (Fig 1B). The importance of TREM2 is highlighted by genetic studies linking TREM2 variants to various neurodegenerative diseases, including Alzheimer’s disease (AD) [4, 5]. TREM2 has been implicated in a wide array of functions including cell maturation, survival, proliferation, activation, phagocytosis, and the regulation of inflammation [1]. Accompanying this diverse set of functions is an even longer list of potential TREM2 ligands. Indeed, since its discovery, the identification of bona fide endogenous TREM2 ligands has proven elusive, although there is an emerging pattern of ligands that are anionic and/or lipidic in nature. It is unclear if the TREM2 ligand is among the suggested ligands or whether TREM2 is simply a highly promiscuous receptor that can engage a wide array of ligands. The goals of this review are to: 1) highlight functions for TREM2 evident from animal studies of homeostasis and disease; 2) identify facets of TREM2 signaling that may be influenced by different ligands; 3) discuss the evidence for the various TREM2 ligands; and 4) suggest where and how different ligands may be involved in TREM2 function. We develop the model that distinct ligands may mediate different components of TREM2 signaling and that a complex of ligands interacting with various portions of TREM2 may be required to assemble a productive signaling complex.
Figure 1. Schematic for TREM2 domain bounderies and structure.
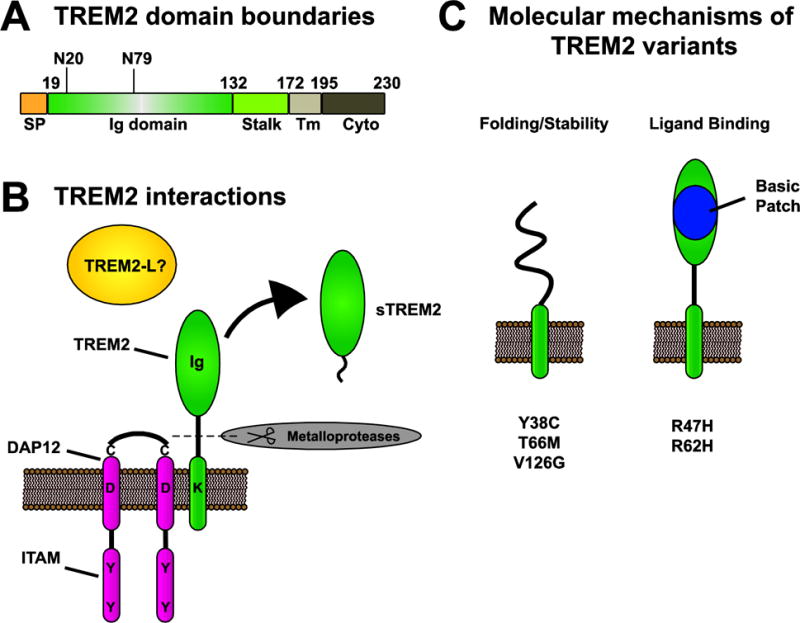
A) Schematic of TREM2 domain boundaries. SP = signaling peptide, Ig = immunoglobulin, Tm = transmembrane, and Cyto = cytosolic tail. N20 and N79 indicate position of N-linked glycosylation sites. Numbers indicate domain boundaries. B) Schematic representing TREM2 structure and interactions with DAP12. TREM2 is a single-pass transmembrane domain that interacts with DAP12 through polar interactions between transmembrane domains. TREM2 contains an Ig domain to interact with ligands. DAP12 exists as a disulfide-linked homodimer and each monomer contains an ITAM motif. C) Molecular impact of disease variants. Coding variants in the Ig domain of TREM2 have been found to either impact protein folding and stability (Y38C, T66M, V126G) or ligand binding (R47H, R62H). The major AD risk variants R47H and R62H impact binding to multiple ligands (Table 2) and are contained within a basic patch on the protein surface that likely mediates ligand binding.
1. TREM2 in disease
TREM2 variants are risk factors for neurodegenerative diseases
The importance of TREM2 in neuronal health was first demonstrated by genetic studies that identified TREM2 variants in families with Nasu-Hakola disease (NHD, also known as Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, or PLOSL) a fatal disease characterized by presenile dementia and bone cysts [6–8]. NHD patients are homozygous for loss-of-function DAP12 or TREM2 variants. In some cases, TREM2 mutant carriers present a fronto-temporal lobar form of dementia lacking the bone phenotype. The TREM2 variants include splice site [7, 9] early stop sites [7, 10–12], and coding ectodomain mutations [7, 8, 13–15]. These mutations are all believed to produce nonfunctional proteins. More recently, separate coding variants in the Ig domain of TREM2 were linked to an increase risk for late onset AD (LOAD) [4, 5]. The link between TREM2 variants and LOAD, particularly the R47H and R62H variants, is now well-established [3, 16–18]. TREM2 AD risk variants are rare, but carry roughly the same risk as a copy of the apolipoprotein E4 (APOE4) allele and clearly link the innate immune system to neurodegenerative disease [19]. Beyond AD, TREM2 variants have been linked to other neurodegenerative diseases, including Parkinson’s disease [20, 21] and sporadic amyotrophic lateral sclerosis (ALS) [22], and fronto-temporal dementia [23, 24], though these non-AD associations have not been as widely reproduced [16]. The association of distinct variants with different diseases is born out on the protein level. Structural, biophysical, and cellular studies have shown the NHD coding mutants are misfolded with immature glycosylation patterns while the AD variants are properly folded and have mature glycosylation patterns but likely impact ligand binding [25, 26] (Fig 1C and Table 2). Slightly altered glycosylation patterns have been observed for the R47H variant [27, 28], but it is unclear if these have functional ramifications. Finally, a study examined the levels of sTREM2 in human variant carriers and found reduced levels of protein with NHD variants but normal or perhaps even elevated detection of AD variants, showing a functional divergence in protein stability with the different variants [29].
Table 2.
Reported TREM2 ligands
| Ligand | Techniques | Variants | Ref |
|---|---|---|---|
| Bacteria/bacterial components | |||
| Whole Bacteria | CS, P, FC, RC, CB, E | [75, 121, 122] | |
| C. jejuni lysate | E, RC | [123] | |
| N. gonococcus lipooligosaccharides | E, SPR, RC | [124] | |
| Anionic Bacterial Carbohydrates | CB | [121] | |
| Cholera toxin B | E, RC | [156] | |
| Mammalian Cells | |||
| THP-1 monocytes | FC | [26] | |
| BMDMs | FC | [98] | |
| BMDCs | FC | [100] | |
| Astrocytes | RC, CS | [121, 125, 126] | |
| Neuronal Cells | CS, RC, FC | [26, 37, 39, 125] | |
| Apoptotic cells | FC, RC, P | [30, 37] | |
| Anionic molecules | |||
| Phospholipids & Sulfolipids | E, DB, RC, LB | R47H, R62H ↓ D87N, T96K ↑ | [26, 30, 65, 127, 128, 132, 157] |
| DNA | IP, RC | [39] | |
| Sulfated proteoglycans | FC | [26] | |
| Mammalian proteins | |||
| HSP60 | E, CS. | [125] | |
| Plexin-A1 | FRET, IP | [113] | |
| TREML1 (short transcript) | IP | [96] | |
| Apolipoproteins (A,B,E,J) | DB, P, IP, E, BLI, PM | R47H ↓[128, 134] R47H, R62H, D87N↓[135] | [128, 134, 135] |
| Lipoparticles | BLI, RC, PM | R47H, R62H ↓ D87N↑↓, T96K ↑ | [132, 135] |
| Negative results | |||
| Certain Mammalian Cells | CS | [121] | |
| Apoptotic Jurkat cells | FC | [128] |
Key: E = ELISA, RC = reporter cell, FC = flow cytometry P = phagocytosis/cellular uptake, IP = Co-immunoprecipitation, CB = competitive binding, CS = cell staining, DB = dot blot, LB = liposome binding, BLI = biolayer interferometry, PM = protein microarray, SPR = surface plasmon resonance.
Animal models and human studies of CNS disease indicate a crucial role for TREM2 in microglia function
Most studies of TREM2 in neurodegenerative diseases have used mouse models of AD. We will highlight key findings from those models and draw connections between the AD phenotypes and those observed in other neurodegenerative models such as stroke and MS to identify functions TREM2 may be contributing to during disease (Table 1).
Table 1.
Summary of TREM2 links to human disease and animal models of disease
| Disease | Observation or model | Phenotype | Possible role of TREM2 | Reference: |
|---|---|---|---|---|
| Neurodegenerative disease | ||||
| Nasu-Hakola Disease | NHD patient | TREM2 mutations co-segregate with NHD | TREM2 mutations cause NHD | [7] |
| Osteoclast maturation from PBMCs | Monocytes from NHD TREM2 carriers have impaired osteoclast differentiation. | TREM2 involved in myeloid maturation | [10, 89] | |
| Osteopenic mice | Reduced proliferation of osteoclast precursors, accelerated osteoclast maturation and apoptosis. | Myeloid proliferation and survival | [117] | |
| Alzheimer’s disease Genetic links | Sequencing & GWAS studies | First two studies to identify a link between TREM2 variants and AD | R47H is a risk variant. Others possible. | [4, 5] |
| TREM2 R47H correlated with CSF tau and p-tau | R47H is an AD risk variant | [18] | ||
| R47H did not fully explain TREM2 disease risk | R62H is an AD risk variant | [3] | ||
| Alzheimer’s disease Aβ Models |
APPPS1 mice | Decreased number and size of plaque-associated microglia in Trem2 heterozygotes. No change in total Aβ. | Microgliosis | [47] |
| 5XFAD mice | Trem2−/− mice have Increased Aβ and impaired microglial response at 8mo. Decreased microglial survival. | Microglia survival, microgliosis. Protective in Aβ | [30] | |
| 5XFAD, APPPS1 mice | 5XFAD Trem2−/− mice have fewer microglia (8mo) and less plaque clustering (4,8mo), producing diffuse plaque morphology and increased neuronal pathology. No change in total Aβ. | Microgliosis, microglial proliferation. Protective in Aβ | [42] | |
| APPPS1 mice | Trem2−/− mice have reduced Aβ plaque burden at 4mo. Decreased plaque clustering of Iba1+ cells. | Plaque response. Possible role in proliferation, survival, and/or trafficking Detrimental in Aβ | [31] | |
| 5XFAD, CRND8, APPPS1 mice Human R47H carrier samples | TREM2 is required for microglial barri er function around Aβ plaques. Human R47H carriers mirror Trem2 haploinsufficient mice. | Microgliosis and protective plaque response | [45] | |
| APPPS1 mice | TREM2 increases amyloid burden early, but reduces Aβ burden late in disease. | Microglial/myeloid proliferation, inflammation, and plaque response | [43] | |
| APPswe/PS1dE9 mice | In vivo overexpression reduced plaque load and inflammation. | Anti-inflammatory, Phagocytosis of Aβ | [32] | |
| APPswe/PS1dE9 mice | Overexpressing Trem2 in old mice (18mo) had no effect on AD pathology. | Age-restricted protective role | [146] | |
| Alzheimer’s disease Tau models |
P301S mice | Trem2 overexpression ameliorated tau pathology. | Protective, anti-inflammatory | [50] |
| P301S mice | Lentiviral knockdown increased tau pathology. | Protective, anti-inflammatory | [51] | |
| Multiple Sclerosis | Cuprizone | TREM2 sustains microglia during aging and cuprizone challenge. Reduces myelin debris. | Microglial survival and proliferation during aging myelin response | [65] |
| Cuprizone | Trem2−/− mice have increased myelin debris, decreased microglial proliferation and activation. | Microgliosis and proliferation | [64] | |
| EAE | Antibody blockade of TREM2 signaling exacerbates symptoms. | Beneficial in MS | [126] | |
| EAE | EAE symptoms were reduced by Trem2-transduced macrophages. | Anti-inflammatory, protective | [152] | |
| Prion Pathogenesis | Prion infection | No effect on prion pathogenesis, but reduced microglial activation. | Microgliosis and proliferation | [66] |
| Cardiovascular disease | ||||
| Stroke | Experimental Ischemia | Trem2−/− mice had increased infarction, decreased microglia activation, and fewer phagocytic microglia. | Microgliosis and phagocytosis Protective following stoke | [39] |
| Experimental Ischemia | Trem2−/− mice have fewer microglia at glial scars and decreased microglial activation. No change in scar size. | Microgliosis | [67] | |
| Chronic pulmonary disease | ||||
| Viral-induced COPD | SeV-induced chronic airway disease | Trem2−/− mice have reduced macrophage expansion which reduced chronic disease. IL-13 stimulation produces sTREM2 which prevents macrophage apoptosis. | Survival and proliferation. sTREM2 prevents macrophage apoptosis, allowing conversion to alternatively activated macrophages, which promote airway disease | [71] |
| Wound Healing | ||||
| Colon injury | Chemical colon injury | Trem2−/− mice had reduced inflammation and wound damage. | Inflammatory, bacterial killing. | [73] |
| Biopsy injury | TREM2 required for wound healing and promotes M2 phenotype | Promote anti-inflammatory M2 phenotype | [72] | |
| Infection | ||||
| Animal infection | E. coli endotoxemia. | TREM2 was protective in sepsis infection. | Phagocytic, anti-inflammatory | [153] |
| LPS and E. coli endotoxemia. | Trem2−/− mice had increased early inflammation and faster resolution. | Phagocytic. Anti-inflammatory. | [154] | |
| P. aeruginosa | TREM2 activated PI3K/Akt pathway to control inflammation. | Anti-inflammatory | [155] | |
| S. pneumoniae | TREM2 decreased C1q expression in alveolar macrophages and TREM2 was detrimental to survival in intranasal challenge. | Anti-phagocytic, pro-inflammatory in AMs. Opposite results in BMDMs | [150] | |
| In vitro cellular infection | P. aeruginosa | TREM2 increased ROS production through Akt/PI3K pathway. No change in phagocytosis. | Bacterial killing | [76] |
| S. Typhimurium | TREM2/DAP12 required for ROS response following Salmonella infection. | Bacterial killing | [75] | |
| B. abortus | TREM2 inhibited NO production and increased bacterial load. | Promotes bacterial growth | [77] | |
Key:
5XFAD = transgenic for mutant human APP K670N/M671L + I716V+ V717I and PSEN1 M146L+ L286V
APPPS1 = transgenic for mutant human transgenes APP KM670/671NL and PSEN1 L166P
APPswe/PS1dE9 = transgenic for mutant human APP KM670/671NL and PSEN1 delta exon 9
CRND8 = transgenic for mutant human APP KM670/671NL+V717F
EAE = experimental autoimmune encephalomyelitis
P301S = Tau P301S transgenic mice
SeV = Sendai virus
Animal model studies by several groups have consistently found Trem2 expression in the CNS restricted to microglia and increasing with age and disease progression [4, 30–33]. Within the normal human brain, TREM2 is expressed most highly in white matter, with transcripts detected in all major areas [34]. Pathway analysis identified the TREM2 co-receptor DAP12 as a key regulator of expression changes in LOAD [35]. Although there have been reports of TREM2 detection on non-myeloid/microglial cells in the CNS [4, 36], these cases have largely used immunostaining methods to identify neuronal expression of TREM2, which may identify sTREM2 produced by microglia that is released and bound to neurons which express a TREM2 ligand [26, 37–39]. RNA expression analyses have confirmed Trem2 is expressed selectively on microglia in the brain [40, 41], and parabiosis experiments show these microglia are most likely resident microglia and not infiltrating monocytes [42], although this is still controversial [43]. Within human AD brain, laser microdissection showed Trem2 is highly expressed on plaque-associated microglia [44], and high resolution microscopy techniques showed that TREM2 protein is concentrated on microglia processes that are in contact with amyloid beta (Aβ) plaques [45]. Thus, TREM2 is expressed in the brain and its expression increases both with aging and in response to disease. Moreover, during AD, TREM2 is present on microglia at sites of Aβ deposition. Consistent with its expression on microglia, studies using Trem2-deficient mouse models of AD have shown a role for TREM2 in microglial survival and activation (Table 1).
Impacts of TREM2 function on Amyloid beta pathology
The amyloid cascade hypothesis posits that Aβ is the cause of AD, whether directly by mediating neurotoxicity or indirectly by triggering other neurotoxic events, such as tau aggregation [46]. It is based on the observation that autosomal dominant mutations causative for AD are found in proteins responsible for the production and maturation of the Aβ peptide. Common animal models of AD are genetically engineered to carry these mutant proteins and/or overexpress Aβ. To date, all AD studies involving Trem2 knockout mice have utilized such models. However, the Aβ phenotypes have been inconsistent in these studies. Some studies using Trem2−/− mice found no change in the overall plaque load [42, 47], while further studies found increased [30, 43] or even decreased burden [31, 43]. Some of these differences are perhaps explained by the timing of data collection [43], although they could suggest Aβ clearance is not the only role for TREM2.
While there have been mixed results on the accumulation of Aβ, all Trem2−/− studies have shown decreased clustering of myeloid or microglial cells around plaques, suggesting a defect in microgliosis, or microglia activation [30, 31, 42, 43, 45, 47]. These studies have also found that Trem2−/− mice have fewer microglia and increased microglia apoptosis. Importantly, samples from human R47H carriers recapitulate the microglia- Aβ plaque phenotype observed in Trem2 haploinsufficient mice, with fewer microglia clustering around the plaques, and the plaques displaying a diffuse morphology, which is more toxic to neurons [45]. Therefore, although TREM2 may not have a dramatic effect on the overall Aβ content of the diseased brain, it strongly affects the ability of microglia to respond to and contain the plaques. In support of a role for TREM2 in microgliosis, a recent study found that production of sTREM2 is increased prior to the onset of symptoms but after initial markers of Aβ accumulation and neuronal injury [48]. This sTREM2 continues to increase until after the onset of symptoms and highlights the role for TREM2 in microgliosis at a critical point for the response to accumulating damage from disease.
TREM2 in models of Tau pathology
Besides Aβ, intracellular tangles of hyperphosphorylated tau proteins are the other major pathological component of AD [49]. While no studies using Trem2−/− mice have been published in tau models, Jiang and colleagues have conducted studies altering TREM2 expression in vivo by lentiviral overexpression (under a Cd11b promoter) or shRNA knockdown [50, 51] in the P301S tau transgenic model. These studies found that TREM2 reduces inflammation, decreases tau kinase activity, and improves cognitive function. Future studies should utilize Trem2 knockout and mutant Trem2 knock-in models on this background to confirm the role of TREM2 in tau-mediated neuropathies (if any).
A role for soluble TREM2 during AD?
While potential functions for sTREM2 are discussed below, it is notable that several studies have reported a correlation between CSF tau/p-tau levels and sTREM2 [29, 48, 52–54]. Interestingly, in these studies, sTREM2 is more strongly correlated with CSF tau than Aβ. Given the conflicting reports of TREM2 on Aβ burden, it is tempting to speculate that TREM2 is perhaps involved with the taupathy stage of AD progression. However, a recent paper showed that CSF sTREM2 levels follow both changes to Aβ and tau, and peak during early stages of clinical impairment [48]. This finding suggests that sTREM2 is accumulating during microglial activation in response to these events, and does not produce them.
In the future, it will be important to understand both sTREM2 function and how its production is regulated. While it is well-established that metalloproteases can cleave the TREM2 stalk to liberate the Ig domain [2, 25, 55], whether this function belongs to a specific protease in the human brain is unclear. Moreover, it is also unclear whether protease activity or alternative isoform expression account for the majority of sTREM2. Understanding the regulation of sTREM2 production will be important to understand its role in microglial activation and disease progression.
The role of inflammation in AD
The mechanisms by which TREM2 regulates inflammation are discussed in more detail below, but since both TREM2 and sTREM2 have been implicated in inflammation or its regulation, we will briefly comment on its role in AD here. (A full discussion is beyond the scope of this review, and readers are referred to these excellent reviews on the topic [56–59]). In healthy conditions, certain inflammatory processes are important to induce microglia to respond to and clear debris, and factors such as complement are involved in nonpathogenic functions including synapse pruning during neurodevelopment [56, 60]. However, many neurodegenerative diseases, including AD, are increasingly recognized as having an inflammatory component. Therefore, preventing or containing disease will require a more sophisticated understanding of the protective and pathogenic features of the inflammatory process [57]. In human patients, microglia activation can be assessed indirectly through positron emission tomography (PET) studies using radioligands for TSPO, a receptor upregulated on activated microglia. These studies suggest that microglia activation occurs early, before clinical symptoms, then perhaps levels off temporarily before increasing again later in disease [58]. Locally, the Aβ plaques are foci for immune interactions. Complement system protein are deposited on plaques and recruit microglia which attempt to phagocytose and clear the plaques. However, nearby neurons may be damaged as an off-target consequence of the complement cascade [61]. Whether by contact with plaques or other insults, microglia become primed for excessive inflammatory responses [62]. Indeed, TLRs and many other inflammatory receptors are stimulated directly by Aβ, causing the production of inflammatory cytokines which create a cytotoxic, inflammatory environment [63]. This creates a vicious cycle where microglia are exposed to stimuli, and although they may first respond with phagocytic and less inflammatory results [58], they will fail to resolve the plaques and create an inflammatory milieu that is primed to additional inflammation with subsequent insults until the microglia eventually become exhausted and dystrophic, as observed in Trem2-deficient animals [56].
TREM2 in other neurodegenerative diseases
Similar to what has been observed in AD models, Trem2-deficient mouse models of aging, prion pathogenesis and the cuprizone model of multiple sclerosis (MS) demonstrated fewer activated microglia in the course of the disease [64–66]. In MS models, there was less myelin clearance by microglia [64, 65] although there was no obvious deficiency in myelin uptake in vitro. Instead, Trem2 deficiency was associated with decreased expression of lipid metabolism enzymes and a failure to degrade myelin debris [64, 65]. These findings suggest a more general defect in activation [64–66]. Other studies examining the role of TREM2 in stroke have found increased expression following ischemic attack [39, 67, 68]. As in the other cases, these studies found that Trem2 deficiency decreased inflammation, impaired microglial activation, and resulted in a failure of microglia to cluster at scar tissue [39, 67].
Summary TREM2 in neurodegenerative diseases
The consensus from the neurodegenerative disease studies is that TREM2 is important for enabling the response of myeloid cells (primarily microglia in the CNS) to disease. Mice lacking TREM2 have fewer microglia, and these microglia fail to activate and respond to damage during disease. These observations suggest a general deficit in microgliosis. Additionally, sTREM2 in CSF seems to be a general feature of inflammatory neurodegenerative disease. Soluble TREM2 was first identified in the CSF of MS patients and neuroinflammatory patients [69], and has since been identified in a number of AD cohorts [52–54, 70].
Non-CNS diseases – the role of TREM2 in COPD, gut injury, and infection
Because of the genetic link between TREM2 and neurodegenerative diseases, the majority of TREM2 research in recent years has focused on its role in the CNS. However, TREM2 has important roles outside the CNS, particularly in models where the activation and phenotype of myeloid/macrophage cells is pivotal to disease development. In a model of virus-induced chronic obstructive pulmonary disease (COPD), TREM2 is required for survival and proliferation of alternatively activated (M2) macrophages [71]. Unlike the healthy microglial response in AD, these macrophages promote a COPD-like disease. However, the role of TREM2 in preventing macrophage apoptosis and permitting macrophage proliferation bears a striking similarity to the TREM2 phenotype in CNS diseases (where TREM2 contributes to microglia survival and proliferation). Additionally, this study was the first to identify a potential role for sTREM2 as a signaling molecule, as application of recombinant sTREM2 prevented macrophage apoptosis following M-CSF withdrawal.
In a gut injury model, TREM2 also promoted an M2 macrophage phenotype, which was important for promoting wound healing [72]. TREM2-expressing macrophages infiltrated the wound area, inhibited the expression of inflammatory cytokines, and promoted the expression of Th2 cytokines IL-4 and IL-13. However, in a chemical injury model of inflammatory bowel disorder (IBD), TREM2 promoted an inflammatory phenotype that was detrimental to healing [73]. The differences in these studies may be due to the environment. In the biopsy injury model, the first step in healing was for epithelial cells to form a layer over the wound, which the authors hypothesized would reduce exposure to bacteria and therefore reduce strong TLR signals that are known to down-regulate TREM2 [72]. The healing response may be different in the chemical model, and the interaction between TREM2 and TLR pathways may result in the increased inflammation observed in that model [73].
While the initial characterizations of NHD patients with DAP12 and TREM2 mutants did not report immune deficiencies [7, 74], the role of TREM2 in infections has been studied in various models of bacterial infection. In general, these models are more acute than those discussed above, and therefore may highlight different roles for TREM2 in the innate response to bacteria. These studies have mostly found TREM2 to be phagocytic and anti-inflammatory, although there are some exceptions (Table 1). Cellular studies have shown that TREM2 regulates the response to bacteria, in two examples by promoting killing of bacteria through production of reactive oxygen species (ROS) [75, 76] – even against bacteria that were not directly detected by TREM2 [75]. The role of TREM2 may vary with different pathogens, as it did not promote killing of B. abortus, [77] a pathogen that specializes in invading and surviving in macrophages.
Conclusions from disease models of TREM2
Recent years have seen substantial progress in identifying an important role for TREM2 in animal models of disease. Studies using Trem2-deficient mice highlight its role in sustaining the microglial response to disease. However, important questions remain about what precise function TREM2 is performing, how different ligands may stimulate these functions, and how disease variants or mutations affect function. Mechanistically, there are several possibilities for TREM2 functions, and the answers to these questions depend upon understanding the interaction between TREM2, its signaling mechanisms, and its ligands.
2. TREM2 Signaling
DAP12 mediates intracellular signaling of TREM2 and other receptors
DAP12 is the co-receptor for TREM2 and a number of other single-pass transmembrane receptors. DAP12 contains an Immuno-Tyrosine Activation Motif (ITAM, consensus sequence: YxxI/Lx(6–12)YxxI/L). Broadly, TREM2 signaling through DAP12 results in ITAM phosphorylation which leads to Ca2+ mobilization, ERK (MAPK) phosphorylation, and actin remodeling. However, TREM2 also negatively regulates TLR signaling and has roles in signaling pathways for other receptors [78]. Here we review the TREM2 signaling pathway and highlight areas where TREM2 signaling may have influence outside of the canonical “activation” pathway.
DAP12 associates with receptors through transmembrane interactions
DAP12 forms a disulfide-bonded homodimer with two intermolecular disulfide bonds contained within in a short ectodomain (14 residues in human aa 28–41 [79], 16 in mouse aa 28–43 [80]). Within the transmembrane domain, an electronegative Asp from each DAP12 monomer forms a 2:1 interaction with a Lys from the receptor. A Thr four residues below the Asp is also involved in the electrostatic network [81–83]. The interaction with the receptor is important to shield the polar residues from the lipophilic membrane environment and to allow trafficking to the cell surface (reviewed in [84]). While it is established that DAP12 requires complexing with a receptor for trafficking to the cell surface, there have been multiple reports of TREM2 surface expression in Dap12 and Dap12/Dap10-deficient cells [71, 85, 86]. Other DAP12-coupled receptors require DAP12 for surface expression [86], so it is possible that DAP12 could traffic to the surface with another receptor and engage TREM2 there.
The canonical TREM2-DAP12 signaling pathway
The standard DAP12-receptor signaling pathway was delineated using Ly49D as a model DAP12-coupled receptor on NK cells [87]. Ly49D ligation results in ITAM phosphorylation by Src kinases. The two phosphorylated Tyr residues in the ITAM create a binding site for the SH2 domain of the kinase Syk. Activation of Syk leads to the phosphorylation of ERK1/2, PLCγ1, and Cbl. These same features were observed with TREM2. Stimulation with an anti-TREM2 antibody produced calcium flux and ERK phosphorylation while monovalent stimulation by anti-TREM2 Fab did not result in calcium flux [88]. Functionally, TREM2 stimulation prolonged DC survival following withdrawal of GM-CSF and IL-4. TREM2 has also been implicated in actin mobilization [10, 89]. Monocytes from TREM2-null NHD patients had defective actin rearrangement following RANKL/MCSF stimulation. These initial studies linked TREM2 to the hallmark ITAM signaling events: Ca2+ mobilization, ERK phosphorylation, and actin remodeling (Fig. 2A). Further studies extended the TREM2 signaling pathway, and began to identify nuances that are key to fully understanding TREM2 function.
Figure 2. TREM2 signaling mechanisms.
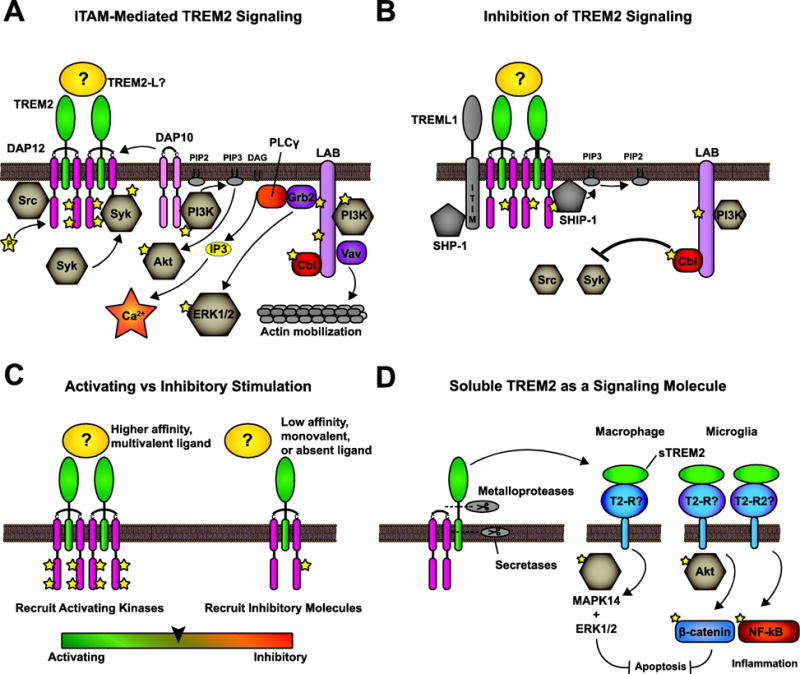
A) Canonical ITAM-mediated TREM2 signaling occurs following cross-linking of TREM2. Src phosphorylates the DAP12 ITAM. This recruits and activates Syk. Syk activity leads the phosphorylation and activation of downstream effectors including PI3K, PLCγ, LAB or LAT, Cbl, Grb2, and Vav. DAP10 also recruits PI3K. The end result of these pathways is Akt activation, calcium flux, ERK/MAPK activity, and actin mobilization. B) TREM2 signaling is inhibited through multiple mechanisms. TREML1 associates with TREM2 signaling complexes and recruits SHP-1 through its ITIM. The scaffolding protein LAB recruits the E3 ubiquitin ligase Cbl, which downregulates Src and Syk. Incomplete DAP12 phosphorylation results in a “hidden ITIM” which can recruit SHIP-1. C) The most prevalent model for multifaceted TREM2 signaling is activating vs tonic receptor engagement by multivalent, high-affinity ligands vs low affinity, monovalent ligands. D) Soluble TREM2 is produced by proteolytic cleavage and may have signaling functions separate from transmembrane TREM2.
Although DAP12 is the primary adaptor for TREM2, DAP10 can also interact with DAP12-TREM2 complex, possibly through the formation of DAP12-DAP10 heterodimers [90]. DAP10 contains a “YINM” motif that can recruit PI3K. Accordingly, DAP10 was required for effective activation of PI3K, Grb2, and therefore Akt and ERK signaling. However, calcium mobilization was surprisingly unchanged in DAP10−/− bone marrow-derived macrophages (BMDMs) [90]. Beyond DAP12, signaling cascades in hematopoietic cells that begin with ITAM phosphorylation use scaffolding proteins to recruit downstream effector proteins such as Grb2, Cbl, PI3K, and Vav. Two such proteins are the Linkers for the Activation of T and B cells (LAT and LAB; LAB is also known as LAT2 and NTAL) [91]. While they share most functions, a key difference is that LAT stimulates calcium mobilization more efficiently than LAB. DAP12 can signal using either LAT or LAB, however TREM2 signaling through DAP12 utilizes LAB in osteoclasts and BMDMs [92].
TREM2-DAP12 signaling is inhibited by phosphatases and ubiquitin ligases
Once activated, how is TREM2 signaling terminated? Following TREM2 stimulation by anti-TREM2 antibodies, the phosphatidylinositol phosphatase SHIP-1 binds the phosphorylated membrane-proximal tyrosine, pY65, within the ITAM motif of DAP12 to inhibit DAP12 signaling and TREM2-dependent osteoclast multinucleation [90]. This tyrosine is part of a “closet ITIM” sequence which was previously hypothesized to mediate inhibitory signals (SPYQEL, canonical ITIM is S/I/V/LxYxxI/V/L) [93]. Furthermore, as noted above, the E3 ubiquitin ligase Cbl is recruited to LAB and forms a complex with LAB and PI3K [92] to target Src and Syk for ubiquitination and degradation [94]. This activity requires LAB, as LAB−/− cells have increased Syk activation but decreased ERK activation [92]. Therefore, DAP12 signaling directly leads to its own inhibition by the recruitment of these two molecules. Finally, TREM-like transcript 1, (TREML1, also TLT-1), an ITIM-containing member of the TREM family is expressed in bone tissue [95] and associates with the tyrosine phosphatase SHP-1 upon phosphorylation. A short alternatively-spliced transcript lacking much of the ectodomain (TREML1s) is also expressed in osteoclast precursors and macrophages. TREML1s co-IPs with TREM2, SHP-1, and SHIP-1 and decreases ERK, Akt, and calcium signals following RANKL stimulation [96]. Therefore, TREML1 (or TREML1s) may inhibit TREM2 signaling (Fig. 2B).
TREM2 regulates inflammation through tonic signaling
In contrast with TREM1, which signals through DAP12 to amplify TLR responses [97], TREM2 inhibits inflammatory cytokine production following TLR stimulation in cultured BMDMs and DCs [98–100]. TREM2-dependent modulation of cytokine production did not require exogenous stimulation of TREM2, suggesting TREM2 could be recognizing endogenous ligands to produce an inhibitory signal. There is precedence for inhibitory ITAM signaling in a similar system [101]. In this example, IgA binds its receptor FcαRI, which signals through the ITAM-containing co-receptor FcRγ to produce either inhibitory (SHP-1 recruiting) or activating ITAM signals based on whether IgA is monomeric or aggregated by antigens. The transient monomeric interaction produces inefficient ITAM phosphorylation, leading the recruitment of the inhibitory tyrosine phosphatase SHP-1. This IgA-FcαRI-FcRγ system is analogous to ligand-TREM2-DAP12 and previous commentaries have suggested monovalent or low-affinity TREM2 ligands induce tonic signals that are less activating and anti-inflammatory through such a mechanism [102–104] (Fig. 2C). However, the endogenous, inhibitory TREM2 ligand has not been identified. Along these lines, it was recently observed that expression of DAP12 with a C-terminal fragment of TREM2, which lacks the ectodomain but contains the DAP12-interacting transmembrane domain, is sufficient to inhibit cytokine production following LPS stimulation [105]. The C-terminal fragment may occupy DAP12 similarly to TREM2 with a monovalent ligand.
Soluble TREM2 (sTREM2) as a signaling molecule
The TREM2 receptor signals through DAP12. However, release of the soluble TREM2 ectodomain (sTREM2) via proteolysis is well-documented [2, 25]. The production of soluble TREM ectodomains appears to be a general feature of the family as soluble forms of TREM1 [106] and TREML1 [107] have also been reported. While sTREM1 is thought to be a decoy receptor [108], current evidence suggests sTREM2 is a signaling molecule. In the first study of sTREM2 function, application of sTREM2 (residues 19–136) to cultured BMDMs resulted in ERK and MAPK14 activation that persisted several hours and prevented apoptosis following M-CSF withdrawal [71]. A similar phenotype was recently observed using microglia [109], although the signaling mechanism differed (Fig. 2D). In the macrophage study, sTREM2 did not detectably increase pAKT, while in microglia, the anti-apoptotic effect was blocked with PI3K inhibitors and was mediated by AKT inhibition of GSK3-β to increaseβ-catenin. Soluble TREM2 also increased transcription of inflammatory cytokines in microglia through NF-κB activation [109], a pathway that was not previously reported to be activated by cellular TREM2 signaling [88]. The sTREM2 receptors have not been identified, so it is unknown whether the differences between microglia and macrophages are due to different receptors or downstream cofactors. Additionally, it is unknown whether Akt and NF-κB activation are mediated through the same receptor pathway. However, both studies observed sTREM2 phenotypes using WT and Trem2−/− cells, showing that sTREM2 is acting independently of transmembrane TREM2 (i.e. sTREM2 is not the TREM2 ligand). It is interesting that only some functions associated with receptor TREM2, such as survival but not proliferation have been recapitulated with sTREM2, which suggests discrete roles for TREM2 both as a cellular receptor and soluble signaling molecule.
TREM2 regulates TLR signaling using inhibitory adaptors
TREM2 also uses adaptor proteins to influence the inflammatory environment. LAB−/− macrophages display increased ERK phosphorylation following TLR stimulation [92]. It could be that TREM2 signaling through LAB restricts ERK or other signaling molecules needed for TLR signaling, or that LAB-dependent Cbl-mediated ubiquitination reduces the level of kinases needed to activate TLR pathways [91]. Another pathway for inhibiting TLR signals is the recruitment of the anti-inflammatory adaptor protein DOK3 to DAP12 following TLR stimulation with LPS. Following LPS stimulation, DOK3 becomes phosphorylated, associates with DAP12, and interacts with Grb2 and Sos1 to inhibit the TLR response. In this system, SHIP-1 was not required to inhibit TLR signals, demonstrating that the inhibition of ITAM signaling by SHIP-1 is separate from the inhibition of TLR signaling by DAP12 [110] (Fig. 3A).
Figure 3. Role for TREM2 in regulating other signal pathways.
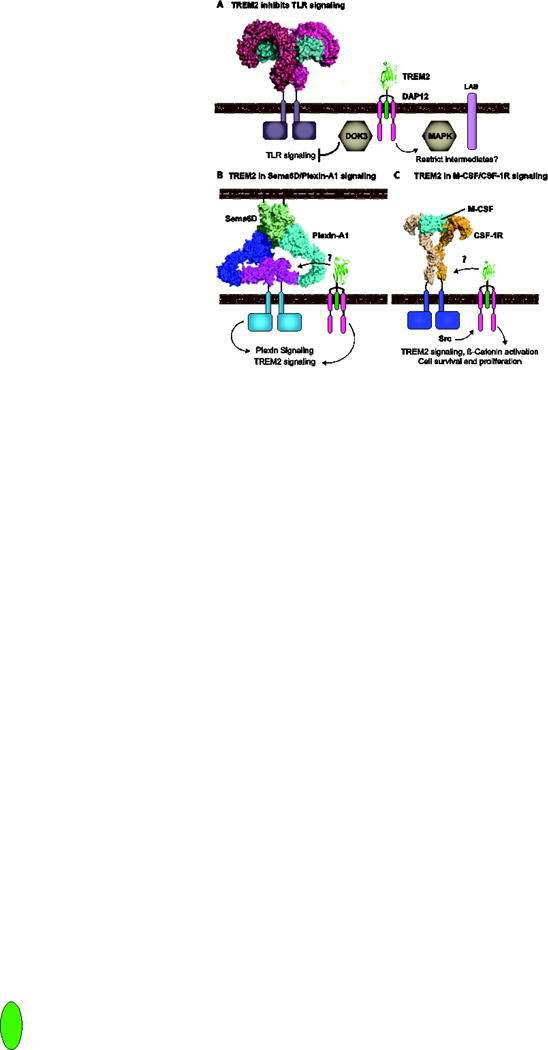
A) TREM2 inhibits TLR responses through the recruitment of DOK3 to the DAP12 ITAM at the plasma membrane and potentially by sequestering signaling intermediates such as ERK or creating a less activating environment with scaffolding proteins such as LAB. (Structures PDBs: TREM2 5ELI [26] and 3FXI [158] - signaling complex of TLR4 (dark and light red), MD2 (cyans) complexed with LPS). B) TREM2 is required for the Plexin-A1 response to Sema6D. Two molecules of Plexin-A1 dimerize upon binding to a dimer of Sema6D. The Ig-like domains of Plexin-A1 colored in magenta were required for the interaction with TREM2. Structure PDBs: TREM2 5ELI, Plexin-A4 dimer 5L5K [159] - (Blue and cyan with magenta highlights) superimposed on Sema6D (greens, 3OKY, [160]). C) TREM2 signaling is required for β-catenin activation during M-CSF/CSF-1R signaling. (Structure PDB: 4WRM [161] - CSF-1R orange and M-CSF cyan with signaling dimer shown in lighter shading).
The role of TREM2 and DAP12 in co-stimulatory signaling
Plexin-A1 is expressed in a number of tissues including DCs and osteoclasts and is a receptor for two separate Semaphorin proteins. When expressed on osteoclast precursors, Plexin-A1 constitutively interacts with Neuropilin-1 (Nrp1) to mediate Semaphorin-3A (Sema3A) signaling, which prevents osteoclast maturation [111]. However, when osteoclast precursors are stimulated with RANKL, Nrp1 is downregulated and Plexin-A1 can be bound by Sema6D to drive osteoclast maturation [112]. Unexpectedly, this Sema6D signaling required TREM2 and DAP12 [113]. When Plexin-A1−/− mice were first characterized, they were discovered to have an osteopetrotic phenotype with decreased osteoclast counts as well as deficits in activation of T cells by DCs, similar to what had been reported for DAP12−/− mice [114, 115]. Takegahara et al. found that stimulation of BMDCs with a soluble form of Sema6D resulted in DAP12 phosphorylation and also found an interaction between TREM2 and Plexin-A1 using co-IP and intracellular FRET. DAP12 and Plexin-A1 did not co-IP without TREM2, suggesting TREM2 connects DAP12 to Plexin-A1 signaling [113]. This Plexin-A1/TREM2 interaction is specific for Sema6D signaling, because the Nrp1-Plexin-A1 interaction displaces TREM2 in co-IP experiments [111, 116] (Fig. 3B).
TREM2 is also involved in M-CSF-mediated survival and proliferation in macrophages and osteoclast precursors. M-CSF signals through its receptor CSF-1R to activate β-catenin through phosphorylation of β-catenin by calmodulin-dependent kinases downstream of DAP12 in a TREM2-dependent mechanism [117, 118]. In Trem2−/− mice, the β-catenin deficit in osteoclast precursors reduces proliferation and prematurely accelerates osteoclast maturation. The increase in osteoclasts produces an osteopenic phenotype in the Trem2−/− mice, similar to NHD patients [117]. How are TREM2 and DAP12 activated by M-CSF signaling? A separate study showed M-CSF stimulation resulted in DAP12 phosphorylation by Src kinases activated by CSF-1R [119]. This mechanism suggests these receptor pairs are in close physical proximity; however, a direct interaction between TREM2 and CSF-1R has not been reported (Fig. 3C). Very recently, a study using mouse microglia confirmed a role for TREM2 in Wnt/β-catenin signaling in these cells. Trem2−/− microglia had suppressed β-catenin signaling and suffered G1/S cell cycle arrest [120], as predicted from the studies in osteoclast precursors. This results in fewer microglia and impaired microgliosis.
3. TREM2 Ligands
Many potential TREM2 ligands have been proposed. These are listed in Table 2 and the major findings are discussed in this section.
TREM2 binds anionic bacterial ligands
Bacteria and poly-anionic molecules were the first potential ligands identified for TREM2 [121]. TREM2 interacted with Gram-positive and Gram-negative bacteria in direct binding and signaling studies. In another study, CHO cells transfected with a chimeric TREM2-DAP12 construct became competant to phagocytose bacteria [122]. Phagocytosis was dependent on Syk activation and the activity of the Rho GTPases Rac and Cdc42. Additional studies confirmed TREM2 can bind E. coli, but not Salmonella [75]. TREM2 also interacts with an unknown ligand in C. jejuni, a gram-negative bacteria which is a causative agent for food poisening [123].
The study by Daws et al. also found that anionic bacterial carbohydrates, particularly dextran sulfate, LPS, and LTA, but not chondroitin sulfate (CS), could block TREM2 binding to bacteria. Dextran sulfate could also activate TREM2 reporter cell lines similarly to E. coli [121]. Another study found that TREM2 can directly bind lipooligosaccharides (LOS) from N. gonococcus. Enzymatic removal of the acyl chains reduced binding, suggesting the lipidic acyl chains mediate binding to TREM2. SPR experiments were used to quantify the interaction (Kd ≈ 350 nM) [124].
TREM2 binds myeloid and non-myeloid mammalian cells
Because myeloid cells have TREM2-dependent phenotypes when cultured without other cell types, it was hypothesized they would express a TREM2 ligand. Indeed, TREM2 can bind primary macrophages [98] and DCs [100]. With DCs, stimulation with LPS, CpG DNA, or Zymosan did not alter staining levels. However, TREM2 staining of THP-1 monocytes was shown to be ablated when cells were pre-treated with PMA+ionomycin [26], the first time a cellular TREM2 ligand was shown to be regulated in viable cells. These findings suggest a ligand that interacts with TREM2 in cis, perhaps to provide tonic signals or to function as a co-receptor with TREM2 to recognize ligands in trans from extracellular stimuli. Importantly, the AD-risk mutations R47H and R62H had decreased binding to this cell-surface ligand, while a T96K variant had enhanced binding [26]. This last variant has unclear disease risk, but may be protective. If these mutations affect binding to a cis ligand required for signaling, it will be important to experimentally distinguish deficits in ligand binding from signaling.
Given the role of TREM2 in CNS homeostasis, several studies have also investigated the potential for a direct interaction between TREM2 on microglia and the other cells of the CNS, namely astrocytes and neurons. The first report of TREM2 ligands used Fc-TREM2 proteins to bind a variety of astrocyte lines [121]. Binding could be blocked by competition with LPS or LTA, showing a role for anionic recognition in the cell-surface TREM2 ligand. Other groups have also shown TREM2 binds primary cultured rat astrocytes [125], and astrocytes can activate TREM2 reporter cells [126]. In addition to astrocytes, cytochemistry and reporter cell activation demonstrated the presence of a TREM2 ligand on cultured neurons [37], and flow cytometry experiments confirmed Neuro2A cells express a TREM2-specific ligand, as other TREM family proteins (TREML2 and TREML4) had minimal binding [26].
In addition to binding viable neurons, Fc-TREM2 staining of Neuro2A and other cell lines increased when the cells were apoptotic [37] and TREM2 staining of neurons was increased following iscemic attack [39]. Apoptotic neurons [37] and thymocytes [30] have also been shown to stimulate TREM2 signaling, suggesting a common ligand on apoptotic cells. It is unclear whether this increased binding to apoptotic cells is from increased exposure of one ligand or nascent exposure of an additional ligand. Functionally, downregulating Trem2 by shRNA in BV2 microglial cells reduced phagocytosis of apoptotic neurons, and transfecting CHO cells with TREM2 made the cells competant to phagocytose apoptotic cells [37]. Altogether, these studies have identified TREM2 interactions with mammalian cells that do and do not express TREM2.
TREM2 binds anionic ligands on mammalian cells
TREM2 has been shown to bind a number of anionic molecules from mammalian cells. Cannon et al. performed the first demonstration of TREM2 proteins binding to phospholipids. Solid-state lipid ELISAs using a variety of plated phospholipids showed TREM2 binding was strongest with phosphatidylethanolamine (PE), phosphatidylserine (PS), and cardiolipin (CL), while phosphatidylcholine (PC) or sphingomyelin (SM) displayed greatly diminished binding [127]. Two other groups have obtained similar results showing strong binding to anionic lipids such as phosphatidic acid (PA) and PS using dot blots [128] or ELISA and liposome sedimentation [26]. Unlike assays with CD300 or [127, 129] or TIM [130, 131] family proteins, additon of calcium or magnesium did not enhance TREM2 binding, suggesting a different mode of recognition [26, 127]. The TREM2 crystal structure also showed that TREM2 does not contain structural features associated with selective phospholipid binding by other Ig domains [26].
Studies using TREM2 reporter cells also support the concept of phospholipids as TREM2 ligands. Plated sulfatide, PC, and SM all strongly stimulated mTREM2 [65] and similar results were obtained with hTREM2 [30]. However, the recognition profile is different than what has been observed in direct binding assays, where PC has minimal binding and anionic moieties have higher binding [26, 127, 128]. The R47H variant had minimal signaling to lipids other than PC and SM [30]. In a followup study with other TREM2 variants, R62H behaved similarly to R47H, while D87N and T96K increased signaling in response to PS and Sulfatide, but not PC [132]. These findings further support the hypothesis that TREM2 variants alter ligand binding and, hence, signaling.
While TREM2 certainly binds phospholipids in the context of solid-state ELISA and liposome assays, and phospholipids can stimulate signaling in reporter cells, they are unlikely to represent the complete TREM2 ligand. In one study, apoptotic Jurkat cells (which expose PS on the cell surface) did not bind Fc-TREM2 [128]. This suggests that phospholipids are not sufficient for TREM2-cell binding and may need co-presentation with other molecules. Second, while results in our lab showed TREM2 binding to phospholipids by ELISA and liposome sedimentation, the disease variants R47H, R62H, and T96K did not grossly alter phospholipid binding [26]. Therefore, it will be important to understand how multiple interactions may influence TREM2 to recognize ligands and facilitate signaling.
In addition to phospholipids, nucleic acids released by damaged cells are a possible TREM2 ligand. TREM2 can IP nucleotides from the supernatant of ischemic brains and purified DNA activated TREM2 reporter cells [39]. Given the common anionic theme for TREM2 ligands, it would not be surprising that nucleotides released from damaged cells in the brain could stimulate TREM2.
Finally, by using enzymes that selectively cleave either heparan sulfates (HS) or CS, it was shown that TREM2 binds cell-surface proteoglycans and these are a major component of the cell-surface ligand [26]. Staining to THP-1 and Neuro2A cells was largely ablated by pre-treating the cells with heparan sulfatases, but not chondroitinase. This selectivity for HS over CS is consistent with the earlier studies showing heparin (but not CS) could block TREM2 binding to bacteria [121]. In addition, proteoglycan binding would explain why TREM2 can bind such a wide range of cells. The AD disease risk variants lose binding to these cells while the T96K variant gained binding, suggesting proteoglycans are an important TREM2 ligand. Future work will be needed to address whether HS is a tonic or activating ligand, whether it is required for presenting another ligand, or whether it is necessary to orient TREM2 to signal upon ligation of an activating ligand such as phospholipids.
TREM2 binds cellular mammalian proteins
Beyond the anionic biomolecules, TREM2 has been linked to other mammalian proteins. Pulldown experiments of surface-biotinylated neurons identified the cytosolic chaperone protein HSP60 [125]. ELISA assays demonstrated a direct interaction between these proteins with a low micromolar affinity. While it is counter-intuitive for an intracellular chaperone to mediate extracellular signals, there is a body of research characterizing the role of various heat-shock chaperones as alarmins for the innate immune system [133].
Additionally, as discussed above, the receptor Plexin-A1 interacts with TREM2 as part of a signaling complex. These proteins were co-immunoprecipitated and the interaction was ablated by deleting certain domains in Plexin-A1 [113]. In addition to Plexin-A1, TREML1s co-IPs with TREM2. The structural basis for these interactions are not known, and they may be indirect through association with signaling complexes [96].
TREM2 binds lipoproteins
In 2015, two groups independently identified a direct interaction between TREM2 and apolipoproteins, including ApoE. APOE and APOA were identified in protein bands precipitated from CSF by TREM2 [128] and interaction was verified by co-IP, ELISA, and dot blots [128, 134]. Both groups found that TREM2 binds all ApoE isoforms, lipidated and non-lipidated, and that the R47H variant reduced binding to ApoE. A separate study used a protein microarray to identify ligands for TREM2 from a library of extracellular and secreted proteins and investigated potential hits with bio-layer interferometry (BLI) [135]. TREM2 bound apolipoproteins, including APOA, APOB, APOE, CLU, and LDL. In contrast to the other reports, non-lipidated proteins had reduced binding as assessed by the response amplitude. The kinetics of these responses were not quantified, so it is unclear if the differences are reduced affinity or because the lower molecular weight non-lipidated proteins produce a smaller signal by BLI. The R47H, R62H, and D87N variants had reduced signal compared to WT. Functionally, ectopic expression of TREM2 in 293T cells enabled phagocytosis of APOE and Aβ that was bound to APOE [135]. Finally, in a study with reporter cells, TREM2 signaled in response to LDL and HDL [132]. However, in contrast with the BLI data, the D87N had about three-fold increase in signaling.
The link between APOE and TREM2 connects these two key AD-risk proteins. The risk associated with TREM2 variants is comparable with the APOE4 allele. Other evidence suggested TREM2 and APOE could have overlapping areas of function. TREM2 has been linked to lipid metabolism: Trem2 transgenic mice gain more weight on a high fat diet [136], and expression analysis connected Trem2 to lipid metabolic pathways [29, 65]. Furthermore, APOE interacts with Aβ, lipids, and heparan sulfates [137], and is important for microglial functions including phagocytosis, inflammation, and cell health [138, 139]. Lipoproteins are highly abundant in serum, and the presence and role of lipoproteins will need to be considered in cellular experiments with TREM2.
4. TREM2 function and model for ligand engagement
To conclude, we summarize the reported TREM2 functions and try to arrange them through the context of various ligands. The reported ligands for TREM2 are broad and include ligands that could be thought to represent steady-state tonic stimuli or disease-specific stimuli. Therefore, the functions of TREM2 signaling could include specific responses such as phagocytosis, cell activation, or collaboration with other signaling pathways to maintain cell survival and proliferation (Fig. 4). These possibilities are not mutually exclusive; on the contrary, it may be useful to conceptualize the different ligands as either tonic, homeostatic signals or damage-associated signals which produce different kinds or strengths of stimuli in order to accomplish the wide variety of functions reported for TREM2.
Figure 4. Possible roles for TREM2 resulting from different ligands.
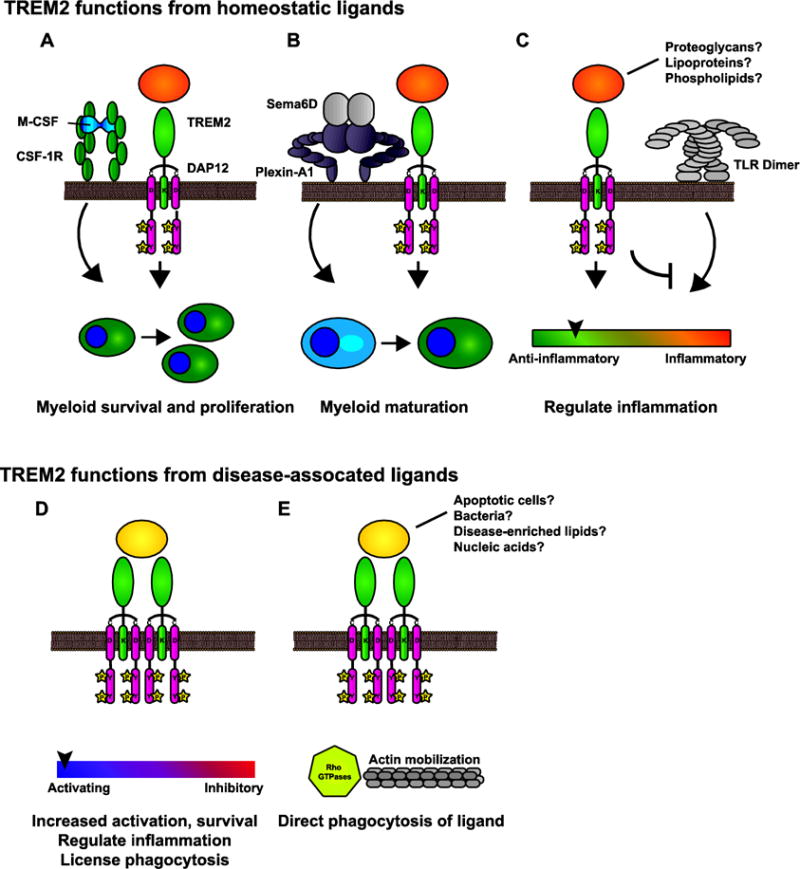
A–C) During normal conditions, TREM2 signaling from endogenous ligands permits a number of functions. A) TREM2 is a co-receptor or co-stimulatory molecule for signaling pathways such as CSF-1R involved in myeloid proliferation and survival. B) TREM2 signals (perhaps in collaboration with Plexin-A1) are important for maturation. C) TREM2 regulates inflammatory responses by influencing TLR and other activating networks to maintain homeostasis. D–E) TREM2 recognizes additional ligands associated with disease. D) These signals may increase activation and survival so that myeloid cells can respond in a context-dependent manner. E) TREM2 can induce phagocytosis of certain ligands, such as bacteria.
TREM2 functions from endogenous signals maintain myeloid cell homeostasis, regulate inflammation, and enable phagocytosis
During development and homeostatic conditions, TREM2 is important for myeloid cell survival, proliferation, and maturation. TREM2 signals ensure myeloid cells remain ready and competent to respond to damage and disease.
1. TREM2 responds to tonic stimuli as a receptor and/or co-receptor for myeloid survival and maturation
This is an attractive, but still undefined mechanism where TREM2 signaling sustains myeloid cell survival and proliferation in order that the myeloid cells can respond to other stimuli in a condition-specific manner. One possible mechanism is a role for TREM2 in β-catenin activation [120], which probably involves interactions with M-CSF signals [117]. Another possibility is TREM2 is not engaging an activating ligand, but is preferentially associating with DAP12 to localize it at the cell surface where it can be phosphorylated by Src or other kinases activated by CSF-1 or other survival pathways (Fig. 4A).
2. TREM2 signals are important for myeloid cell maturation
Studies using human monocytes from NHD patients with mutations in TREM2 observed decreased capacity to mature into osteoclasts and DCs [10, 89]. Similar to collaboration with M-CSF, TREM2 connects Plexin-A1 to DAP12 for osteoclastogenesis [113]. Therefore, TREM2 plays a role in the ability of myeloid cells to mature following stimulation by cytokines (Fig. 4B).
3. TREM2 responds to tonic stimuli to regulate inflammation
TREM2 and DAP12 reduce inflammatory TLR signals [98, 99] in cultured BMDMs. This role did not require stimulation with an exogenous ligand, suggesting it is mediated by a ligand on the BMDMs or perhaps from the culture media. This function likely also involves co-regulation of adaptor proteins that dampen TLR signaling (Fig. 4C).
Which ligands could be important for these functions? Many of these in vitro results did not require antibody stimulation of TREM2, suggesting a ligand present on the myeloid cells or readily available in culture. Phospholipids, proteoglycans, or lipoproteins make attractive candidates as these ligands would be ubiquitous. For example, APOE is highly abundant in serum and CSF, with concentrations in the μg/mL range [140]. However, because these ligands are so ubiquitous, understanding how signaling could be regulated is a challenge.
TREM2 functions during disease
In addition to its role as a tonic receptor, TREM2 recognizes a variety of ligands associated with damage, disease, and infection. These ligands may mimic tonic stimuli or instead may produce an activating signal above the tonic threshold.
1. TREM2 responds to damage stimuli to drive survival and activation of myeloid cells
During disease, TREM2 responds to DAMPs such as ligands exposed on apoptotic membranes, myelin lipids, DNA exposed by necrotic cells, ligands presented by Aβ plaques, or perhaps lipoparticles enriched in apoptotic phospholipids. TREM2 stimulation by these ligands could be important for TREM2 function under the additional stress from damage. In this scenario, TREM2 signaling licenses the response to pathogens and other damage. This cellular activation program can take a number of forms, including sustaining cell survival, regulating cytokine production, and/or permitting phagocytosis by other receptor pathways. For example, TREM2 was required for full phagocytic capacity of antibody-Aβ complexes (which are recognized by FcR for phagocytosis), showing TREM2 enables the phagocytic functions of other receptors [141]. TREM2 inhibits inflammatory cytokine production in cultured microglia following exposure to apoptotic neurons [142], and Trem2-deficient and haploinsufficient microglia have impaired microgliosis and don’t properly respond to plaques [30, 42, 45, 47, 143] (Fig. 4D).
2. TREM2 directly binds DAMPs/PAMPs for phagocytosis
While the primary role of TREM2 may not be phagocytosis, there have been reports of TREM2-mediated phagocytosis of a direct ligand, including phagocytosis of apoptotic neurons that express a TREM2 ligand [37, 125, 142], and bacteria [122] likely through recognition of anionic bacterial components [121]. These examples did not require TREM2 stimulation by antibodies or separate ligands. Instead, cross-linking by bacterial carbohydrates or apoptotic membrane-exposed ligands activate TREM2 signaling, resulting in microtubule mobilization and phagocytosis (Fig. 4E).
Phagocytosis is a good example for how different types of TREM2 signaling and ligands may induce different functions. Studies generally ascribe a pro-phagocytic role for TREM2. For example, some studies have shown ectopic expression of TREM2 is sufficient to promote phagocytosis in non-myeloid cells [25, 122]. However a most interesting study from Knapp and colleagues demonstrated Trem2 deficiency had opposite effects on phagocytosis by bone marrow and alveolar macrophages [144]. In the context of neurodegenerative disease, it has often been assumed that TREM2 has a major role in phagocytosis of debris. However, given that studies have observed increased [30], unchanged [145], and decreased [31] levels of Aβ plaque levels in Trem2-deficient mice, phagocytosis may not be the principal role of TREM2 in AD. Indeed, despite the defect in microgliosis and an apparent role for the plaques in stimulating TREM2 [45], Trem2-haploinsufficient mice did not have a phagocytic deficit. Trem2 deficient microglia display impaired phagocytosis, but this may be an indirect result of even more severe deficits in microglia activation [42, 45]. Another study noted that phagocytosis of Aβ is increased in Trem2-transduced microglia isolated from 7-month-old, but not 18-month-old APPswe/PS1dE9 mice [146], suggesting that TREM2 itself is not sufficient for phagocytosis if other factors lost with age are not present.
The TREM2 crystal structure provides insights into ligand engagement
The human TREM2 Ig domain crystal structure provides some clues as to how TREM2 may interact with various ligands. TREM2 has a pronounced basic patch on its surface that is involved in the recognition of cell-surface proteoglycans and is most likely important in the recognition of additional anionic ligands [26]. The major AD-risk variants (R47H, R62H) are contained in this surface and additional mutations that increase the basic surface (T96K) increase binding to ligands while other R to D mutations in the basic surface decrease cellular binding. Therefore, this surface is involved in recognition of cell-surface proteoglycans. Furthermore, this surface is not observed on TREM1 or the other TREM family crystal structures. Also noteworthy is that circular dichroism spectroscopy studies revealed a subtle conformation change for the R47H variant, but not the R62H or T96K variant. The portion of the CD spectra altered is associated with tryptophan residues within Ig folds. Therefore, it is possible that the R47H variant specifically disrupts another part of the protein structure whereas the other residues likely only impact the basic patch. There is also a hydrophobic surface on the protein that may be involved with recognizing lipidous ligands (Fig 5A). In particular, cell-surface proteoglycans on myeloid cells may act in cis as a tonic ligand for TREM2 while simultaneously orienting or clustering the protein to receive activating signals from other ligands. These signals may be induced through interactions with other surfaces on TREM2 or may result by “hand-off” from GAGs to a higher-affinity ligand that recognizes the basic surface (Fig. 5B). In this model, the basic patch mutations could impact binding to proteoglycans, stimulating ligands, or both to result in impaired signaling (Fig 5C). There are several precedents for receptor-GAG interactions mediating immune signaling, such as CD14-TLR4 [147], CXCL12 chemokine [148], and acetylcholine receptors [149]. To answer these questions, experiments must be performed with the different TREM2 ligands to dissect their effect on the different modules of TREM2 signaling and interplay with other pathways. Epitope-mapping experiments will be needed to confirm binding sites between TREM2 and ligands to understand overlap in TREM2 ligands and also to understand how different ligands interact with TREM2 to result in inhibitory or activating signals. Given that different TREM2 variants produce different results on protein structure and function [25, 26, 28], it will be also important to conduct studies using knock-in models of the mutant proteins. Finally, it is important to consider the role of sTREM2 in disease and to identify its signaling receptors.
Figure 5. Model for TREM2 signal by WT and variant proteins.
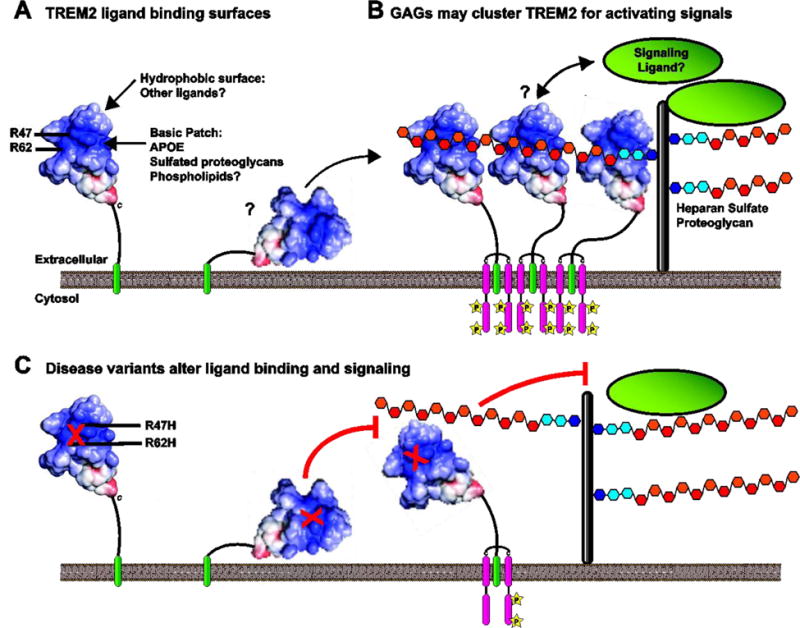
A) TREM2 has a pronounced basic patch, and the disease variants R47 and R62 are part of this surface. TREM2 also has separate surfaces that are potentially involved in ligand binding, such as a hydrophobic surface above the basic patch. B) Role of GAGs in TREM2 signaling. Heparan sulfate proteoglycans interact with TREM2, likely in cis on TREM2-expressing cells. These may function to orient or cluster TREM2 to receive activating signals such as phospholipids or APOE. C) The R47H and R62H variant have reduced binding to cell-surface proteoglycans and have reduced signaling to phospholipids and lipoparticles. The decreased affinity for GAG may impair the ability of these variants to recognize and/or signal in response to these ligands.
Future perspectives: Outstanding questions regarding TREM2
Is there a role for TREM2 in regulating C1q and complement expression in microglia? TREM2 was reported to inhibit expression of C1q in alveolar macrophages [150] and C1q is expressed by microglia during development and improperly during AD to prune neuronal synapses [151].
Is surplus TREM2 signaling harmful? The T96K variant has been shown in two studies to have increased ligand binding or signaling [26, 132], but this variant has not been robustly linked to disease. If gain-of-function TREM2 variants are indeed disease-risk, it would mean therapies that stimulate TREM2 must be carefully assessed.
When is TREM2 function most critical during disease and when could TREM2 agonists or antagonists have the most benefit? TREM2 deficiency is reported to have opposing effects on Aβ levels and disease pathology during early and late disease [43].
What is the role of sTREM2 in AD, and can it be exploited therapeutically? A decoy function would make stimulation of transmembrane TREM2 more difficult. However, if sTREM2 has a beneficial function as a signaling molecule, recombinant sTREM2 could be considered as a therapeutic. Given that sTREM2 is reported to increase both survival and inflammation in cultured microglia [109], it may be necessary to pair sTREM2 with an anti-inflammatory drug to isolate the pro-survival effect. While these possibilities are explored, CSF sTREM2 certainly has potential as a biomarker for microgliosis during AD onset [48]
How do culturing conditions influence the interpretation of cellular assays? TREM2 is likely to be highly influenced by cell density, health of neighboring cells, and perhaps most importantly, the concentration and activity of lipoparticles.
Highlights.
TREM2 is an innate immune receptor with neurodegenerative disease variants
TREM2 contributes to myeloid cell survival and activation
TREM2 is a receptor, but may also act as a soluble signaling molecule
The endogenous ligand is unknown, although many have been suggested
This review suggests how different ligands may contribute to TREM2 signaling
Acknowledgments
This work was supported in part by funding from NIH R01-HL119813 (T.J.B), Knight Alzheimer’s Disease Research Center pilot grant P50-AG005681-30.1 (T.J.B.), Alzheimer’s Association Research Grant AARG-16-441560 (T.J.B.), NIH T32-GM007067 (D.L.K.), and American Heart Association Predoctoral Fellowships 15PRE22110004 and 17PRE32780001 (D.L.K.).
Abbreviations used
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- Akt
Protein kinase B
- ALS
amyotrophic lateral sclerosis
- AM
alveolar macrophage
- APOE
Apolipoprotein E
- BLI
BioLayer Interferometry
- BMDM
Bone marrow-derived macrophage
- C1q
component of the C1 complement protein complex
- CL
cardiolipin
- COPD
chronic obstructive pulmonary disease
- CS
Chondroitin sulfate
- CSF-1R
Colony stimulating factor 1 receptor
- DAG
diacylglycerol
- DAMPs
Damage-associated molecular patters
- DAP12
DNAX-activation protein 12
- DC
Dendritic cell
- ELISA
Enzyme-linked immunosorbent assay
- ERK
extracellular signal-related kinase
- FRET
Förster resonance energy transfer
- GAG
glycosaminoglycan
- GM-CSF
Granulocyte macrophage colony-stimulating factor
- Grb2
Growth factor receptor-bound protein 2
- GSK3-β
Glycogen synthase kinase 3
- HDL
high-density lipoprotein
- HS
Heparan sulfate
- hTREM2
human Triggering receptor expressed on myeloid cells-2
- IBD
inflammatory bowel disorder
- Ig
immunoglobulin
- IL-4
interleukin 4
- IP
immunoprecipitate
- ITAM
Immuno-Tyrosine Activation Motif
- ITIM
Immuno-Tyrosine Inhibitor Motif
- LAB
Linker for the activation of B cells
- LAT
Linker for the activation of T cells
- LDL
low-density lipoprotein
- LMIR5
Leukocyte mono-immunoglobulin (Ig)–like receptor-5 (Also CD300b)
- LOAD
late-onset Alzheimer’s disease
- LOS
Lipooligosaccharides
- LPS
Lipopolysaccharide
- LTA
Lipoteichoic acid
- M-CSF
macrophage colony-stimulating factor
- MAPK
Mitogen-activated protein kinase
- MS
multiple sclerosis
- mTREM2
mouse Triggering receptor expressed on myeloid cells-2
- NF-kB
Nuclear factor kappa B
- NHD
Nasu-Hakola Disease
- NK
Natural killer cell
- NO
nitric oxide
- Nrp1
Neuropilin-1
- PA
phosphatidic acid
- pAkt
phosphorylated-Protein kinase B
- PAMPs
Pathogen-associated molecular patterns
- PBMC
peripheral blood mononuclear cell
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PET
positron emission tomography
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphatidylinositol-3,4,5-trisphosphate
- PLCγ1
phospholipase C γ
- PMA
phorbol 12-myristate 13-acetate
- PS
phosphatidylserine
- RANKL
Receptor activator of nuclear factor kappa-B ligand
- ROS
reactive oxygen species
- Sema3A
Semaphorin-3A
- Sema6D
Semaphorin-6D
- SHIP-1
phosphatidylinositol phosphatase
- SHP-1
Protein tyrosine phosphatase
- SM
sphingomyelin
- SOS
son of sevenless
- SPR
surface plasmon resonance
- sTREM2
soluble TREM2
- TIM
T cell Ig mucin
- TLR
Toll-like receptor
- TREM2
Triggering receptor expressed on myeloid cells-2
- TREML
Triggering receptor expressed on myeloid cells like
References
- 1.Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. 2016;17:201–207. doi: 10.1038/nrn.2016.7. [DOI] [PubMed] [Google Scholar]
- 2.Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J Biol Chem. 2013;288:33027–33036. doi: 10.1074/jbc.M113.517540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. 2014;23:5838–5846. doi: 10.1093/hmg/ddu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paloneva Bm J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, et al. CNS manifestations of Nasu-Hakola disease: A frontal dementia with bone cysts. Neurology. 2001;56:1552–1558. doi: 10.1212/wnl.56.11.1552. [DOI] [PubMed] [Google Scholar]
- 7.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klunemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005;64:1502–1507. doi: 10.1212/01.WNL.0000160304.00003.CA. [DOI] [PubMed] [Google Scholar]
- 9.Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL, Megarbane A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Human mutation. 2008;29:E194–204. doi: 10.1002/humu.20836. [DOI] [PubMed] [Google Scholar]
- 10.Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198:669–675. doi: 10.1084/jem.20030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soragna D, Papi L, Ratti MT, Sestini R, Tupler R, Montalbetti L. An Italian family affected by Nasu-Hakola disease with a novel genetic mutation in the TREM2 gene. J Neurol Neurosurg Psychiatry. 2003;74:825–826. doi: 10.1136/jnnp.74.6.825-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, Schrauwen I, Carvajal J, et al. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiol Aging. 2013;34:2077 e2011–2078. doi: 10.1016/j.neurobiolaging.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, et al. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging. 2013;34:2890 e2891–2895. doi: 10.1016/j.neurobiolaging.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Ber I, De Septenville A, Guerreiro R, Bras J, Camuzat A, Caroppo P, et al. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging. 2014;35:2419 e2423–2415. doi: 10.1016/j.neurobiolaging.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2015;11:1407–1416. doi: 10.1016/j.jalz.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol Neurodegener. 2015;10:19. doi: 10.1186/s13024-015-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruchaga C, Kauwe JSK, Harari O, Jin SC, Cai Y, Karch CM, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78:256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golde TE, Streit WJ, Chakrabarty P. Alzheimer’s disease risk alleles in TREM2 illuminate innate immunity in Alzheimer’s disease. Alzheimers Res Ther. 2013;5:24. doi: 10.1186/alzrt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu G, Liu Y, Jiang Q, Jiang Y, Feng R, Zhang L, et al. Convergent Genetic and Expression Datasets Highlight TREM2 in Parkinson’s Disease Susceptibility. Mol Neurobiol. 2016;53:4931–4938. doi: 10.1007/s12035-015-9416-7. [DOI] [PubMed] [Google Scholar]
- 22.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 Variant p.R47H as a Risk Factor for Sporadic Amyotrophic Lateral Sclerosis. JAMA neurology. 2014;71:449–453. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging. 2014;35:934 e937–910. doi: 10.1016/j.neurobiolaging.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35:726 e711–729. doi: 10.1016/j.neurobiolaging.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra286. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 26.Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife. 2016;5:e20391. doi: 10.7554/eLife.20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JS, Ji IJ, Kim DH, An HJ, Yoon SY. The Alzheimer’s Disease-Associated R47H Variant of TREM2 Has an Altered Glycosylation Pattern and Protein Stability. Front Neurosci. 2016;10:618. doi: 10.3389/fnins.2016.00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JS, Ji IJ, An HJ, Kang MJ, Kang SW, Kim DH, et al. Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus. Traffic. 2015;16:510–518. doi: 10.1111/tra.12264. [DOI] [PubMed] [Google Scholar]
- 29.Piccio L, Deming Y, Del-Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131:925–933. doi: 10.1007/s00401-016-1533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–295. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2014;39:2949–2962. doi: 10.1038/npp.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang T, Yu JT, Zhu XC, Tan MS, Gu LZ, Zhang YD, et al. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014;35:1243–1251. doi: 10.1016/j.neurobiolaging.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 34.Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013;34:2699–2714. doi: 10.1016/j.neurobiolaging.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F, et al. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci. 2004;20:2617–2628. doi: 10.1111/j.1460-9568.2004.03729.x. [DOI] [PubMed] [Google Scholar]
- 37.Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina Bordignon P, et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. Journal of neurochemistry. 2009;110:284–294. doi: 10.1111/j.1471-4159.2009.06130.x. [DOI] [PubMed] [Google Scholar]
- 39.Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. 2015;35:3384–3396. doi: 10.1523/JNEUROSCI.2620-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nature neuroscience. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nature neuroscience. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213:667–675. doi: 10.1084/jem.20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2016 doi: 10.1523/JNEUROSCI.2110-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008;56:1438–1447. doi: 10.1002/glia.20710. [DOI] [PubMed] [Google Scholar]
- 45.Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016;90:724–739. doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nature neuroscience. 2015;18:800–806. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, et al. Altered microglial repsonse to AB plaques in APS1-21 mice heterozygous for TREM2. Molecular Neurodegeneration. 2014;9 doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016;8:369ra178. doi: 10.1126/scitranslmed.aag1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pimplikar SW. Neuroinflammation in Alzheimer’s disease: from pathogenesis to a therapeutic target. J Clin Immunol. 2014;34(Suppl 1):S64–69. doi: 10.1007/s10875-014-0032-5. [DOI] [PubMed] [Google Scholar]
- 50.Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, et al. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology. 2016;105:196–206. doi: 10.1016/j.neuropharm.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 51.Jiang T, Tan L, Zhu XC, Zhou JS, Cao L, Tan MS, et al. Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol Aging. 2015;36:3176–3186. doi: 10.1016/j.neurobiolaging.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 52.Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8:466–476. doi: 10.15252/emmm.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol Neurodegener. 2016;11:3. doi: 10.1186/s13024-016-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henjum K, Almdahl IS, Arskog V, Minthon L, Hansson O, Fladby T, et al. Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimers Res Ther. 2016;8:17. doi: 10.1186/s13195-016-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glebov K, Wunderlich P, Karaca I, Walter J. Functional involvement of γ-secretase in signaling of the triggering receptor expressed on myeloid cells-2 (TREM2) Journal of Neuroinflammation. 2016;13:17. doi: 10.1186/s12974-016-0479-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 57.Czirr E, Wyss-Coray T. The immunology of neurodegeneration. The Journal of Clinical Investigation. 2012;122:1156–1163. doi: 10.1172/JCI58656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s & Dementia. 2016;12:719–732. doi: 10.1016/j.jalz.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 59.Pimplikar SW. Neuroinflammation in Alzheimer’s Disease: from Pathogenesis to a Therapeutic Target. Journal of Clinical Immunology. 2014;34:64–69. doi: 10.1007/s10875-014-0032-5. [DOI] [PubMed] [Google Scholar]
- 60.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 61.McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126:479–497. doi: 10.1007/s00401-013-1177-7. [DOI] [PubMed] [Google Scholar]
- 62.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 63.Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, et al. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Molecular Neurodegeneration. 2015;10:52. doi: 10.1186/s13024-015-0048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015;129:429–447. doi: 10.1007/s00401-015-1388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015;125:2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu C, Herrmann US, Li B, Abakumova I, Moos R, Schwarz P, et al. Triggering receptor expressed on myeloid cells-2 is involved in prion-induced microglial activation but does not contribute to prion pathogenesis in mouse brains. Neurobiol Aging. 2015;36:1994–2003. doi: 10.1016/j.neurobiolaging.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 67.Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, et al. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8:e52982. doi: 10.1371/journal.pone.0052982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawabori M, Hokari M, Zheng Z, Kim JY, Calosing C, Hsieh CL, et al. Triggering Receptor Expressed on Myeloid Cells-2 Correlates to Hypothermic Neuroprotection in Ischemic Stroke. Ther Hypothermia Temp Manag. 2013;3:189–198. doi: 10.1089/ther.2013.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131:3081–3091. doi: 10.1093/brain/awn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gispert JD, Suarez-Calvet M, Monte GC, Tucholka A, Falcon C, Rojas S, et al. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2016;12:1259–1272. doi: 10.1016/j.jalz.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 71.Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med. 2015;212:681–697. doi: 10.1084/jem.20141732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, Stappenbeck TS. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci U S A. 2009;106:256–261. doi: 10.1073/pnas.0803343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Correale C, Genua M, Vetrano S, Mazzini E, Martinoli C, Spinelli A, et al. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology. 2013;144:346–356 e343. doi: 10.1053/j.gastro.2012.10.040. [DOI] [PubMed] [Google Scholar]
- 74.Paloneva J, Kestila M, Wu J, Salminen A, Bohling T, Ruotsalainen V, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25:357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- 75.Charles JF, Humphrey MB, Zhao X, Quarles E, Nakamura MC, Aderem A, et al. The innate immune response to Salmonella enterica serovar Typhimurium by macrophages is dependent on TREM2-DAP12. Infect Immun. 2008;76:2439–2447. doi: 10.1128/IAI.00115-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu M, Li D, Wu Y, Huang X, Wu M. TREM-2 promotes macrophage-mediated eradication of Pseudomonas aeruginosa via a PI3K/Akt pathway. Scand J Immunol. 2014;79:187–196. doi: 10.1111/sji.12148. [DOI] [PubMed] [Google Scholar]
- 77.Wei P, Lu Q, Cui G, Guan Z, Yang L, Sun C, et al. The role of TREM-2 in internalization and intracellular survival of Brucella abortus in murine macrophages. Vet Immunol Immunopathol. 2015;163:194–201. doi: 10.1016/j.vetimm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–707. doi: 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- 80.Tomasello E, Olcese L, Vely F, Geourgeon C, Blery M, Moqrich A, et al. Gene structure, expression pattern, and biological activity of mouse killer cell activating receptor-associated protein (KARAP)/DAP-12. Journal of Biological Chemistry. 1998;273:34115–34119. doi: 10.1074/jbc.273.51.34115. [DOI] [PubMed] [Google Scholar]
- 81.Call ME, Wucherpfennig KW, Chou JJ. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol. 2010;11:1023–1029. doi: 10.1038/ni.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wei P, Zheng BK, Guo PR, Kawakami T, Luo SZ. The association of polar residues in the DAP12 homodimer: TOXCAT and molecular dynamics simulation studies. Biophys J. 2013;104:1435–1444. doi: 10.1016/j.bpj.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheng X, Im W. NMR observable-based structure refinement of DAP12-NKG2C activating immunoreceptor complex in explicit membranes. Biophys J. 2012;102:L27–29. doi: 10.1016/j.bpj.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Call ME, Wucherpfennig KW. Common themes in the assembly and architecture of activating immune receptors. Nat Rev Immunol. 2007;7:841–850. doi: 10.1038/nri2186. [DOI] [PubMed] [Google Scholar]
- 85.Humphrey MB, Daws MR, Spusta SC, Niemi EC, Torchia JA, Lanier LL, et al. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res. 2006;21:237–245. doi: 10.1359/JBMR.051016. [DOI] [PubMed] [Google Scholar]
- 86.Inui M, Kikuchi Y, Aoki N, Endo S, Maeda T, Sugahara-Tobinai A, et al. Signal adaptor DAP10 associates with MDL-1 and triggers osteoclastogenesis in cooperation with DAP12. Proc Natl Acad Sci U S A. 2009;106:4816–4821. doi: 10.1073/pnas.0900463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McVicar DW, Taylor LS, Gosselin P, Willette-Brown J, Mikhael AI, Geahlen RL, et al. DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J Biol Chem. 1998;273:32934–32942. doi: 10.1074/jbc.273.49.32934. [DOI] [PubMed] [Google Scholar]
- 88.Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cella M, Buonsanti C, Strader C, Kondo T, Salmaggi A, Colonna M. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med. 2003;198:645–651. doi: 10.1084/jem.20022220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3:ra38. doi: 10.1126/scisignal.2000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Orr SJ, McVicar DW. LAB/NTAL/Lat2: a force to be reckoned with in all leukocytes? J Leukoc Biol. 2011;89:11–19. doi: 10.1189/jlb.0410221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Whittaker GC, Orr SJ, Quigley L, Hughes L, Francischetti IM, Zhang W, et al. The linker for activation of B cells (LAB)/non-T cell activation linker (NTAL) regulates triggering receptor expressed on myeloid cells (TREM)-2 signaling and macrophage inflammatory responses independently of the linker for activation of T cells. J Biol Chem. 2010;285:2976–2985. doi: 10.1074/jbc.M109.038398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol. 2006;36:1646–1653. doi: 10.1002/eji.200636195. [DOI] [PubMed] [Google Scholar]
- 94.Rao N, Dodge I, Band H. The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol. 2002;71:753–763. [PubMed] [Google Scholar]
- 95.Washington AV, Quigley L, McVicar DW. Initial characterization of TREM-like transcript (TLT)-1: a putative inhibitory receptor within the TREM cluster. Blood. 2002;100:3822–3824. doi: 10.1182/blood-2002-02-0523. [DOI] [PubMed] [Google Scholar]
- 96.Yoon SH, Lee YD, Ha J, Lee Y, Kim HH. TLT-1s, alternative transcripts of triggering receptor expressed on myeloid cell-like transcript-1 (TLT-1), Inhibits the triggering receptor expressed on myeloid cell-2 (TREM-2)-mediated signaling pathway during osteoclastogenesis. J Biol Chem. 2012;287:29620–29626. doi: 10.1074/jbc.M112.351239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3:445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- 98.Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. 2006;177:2051–2055. doi: 10.4049/jimmunol.177.4.2051. [DOI] [PubMed] [Google Scholar]
- 99.Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177:3520–3524. doi: 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- 100.Ito H, Hamerman JA. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol. 2012;42:176–185. doi: 10.1002/eji.201141679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 102.Hamerman JA, Lanier LL. Inhibition of immune responses by ITAM-bearing receptors. Sci STKE. 2006;2006:re1. doi: 10.1126/stke.3202006re1. [DOI] [PubMed] [Google Scholar]
- 103.Xing J, Titus AR, Humphrey MB. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res Rep Biochem. 2015;5:89–100. doi: 10.2147/RRBC.S58057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Turnbull IR, Colonna M. Activating and inhibitory functions of DAP12. Nat Rev Immunol. 2007;7:155–161. doi: 10.1038/nri2014. [DOI] [PubMed] [Google Scholar]
- 105.Zhong L, Chen XF, Zhang ZL, Wang Z, Shi XZ, Xu K, et al. DAP12 Stabilizes the C-terminal Fragment of the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) and Protects against LPS-induced Pro-inflammatory Response. J Biol Chem. 2015;290:15866–15877. doi: 10.1074/jbc.M115.645986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gomez-Pina V, Soares-Schanoski A, Rodriguez-Rojas A, Del Fresno C, Garcia F, Vallejo-Cremades MT, et al. Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J Immunol. 2007;179:4065–4073. doi: 10.4049/jimmunol.179.6.4065. [DOI] [PubMed] [Google Scholar]
- 107.Gattis JL, Washington AV, Chisholm MM, Quigley L, Szyk A, McVicar DW, et al. The structure of the extracellular domain of triggering receptor expressed on myeloid cells like transcript-1 and evidence for a naturally occurring soluble fragment. J Biol Chem. 2006;281:13396–13403. doi: 10.1074/jbc.M600489200. [DOI] [PubMed] [Google Scholar]
- 108.Haselmayer P, Grosse-Hovest L, von Landenberg P, Schild H, Radsak MP. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood. 2007;110:1029–1035. doi: 10.1182/blood-2007-01-069195. [DOI] [PubMed] [Google Scholar]
- 109.Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017 doi: 10.1084/jem.20160844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Peng Q, Long CL, Malhotra S, Humphrey MB. A physical interaction between the adaptor proteins DOK3 and DAP12 is required to inhibit lipopolysaccharide signaling in macrophages. Sci Signal. 2013;6:ra72. doi: 10.1126/scisignal.2003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature. 2012;485:69–74. doi: 10.1038/nature11000. [DOI] [PubMed] [Google Scholar]
- 112.Kumanogoh A, Kikutani H. Immunological functions of the neuropilins and plexins as receptors for semaphorins. Nature Reviews Immunology. 2013;13:802–814. doi: 10.1038/nri3545. [DOI] [PubMed] [Google Scholar]
- 113.Takegahara N, Takamatsu H, Toyofuku T, Tsujimura T, Okuno T, Yukawa K, et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol. 2006;8:615–622. doi: 10.1038/ncb1416. [DOI] [PubMed] [Google Scholar]
- 114.Bakker ABH, Hoek RM, Cerwenka A, Blom B, Lucian L, McNeil T, et al. DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity. 2000;13:345–353. doi: 10.1016/s1074-7613(00)00034-0. [DOI] [PubMed] [Google Scholar]
- 115.Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, et al. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A. 2004;101:6158–6163. doi: 10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y, Su J, Wu S, Teng Y, Yin Z, Guo Y, et al. DDR2 (discoidin domain receptor 2) suppresses osteoclastogenesis and is a potential therapeutic target in osteoporosis. Sci Signal. 2015;8:ra31. doi: 10.1126/scisignal.2005835. [DOI] [PubMed] [Google Scholar]
- 117.Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, et al. TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol. 2012;188:2612–2621. doi: 10.4049/jimmunol.1102836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol. 2009;10:734–743. doi: 10.1038/ni.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell. 2008;31:422–431. doi: 10.1016/j.molcel.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J Neurosci. 2017;37:1772–1784. doi: 10.1523/JNEUROSCI.2459-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol. 2003;171:594–599. doi: 10.4049/jimmunol.171.2.594. [DOI] [PubMed] [Google Scholar]
- 122.N’Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184:215–223. doi: 10.1083/jcb.200808080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Phongsisay V. Campylobacter jejuni targets immunoglobulin-like receptor LMIR5. Mol Immunol. 2015;63:574–578. doi: 10.1016/j.molimm.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 124.Quan DN, Cooper MD, Potter JL, Roberts MH, Cheng H, Jarvis GA. TREM-2 binds to lipooligosaccharides of Neisseria gonorrhoeae and is expressed on reproductive tract epithelial cells. Mucosal Immunology. 2008;1:229–238. doi: 10.1038/mi.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina Bordignon P, et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem. 2009;110:284–294. doi: 10.1111/j.1471-4159.2009.06130.x. [DOI] [PubMed] [Google Scholar]
- 126.Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007;37:1290–1301. doi: 10.1002/eji.200636837. [DOI] [PubMed] [Google Scholar]
- 127.Cannon JP, O’Driscoll M, Litman GW. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics. 2012;64:39–47. doi: 10.1007/s00251-011-0562-4. [DOI] [PubMed] [Google Scholar]
- 128.Bailey CC, DeVaux LB, Farzan M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem. 2015;290:26033–26042. doi: 10.1074/jbc.M115.677286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Simhadri VR, Andersen JF, Calvo E, Choi SC, Coligan JE, Borrego F. Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood. 2012;119:2799–2809. doi: 10.1182/blood-2011-08-372425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD, et al. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc Natl Acad Sci U S A. 2014;111:E1463–1472. doi: 10.1073/pnas.1320174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2016 doi: 10.1016/j.jalz.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Giuliano JS, Jr, Lahni PM, Wong HR, Wheeler DS. Pediatric Sepsis - Part V: Extracellular Heat Shock Proteins: Alarmins for the Host Immune System. Open Inflamm J. 2011;4:49–60. doi: 10.2174/1875041901104010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) J Biol Chem. 2015;290:26043–26050. doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron. 2016;91:328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 136.Park M, Yi JW, Kim EM, Yoon IJ, Lee EH, Lee HY, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes. 2015;64:117–127. doi: 10.2337/db13-1869. [DOI] [PubMed] [Google Scholar]
- 137.Frieden C. ApoE: the role of conserved residues in defining function. Protein Sci. 2015;24:138–144. doi: 10.1002/pro.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JA, Holtzman DM, et al. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81:740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016;8:992–1004. doi: 10.15252/emmm.201606370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ulrich JD, Holtzman DM. TREM2 Function in Alzheimer’s Disease and Neurodegeneration. ACS Chem Neurosci. 2016;7:420–427. doi: 10.1021/acschemneuro.5b00313. [DOI] [PubMed] [Google Scholar]
- 144.Sharif O, Gawish R, Warszawska JM, Martins R, Lakovits K, Hladik A, et al. The Triggering Receptor Expressed on Myeloid Cells 2 Inhibits Complement Component 1q Effector Mechanisms and Exerts Detrimental Effects during Pneumococcal Pneumonia. PLOS Pathogens. 2014;10:e1004167. doi: 10.1371/journal.ppat.1004167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9 doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jiang T, Wan Y, Zhang YD, Zhou JS, Gao Q, Zhu XC, et al. TREM2 Overexpression has Improvement on Neuropathology and Cognitive Impairment in Aging APPswe/PS1dE9 Mice. Mol Neurobiol. 2017;54:855–865. doi: 10.1007/s12035-016-9704-x. [DOI] [PubMed] [Google Scholar]
- 147.O’Callaghan P, Li JP, Lannfelt L, Lindahl U, Zhang X. Microglial Heparan Sulfate Proteoglycans Facilitate the Cluster-of-Differentiation 14 (CD14)/Toll-like Receptor 4 (TLR4)-Dependent Inflammatory Response. J Biol Chem. 2015;290:14904–14914. doi: 10.1074/jbc.M114.634337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J Biol Chem. 2007;282:10018–10027. doi: 10.1074/jbc.M608796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McDonnell KM, Grow WA. Reduced glycosaminoglycan sulfation diminishes the agrin signal transduction pathway. Dev Neurosci. 2004;26:1–10. doi: 10.1159/000080706. [DOI] [PubMed] [Google Scholar]
- 150.Sharif O, Gawish R, Warszawska JM, Martins R, Lakovits K, Hladik A, et al. The triggering receptor expressed on myeloid cells 2 inhibits complement component 1q effector mechanisms and exerts detrimental effects during pneumococcal pneumonia. PLoS Pathog. 2014;10:e1004167. doi: 10.1371/journal.ppat.1004167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 2007;4:e124. doi: 10.1371/journal.pmed.0040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen Q, Zhang K, Jin Y, Zhu T, Cheng B, Shu Q, et al. Triggering receptor expressed on myeloid cells-2 protects against polymicrobial sepsis by enhancing bacterial clearance. Am J Respir Crit Care Med. 2013;188:201–212. doi: 10.1164/rccm.201211-1967OC. [DOI] [PubMed] [Google Scholar]
- 154.Gawish R, Martins R, Bohm B, Wimberger T, Sharif O, Lakovits K, et al. Triggering receptor expressed on myeloid cells-2 fine-tunes inflammatory responses in murine Gram-negative sepsis. FASEB J. 2015;29:1247–1257. doi: 10.1096/fj.14-260067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sun M, Zhu M, Chen K, Nie X, Deng Q, Hazlett LD, et al. TREM-2 Promotes Host Resistance Against Pseudomonas aeruginosa Infection by Suppressing Corneal Inflammation via a PI3K/Akt Signaling PathwayTREM-2 in Corneal Inflammation. Investigative Ophthalmology & Visual Science. 2013;54:3451–3462. doi: 10.1167/iovs.12-10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Phongsisay V, Iizasa E, Hara H, Yoshida H. Evidence for TLR4 and FcRgamma-CARD9 activation by cholera toxin B subunit and its direct bindings to TREM2 and LMIR5 receptors. Mol Immunol. 2015;66:463–471. doi: 10.1016/j.molimm.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 157.Phongsisay V, Iizasa E, Hara H, Yamasaki S. 3-O-sulfo-beta-D-galactose moiety of endogenous sulfoglycolipids is a potential ligand for immunoglobulin-like receptor LMIR5. Mol Immunol. 2015;63:595–599. doi: 10.1016/j.molimm.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 158.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 159.Kong Y, Janssen BJ, Malinauskas T, Vangoor VR, Coles CH, Kaufmann R, et al. Structural Basis for Plexin Activation and Regulation. Neuron. 2016;91:548–560. doi: 10.1016/j.neuron.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Janssen BJ, Robinson RA, Perez-Branguli F, Bell CH, Mitchell KJ, Siebold C, et al. Structural basis of semaphorin-plexin signalling. Nature. 2010;467:1118–1122. doi: 10.1038/nature09468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Felix J, De Munck S, Verstraete K, Meuris L, Callewaert N, Elegheert J, et al. Structure and Assembly Mechanism of the Signaling Complex Mediated by Human CSF-1. Structure. 2015;23:1621–1631. doi: 10.1016/j.str.2015.06.019. [DOI] [PubMed] [Google Scholar]