

Abstract
p62/sequestosome-1 (SQSTM1) is a multifunctional adaptor protein and autophagic substrate which accumulates in cells with hyperactive mTORC1, such as kidney cells with mutations in the tumor suppressor genes TSC1 or TSC2. Here we report that p62 is a critical mediator of TSC2-driven tumorigenesis, as Tsc2+/− and Tsc2f/f Ksp-CreERT2+ mice crossed to p62−/− mice were protected from renal tumor development. Metabolic profiling revealed that depletion of p62 in Tsc2-null cells decreased intracellular glutamine, glutamate, and glutathione (GSH). p62 positively regulated the glutamine transporter Slc1a5 and increased glutamine uptake in Tsc2-null cells. We also observed p62-dependent changes in Gcl, Gsr, Nqo1 and Srxn1 which were decreased by p62 attenuation and implicated in GSH production and utilization. p62 attenuation altered mitochondrial morphology, reduced mitochondrial membrane polarization and maximal respiration, and increased mitochondrial ROS and mitophagy marker PINK1. These mitochondrial phenotypes were rescued by addition of exogenous GSH and overexpression of Sod2, which suppressed indices of mitochondrial damage and promoted growth of Tsc2-null cells. Finally, p62 depletion sensitized Tsc2-null cells to both oxidative stress and direct inhibition of glutathione biosynthesis by buthionine sulfoximine (BSO). Our findings show how p62 helps maintain intracellular pools of glutathione needed to limit mitochondrial dysfunction in tumor cells with elevated mTORC1, highlighting p62 and redox homeostasis as nodal vulnerabilities for therapeutic targeting in these tumors.
Keywords: p62, mTORC1, Tuberous Sclerosis Complex, glutathione, mitochondria
Introduction
Tuberous sclerosis complex (TSC) is a multisystem disease in which hamartomatous tumors develop in the brain, skin, heart, kidney, and lung caused by germline inactivating mutations in the TSC1 or TSC2 genes (1). The TSC protein complex, which includes TSC1 (also called hamartin), TSC2 (also called tuberin) and TBC1D7, negatively regulates mechanistic/mammalian Target of Rapamycin Complex 1 (mTORC1) via the small GTPase Rheb (2). mTORC1 activates anabolic processes including ribosome expression, protein translation and lipid and nucleotide synthesis, while inhibiting catabolic processes, such as autophagy. In healthy cells, these processes are tightly coordinated in response to multiple factors including growth factor signaling, oxygen tension, and nutrient availability. In contrast, in TSC-null cells, mTORC1 activation is largely uncoupled from upstream regulation (2–4).
mTORC1 hyperactive cells are characterized by metabolic reprogramming, which makes the cells dependent upon glucose and glutamine uptake, as well as autophagy particularly in settings of metabolic stress (5–9). Tsc2-null cells also have HIF1-dependent activation of glycolysis and increased endoplasmic reticulum stress (10,11). Therefore, tumor cells in TSC require extensive adaptation to survive and proliferate with elevated biosynthetic requirements, bioenergetic demands, and low autophagy. Autophagy appears to be a particularly critical node through which TSC2-null cells are vulnerable to bioenergetic stress (7,9).
The autophagic substrate, p62/sequestosome 1, is upregulated in TSC lesions and TSC2-null cells as a result of reduced autophagic flux (7). p62 is a signaling hub and scaffold protein with numerous functional domains and interacting partners (12). p62 is recruited to autophagosomes via direct interaction with the autophagosome membrane component, LC3B, and can promote selective autophagy of ubiquitinated cargo through an ubiquitin binding domain (13). Through the ability of p62 to interact with TRAF6, p62 activates NF-κB (14). Recently, p62 has been shown to promote genomic instability by slowing the repair of DNA double strand breaks, thereby promoting error prone nonhomologous end joining (NHEJ) (15). p62 also sequesters Keap1 from Nrf2 and promotes activation of the antioxidant transcription pathway (16–18), while also acting as a direct positive regulator of mTORC1 through interactions with Raptor and the Rag proteins in the amino acid-sensing pathway (19,20).
Autophagy deficiency is one mechanism by which p62 is upregulated in tumors. p62 was also shown to be upregulated transcriptionally by AP1 and NF-κB downstream of Ras activation, and in renal cell carcinomas following 5q amplification (21–23). p62 drives tumorigenesis in several models, in a cell context dependent manner, through upregulation of antioxidant pathways via Nrf2 activation by chelating Keap1 (24–26), and/or the transcriptional activation of NF-κB (22,23).
Little is known about the impact of p62 on tumorigenesis in cells with hyperactive mTORC1. We report here that p62 promotes renal tumorigenesis in two genetically engineered murine models of TSC. TSC2-null cells depend upon p62 to maintain cellular redox homeostasis and cell survival through preservation of cellular glutathione pools and mitochondrial function. These findings provide evidence for the critical role of p62 in facilitating the growth of mTORC1-hyperactivated tumors. Our data suggest that direct downstream targeting of p62-specific redox pathways will provide innovative therapeutic strategies for tumors with mTORC1-hyperactivation.
Materials and Methods
Cell lines and culture
Tsc2−/−p53−/− and Tsc2+/+p53−/− mouse embryonic fibroblasts (MEFs) were authenticated and provided by David Kwiatkowski (Brigham and Women’s Hospital, Boston, MA). The Tsc2−/−p53−/− and Tsc2+/+p53−/− cells were obtained from the Kwiatkowski laboratory in December 2005 and banked at passage 8. Cells were subsequently thawed in 2013 and p62 shRNAs were introduced. Experiments were then performed between passage 16 and 30. The identity of the MEFs was recently confirmed by multiplex PCR assay targeting short tandem repeat markers. All cell lines were validated for each experiment by immunoblot analysis to confirm Tsc2 and p62 expression status in the cell lines and aberrant mTORC1 hyperactivation by phosphorylation of ribosomal subunit S6 (S235/236) in serum free conditions. All cells tested negative for mycoplasma contamination using MycoAlert (Lonza) and were re-tested monthly. Cells were cultured at 37°C in 5% CO2 in DMEM supplemented with 10% FBS and gentamycin sulfate (50 μg/mL). For serum free conditions, cells were cultured in DMEM without serum. Cells were maintained prior to experiments in puromycin (5 μg/mL) to select for short hairpin RNA expression.
Antibodies, Drugs, and shRNA Reagents
The following antibodies were used: p62 (Sigma-Aldrich), p62 (abcam), TOMM20, Nrf2, Keap1 (Santa Cruz Biotechnology), P-S6 (S235/236), NFkB, GAPDH, and CREB (Cell Signaling Technology), PINK1 (Novus). Pegylated superoxide dismutase (SOD), catalase and L-Buthionine sulfoximine (BSO) were purchased from Sigma-Aldrich and rapamycin was purchased from LC Laboratories. shRNA targeting p62 were obtained from the RNAi Consortium (TRC): shp62-1 (sh133) TRCN0000238133 TAGTACAACTGCTAGTTATTT, shp62-2 (sh615) TRCN0000098615 CCCTTTGTCTTGTAGTTGCAT, shp62-3 (sh616) TRCN0000098616 CCGCATCTACATTAAAGAGAA ORFs for mouse Sod2 (Lenti-05359-152), Catalase (Lenti-34173-152) and empty (Lenti-Neg-152) lentiviral vector were purchased from Genecopoeia.
Immunohistochemistry
Kidneys were isolated, fixed in 10% formalin, embedded in paraffin, and sectioned onto positively charged slides. Sections were deparaffinized in gradient ethanol and xylenes, then heat-induced antigen retrieval was performed. Sections were blocked in 5% goat serum and incubated with primary antibodies overnight at 4°C. Immunoreactivity was detected using the EnVision+HRP-DAB system (Dako, Denmark) with hematoxylin counterstain. Images were acquired using the Olympus BX63 light microscope or Zeiss FSX1000 with 4.2x or 40x objectives.
Immunofluorescence Staining
Cells were fixed with 2% paraformaldehyde in PBS, washed with PBS, then permeabilized with 0.1% Triton-X for 15 minutes. Cells were blocked in 2% bovine serum albumin (BSA) for 45 minutes. Cells were then incubated with primary antibodies for 1h at room temperature, washed and incubated with fluorophore conjugated secondary antibodies for another hour. Nuclei were visualized with DAPI. Cells were imaged using the Olympus Fluoview FV10i confocal microscope with 60× objective.
Metabolite Profiling and Analysis
Metabolites were extracted with 80% aqueous methanol from 5 replicate 60 mm plates. Metabolites were processed and analyzed using Selected Reaction Monitoring (SRM) with polarity switching on a 5500 QTRAP triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) using amide HILIC chromatography (Waters) at pH 9.2. Two hundred fifty-nine endogenous water soluble metabolites were measured at steady state. The resulting raw metabolomics data were normalized to protein concentration of 3 additional replicate plates and uploaded into MetaboAnalyst 2.0 (http://www.metaboanalyst.ca/MetaboAnalyst/) for subsequent processing and metabolite set enrichment analysis.
Ion Torrent Sample Preparation, Sequencing and Analysis
Expression of 3,826 mouse genes was determined with a custom panel ordered from Thermo Fisher Scientific, use ion AmpliSeq library construction kit and run on the Ion Proton System for next generation sequencing according to manufacturer’s protocols (Thermo Fisher Scientific). 10 ng of mRNA for each of 6 samples were isolated using the miRcury mRNA Isolation Cell and Plant Kit (Exiqon) prior to reverse transcription with the Superscript VILO cDNA Sythesis Kit (Thermo Fisher Scientific). Data were analyzed using Torrent Suite Software 4.4 and Ion Reporter software (Thermo Fisher Scientific) and exported to Excel.
For normalization and downstream analysis, the edgeR package (27) was used to compute differential expression. The software’s built-in normalization methods and tagwise dispersion were used to determine the fold-change differential expression. Gene set enrichment analysis (GSEA) was performed on the differentially expressed genes using annotations from The Broad Institutes MSIG database (28) and correcting for false positives using the Benjamini-Hochberg method.
Network Analysis
To generate the oxidative stress network, direct interactions from the consolidated interactome were mapped among the oxidative stress seed genes (identified by Ion Torrent analysis and GSEA; Supplementary Table S1) as well as their direct interactors (29). Network interacting genes were iteratively added, based on a hypergeometric distribution to enrich for those interactors with the most statistically significant connections to the seed network. The resulting oxidative stress network contained 50 genes and 211 interactions.
Glutamine Uptake Assay
Cells seeded 3–5×104 into 12-well plates were incubated with L-[U-14C]glutamine in phenol red free and pyruvate free media supplemented with 5% dialyzed FBS and 1 mM L-glutamine for 45 minutes. Cells were then washed 3 times with PBS and collected with phenol red free trypsin and PBS. Counts per minute (CPM) were determined using a Packard Bioscience liquid scintillation analyzer. Cell number was then counted in replicate wells for data normalization.
Animal Studies
All animal studies were performed in accordance with institutional protocols approved by Boston Children’s Hospital Animal Care and Use Committee. The severity of renal lesions was scored using previously established macroscopic and microscopic scoring methods (30,31). Macroscopic cysts per kidney were scored according to size: < 1mm, score 1; 1–1.5, score 2; 1.5–2, score 5; and > 2, score 10. The sum of the cyst scores were determined and reported per kidney. Microscopic kidney tumor scores were determined in a semi-quantitative fashion by a blinded observer from hematoxylin and eosin (H&E) 5 μm sections of samples prepared by embedding 1 mm-interval sections through the kidney. Each tumor or cyst identified was measured (length, width) and percent of the lumen filled by tumor determined (0% for a simple cyst, and 100% for a completely filled, solid tumor). The measurements were converted into a score using a previously established formula (30).
Statistical Analyses
In vivo data are presented as the mean +/− 95% confidence interval (CI) and in vitro studies are presented as the mean +/− standard deviation (SD). Normally distributed data were analyzed for statistical significance with Student’s unpaired t-test and multiple comparisons were made with one-way and two-way ANOVAs with Bonferroni correction. Alternatively, nonparametric Kruskal-Wallis and Dunn’s correction were used (GraphPad Prism version 6 for Mac; GraphPad Software, www.graphpad.com). Statistical significance was defined as p < 0.05.
Results
p62 promotes kidney cystogenesis and cystadenoma formation
Tsc2+/− mice develop spontaneous renal cysts, cystadenomas, and cystadenocarcinomas as a result of mTORC1 hyperactivation (32). To determine how p62 impacts mTORC1-driven tumorigenesis, Tsc2+/− mice were crossed to p62−/− mice and kidney tumors were assessed (Fig. 1A, B and C) (30,31). The macroscopic cystic index, based on cyst number and size, was significantly reduced in Tsc2+/− p62+/− (mean macroscopic cystic index 5.6, p<0.001) and Tsc2+/− p62−/− (mean macroscopic cystic index 7.8, p<0.05) mice when compared with littermate control Tsc2+/− p62+/+ (mean macroscopic cystic index 14.8) mice (Fig. 1B). Microscopic score, an indication of progression to cystic filling and formation of tumors, was also significantly lower in Tsc2+/− p62−/− mice (microscopic index 0.1) when compared with Tsc2+/− p62+/+ mice (microscopic index 0.8, p<0.001; Fig. 1C). As expected, phosphorylation of ribosomal protein S6 (P-S6, S234/236), a marker of mTORC1 hyperactivation, as well as p62, was elevated in the renal lesions of Tsc2+/− mice compared to adjacent normal kidney (Fig. 1D) (7).
Figure 1.
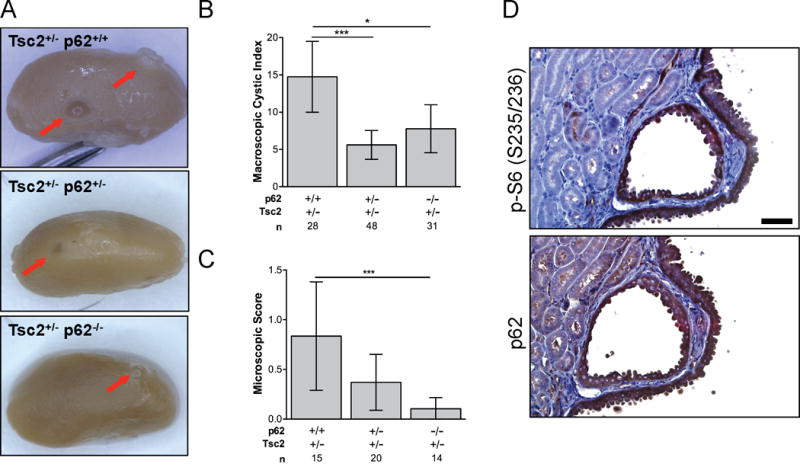
p62 promotes kidney tumorigenesis in Tsc2+/− mice. A, Representative kidneys of 10-month Tsc2+/− p62+/+, Tsc2+/− p62+/− and Tsc2+/− p62−/− littermates. Arrows indicate macroscopic cysts. B, The macroscopic cystic index was assessed for each kidney using an established method based on the number and size of the lesions. C, The microscopic score was determined by an established method for assessing cyst size and tumor filling from H&E sections. D, Phosphorylated S6 (S235/236) and p62 expression in cystadenoma lesions of Tsc2+/− mice. Scale bar, 50 μm. n is number of kidneys analyzed. Data presented as mean +/− 95% CI with *p<0.05, ***p<0.001.
To better understand the impact of p62 on mTORC1-driven renal tumorigenesis in the kidney epithelial compartment, Tsc2-floxed (Tsc2f/f) mice were crossed to KSP-CreERT2+ mice carrying a tamoxifen-inducible Cre recombinase under the kidney specific cadherin promoter, which is expressed in the collecting ducts, loops of Henle, and distal tubules of adult mice. These mice were then crossed to total body p62−/− mice. Recombination was induced by tamoxifen treatment in 8–10 week-old mice with 63 mg/kg tamoxifen given by intraperitoneal injection every other day for 5 days. The kidney phenotype was then assessed in 6 month-old mice. Tsc2f/f Ksp-CreERT2+ p62+/+ mice developed numerous microscopic cystic lesions (Fig. 2A), with a ~2-fold increase in cross-sectional kidney area (p<0.0001; Fig. 2B) and a 2.5-fold increase in kidney weight (p<0.01; Fig. 2C) compared to control Tsc2f/f Ksp-CreERT2− p62+/+ and corn oil injected Tsc2f/f Ksp-CreERT2+ p62+/+ mice. In contrast, the kidney phenotype of Tsc2f/f Ksp-CreERT2+ p62−/− mice was significantly attenuated, by about 20% in kidney area (p<0.05) and 40% in kidney mass (p<0.05). These data demonstrate that p62 is critical for the abnormal proliferation of Tsc2-deficient kidney epithelial cells necessary for kidney cystogenesis, and suggest cooperativity between mTORC1-hyperactivation and p62-dependent tumorigenic pathways in the kidney.
Figure 2.
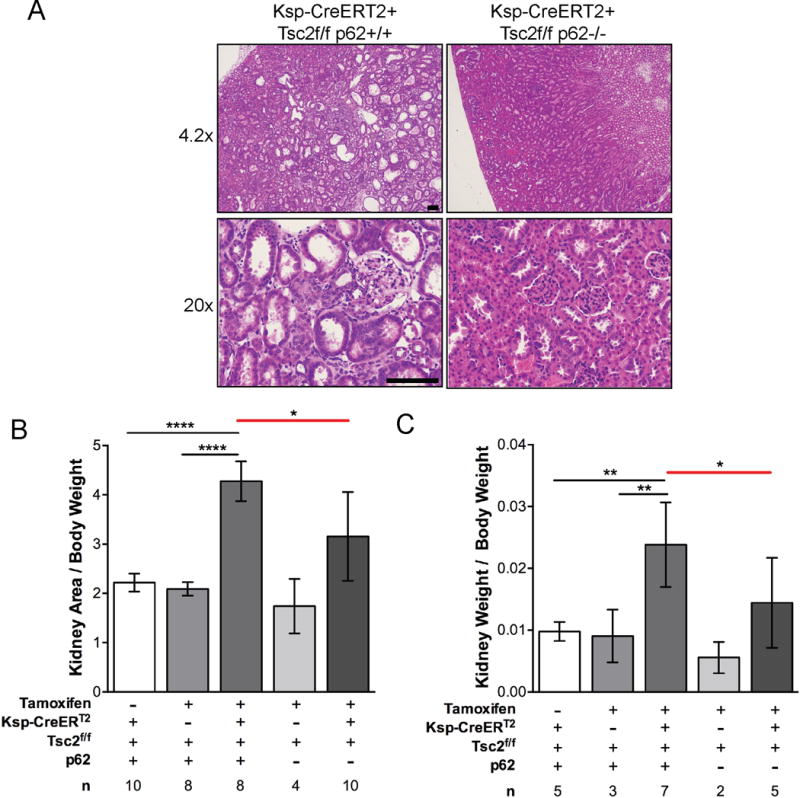
p62 promotes kidney cystogenesis and tumorigenesis in Tsc2f/f Ksp-CreERT2+ mice. A, Mice were treated with tamoxifen 63 mg/kg every other day for 5 days at 8–10 weeks and sacrificed 4 months later. Representative H&E images of Tsc2f/f Ksp-CreERT2+ p62+/+ and Tsc2f/f Ksp-CreERT2+ p62−/− kidneys. Scale bar, 100 um. B, Kidney areas were normalized to body weight. C, Kidney weights were normalized to body weight. n is number of kidneys analyzed. Data presented as the mean +/− 95% CI with *p<0.05, **p<0.01, ****p<0.0001.
p62 mediates anchorage-independent cell growth
To investigate the mechanisms by which p62 promotes the growth of mTORC1 hyperactive cells, we generated Tsc2+/+ and Tsc2−/− MEFs with stable p62 downregulation using three different shRNAs. We confirmed p62 knockdown by immunoblot (Fig. 3A). As expected, p62 levels were higher in Tsc2−/− MEFs, especially in serum free conditions, compared to Tsc2+/+ MEFs. Treatment with rapamycin (20 nM, 24h) decreased levels of p62 in both Tsc2+/+ and Tsc2−/− MEFs. p62 downregulation did not affect cell proliferation of Tsc2+/+ or Tsc2−/− MEFs over 96 hours in either 10% FBS or serum free conditions (Fig. 3B). Treatment of the control shRNA cells with rapamycin (20 nM, 96h replenished at 48h) decreased proliferation by ~50% in the Tsc2−/− MEFs. In contrast, downregulation of p62 in Tsc2−/− MEFs resulted in a 3-fold reduction in soft agar colony formation (p<0.0001; Fig. 3C). These data suggest that p62 is necessary for adaptation to the stress of anchorage-independent growth.
Figure 3.
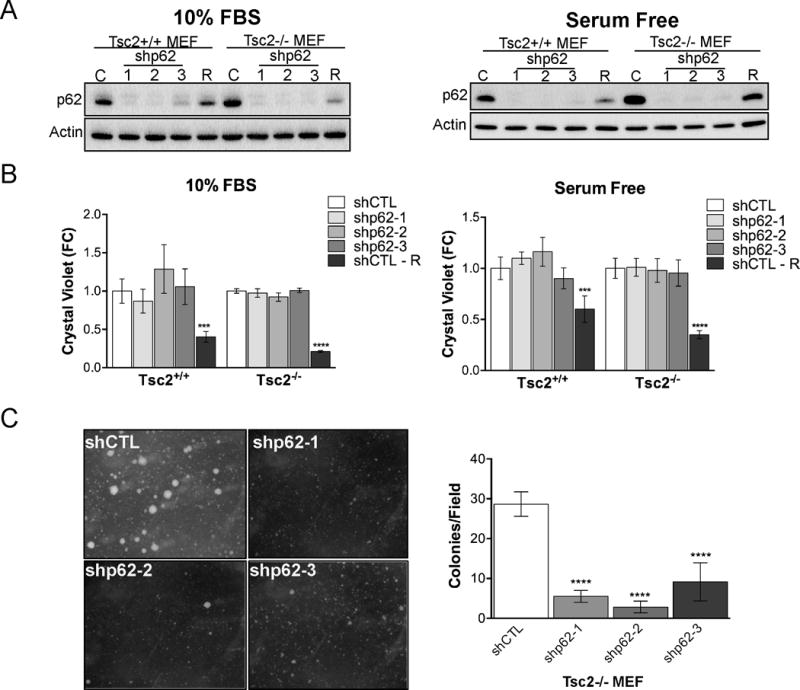
p62 knockdown suppresses Tsc2−/− MEF anchorage independent cell growth. A, Knockdown efficiency of p62 by three shRNA compared to shCTL in Tsc2+/+ and Tsc2−/− MEFs in 10% FBS or serum free conditions. shCTL cells were also treated with rapamycin (R; 20 nM, 24h). B, Cell proliferation assessed by crystal violet was unchanged by p62 knockdown in either 10% FBS or serum free conditions. Normalization was first performed to T=0h then fold change was calculated by normalizing to the untreated shCTL Tsc2+/+ or shCTL Tsc2−/− at 96 h. shCTL cells were treated with rapamycin (R) at 20nM beginning at T=0h. C, Representative images and quantification of colonies after 4 weeks of growth in soft agar. Data are mean +/− SD with ***p<0.001, ****p<0.0001.
p62 regulates glutamine uptake and controls expression of redox genes
To identify metabolic mechanisms through which p62 might impact the growth of Tsc2−/− cells under stress conditions, targeted liquid chromatography-mass spectrometry (33) was used to measure 259 metabolites in Tsc2−/− MEFs with p62 downregulation (shp62-2) versus shCTL in serum free conditions (24h). Twenty-eight metabolites were significantly altered by p62 knockdown (FDR<5%, Supplementary Table S1). Metabolite set enrichment analysis (MSEA) of all measured metabolites identified glutamate and glutathione metabolism as the top differentially impacted pathways in p62 knockdown compared to controls (Fig. 4A). These data implicate p62 in glutamine metabolism and the downstream production of glutamate and glutathione (GSH, Fig. 4B). While GSH precursors, glycine and cysteine, were unaffected by p62 loss (data not shown), the upstream metabolites, glutamine and glutamate, were significantly decreased along with GSH and glutathione disulfide (GSSG) (Fig. 4C). To confirm these results, GSH was measured using a GSH-Glo Assay (Promega). The intracellular glutathione pool was ~20% lower in the p62 knockdown cells, consistent with the metabolomic data (Supplementary Table S1), and was further decreased ~30% by the addition of 500 μM H2O2 and 70% by the addition of 1000 μM H2O2 (serum free, 2 hr; Fig. 4D).
Figure 4.
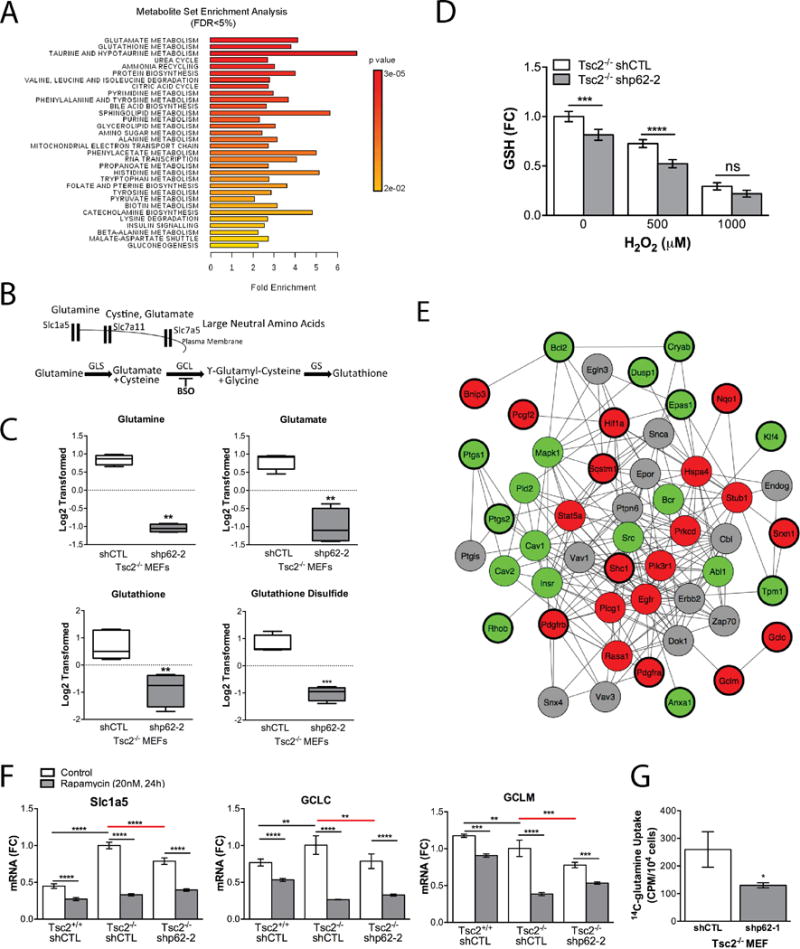
p62 regulates intracellular levels of glutathione. A, Metabolite Set Enrichment Analysis (MSEA) identified differentially regulated metabolic pathways with FDR<5%. B, Cartoon depicting key transporters and enzymes in glutathione biosynthesis. C, Normalized and log2 transformed metabolomic data of GSH related metabolites. D, Total intracellular GSH was measured following 0, 500 and 1000 μM H2O2 (1hr) in shCTL and shp62 Tsc2−/− MEFs. E, Following transcriptional profiling and Gene Set Enrichment Analysis (GSEA), the connectivity of 25 oxidative stress seed genes from Supplementary Table S2 was assessed by network analysis using the consolidated interactome. Seed genes are highlighted with thicker outlines, red indicates genes downregulated and green indicates genes upregulated by p62 knockdown in Tsc2−/− MEF compared to controls. First degree interacting proteins were added of which some did not have available expression data (grey). F, Genes associated with glutamine uptake and glutathione biosynthesis were further quantified by RT-qPCR. Data presented as fold change relative to Tsc2−/− shCTL MEFs. G, Uptake of radiolabled glutamine was assessed in shp62 Tsc2−/− MEFs compared to controls. Data are mean +/− SD with *p<0.05, **p<0.01, ***p<0.001, ****p <0.0001.
To reveal, in an unbiased manner, potential molecular pathways by which p62 knockdown mediated these metabolic changes, expression profiling of ~3,800 cancer-associated genes using an Ion Torrent-based platform was performed. As expected, p62/Sqstm1 was the most robustly downregulated gene (FC=0.09, FDR 3.1E-178). Via Gene Set Enrichment Analysis (GSEA), we found that, in response to p62 knockdown, there was a substantial reprogramming of oxidative stress genes (Supplementary Table S2, GSEA corrected p=0.001). Specifically, genes implicated in intracellular redox or glutathione metabolism that were significantly decreased in Tsc2−/− MEFs by p62 knockdown include: Gclc (FC=0.71, FDR=2.4E-4), Gclm (FC=0.72, FDR=1.6E-4), Gsr (FC=0.58, FDR=1.6E-7), Nqo1 (FC=0.37, FDR=1.5E-20), Srxn1 (FC=0.71, FDR=1.6E-3) and Hmox1 (FC=0.76, FDR=5.0E-3). The high connectivity among these p62-dependent oxidative stress genes was further highlighted by mapping their network connections using a consolidated set of databases cataloguing known functional interactions in mouse cells (the “consolidated interactome”; Fig. 4E).
It was previously shown that p62 regulates components of the glutamine uptake and biosynthesis (34). Therefore, we analyzed the expression of the neutral amino acid transporter (Slc1a5) along with the amino acid transporters Slc7a5 and Slc7a11 (Fig. 4F and Supplementary Fig. S1). The expression of Slc1a5 was 2-fold higher in Tsc2−/− cells compared to Tsc2+/+ cells and reduced significantly by rapamycin (20 nM, 24h). Slc1a5 was reduced by ~20% in cells with p62 knockdown (p<0.0001). Slc7a5 and Slc7a11 were also decreased by rapamycin, but were not dependent on either Tsc2 or p62. In parallel, we measured the expression of the rate-limiting enzymes in glutathione biosynthesis, the catalytic and regulatory subunits of glutamate-cysteine ligase (GCL; GCLC and GCLM), and glutathione synthase (GSS). These enzymes were decreased by rapamycin treatment, but only GCLC was Tsc2-dependent (increased ~20%, p<0.01) and p62-dependent (decreased ~20%, p<0.01) (Fig. 4F and Supplementary Fig. S1). GCLM was p62 dependent (decreased ~20%, p<0.001). These data are in agreement with the expression changes determined by Ion Torrent-based profiling (Supplementary Table S2).
Finally, we found that p62 knockdown in Tsc2−/− MEFs reduced glutamine uptake ~50% (p<0.05) compared to Tsc2−/− shCTL MEFs (Fig. 4G). In sum, these data highlight the critical role of p62 in glutathione biosynthesis and metabolism and suggest a pivotal role for p62 in maintaining redox homeostasis in the context of mTORC1 hyperactivation.
p62 protects mitochondria from oxidative stress
Mitochondria generate ROS during oxidative phosphorylation, and damaged or dysfunctional mitochondria produce more ROS (35). Glutathione is a critical antioxidant that protects cells from mitochondrial ROS (36). p62 has been implicated in cellular responses to mitochondrial stress, acting as a direct adaptor for selective mitophagy or promoting perinuclear localization of mitochondria to enhance clearance efficiency (37–39). Tsc2-null cells have low autophagic flux (7), which would be predicted to decrease mitophagy and possibly increase the levels of damaged mitochondria. We hypothesized, therefore, that p62 may be critical for protecting the integrity mitochondria in Tsc2−/− cells via maintenance of the glutathione pool and possibly promoting mitophagy. To explore this hypothesis, we assessed mitochondrial morphology under basal conditions and after H2O2 treatment. Using immunofluorescence to visualize the mitochondrial protein, TOMM20, we observed increased co-localization of TOMM20 with p62 following oxidative stress in Tsc2−/− shCTL MEFs treated with H2O2 (250 μM, 1h). This finding suggests that oxidative stress may promote an interaction between p62 and mitochondria in Tsc2−/− cells (Fig. 5A). Furthermore, p62 knockdown increased mitochondrial fission upon challenge with oxidative stress (H2O2, 250 uM, 1h; Fig 5B). Consistent with increased H2O2-induced fission, the mean mitochondria length per cell (p<0.05; Fig. 5C) and the mean mitochondria network branching index (a measure of the mitochondria perimeter to area relationship) per cell decreased by p62 knockdown compared to Tsc2−/− shCTL MEFs (p<0.05; Fig. 5D). Mitochondrial fission is a critical initial step of mitophagy since less networked mitochondria are believed to be more efficiently sequestered into autophagosomes (40).
Figure 5.
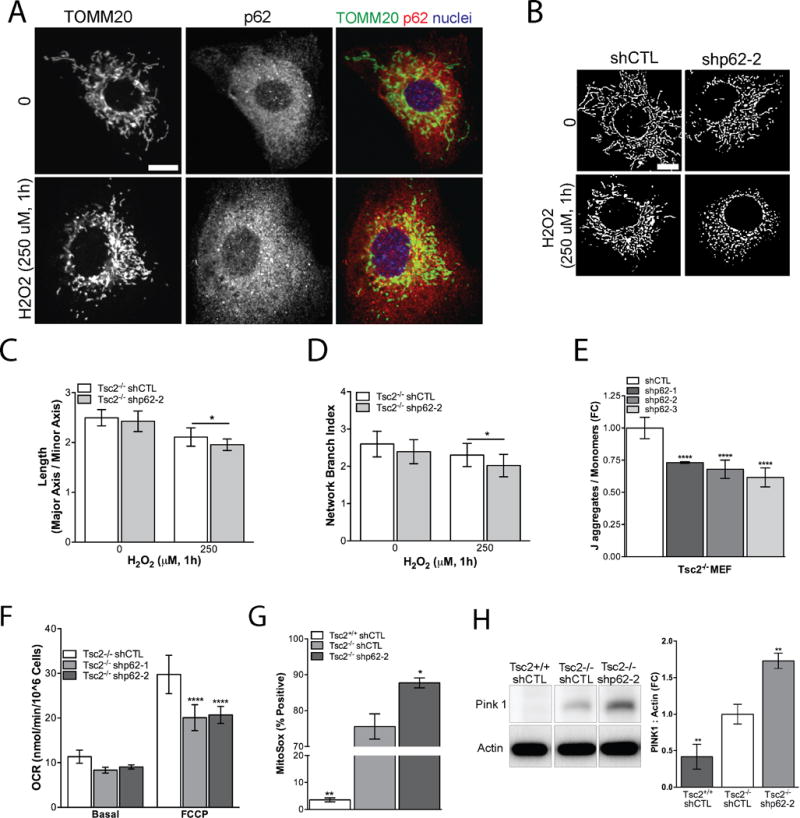
p62 protects mitochondria from oxidative stress. A, In Tsc2−/− shCTL MEFs colocalization (yellow) of p62 and mitochondria was visualized using TOMM20 (mitochondria, green), p62 (red) and nuclei (blue) following H2O2 (250 μM, 1h) treatment. Scale bar, 10 μm. B, Cells were processed to generate a binary image of the TOMM20 stain and mitochondria morphology was assessed by established methods for average mitochondrial C, length per cell and D, network branch index per cell. Fifteen-twenty cells were analyzed per group. Scale bar, 10 μm. E, The ratio of J-aggregates to monomer fluorescence in Tsc2−/− shp62 compared to vehicle treated Tsc2−/− shCTL cells grown in 2% serum and phenol-free DMEM. F, p62 knockdown reduces oxygen consumption rate (OCR) induced by FCCP (1.5 μM) determined by Seahorse XF24 Analyzer. G, p62 knockdown increases production of mitochondrial superoxide by flow cytometry using MitoSOX Assay. H, PINK1 expression is Tsc2- and p62-dependent. Densitometry representative data from three biological replicates. Data are mean+/− SD *p<0.05, **p<0.01, ***p<0.001, ****p <0.0001.
We then assessed the effects of p62 on mitochondria polarization, an index of capacity for oxidative phosphorylation, using the mitochondria membrane potential dependent dye JC-1. p62 knockdown significantly reduced ~25% (p<0.0001) the ratio of red J-aggregates to green monomers, supporting the hypothesis that p62 is critical to the maintenance of mitochondrial polarization in Tsc2-null cells (Fig. 5E). Furthermore, oxygen consumption rate (OCR) was ~30% lower in Tsc2−/− shp62 cells, as measured using the Seahorse Extracellular Flux Analyzer, after treatment with the mitochondrial uncoupler FCCP (1.5 μM). This finding suggests that p62 positively regulates maximal respiratory capacity in Tsc2−/− cells (Fig. 5F). The mitochondrial dye MitoSOX fluoresces red in the presence of superoxide produced by mitochondria. We found significantly more MitoSOX positive Tsc2−/− cells (~70%, p<0.01) compared to Tsc2+/+ cells (5%), consistent with previous reports (8). Knockdown of p62 further increased MitoSOX positive cells by (~15%, p<0.05) in the Tsc2−/− shCTL cells (Fig. 5G). To confirm the specificity of the MitoSOX dye, we overexpressed mnSod/Sod2, which decreased the proportion of MitoSOX positive cells by ~15–20% in shCTL and shp62 Tsc2−/− MEFs (Supplementary Fig. S2). As an additional marker of mitochondrial damage, we assessed expression of PINK1, which recruits ubiquitin ligases to damaged mitochondria. These ubiquitinated proteins are then recognized by selective autophagy adaptors, such as p62, which promote mitophagy. While PINK1 was barely detectable in Tsc2+/+ MEFs, PINK1 was increased in Tsc2−/− MEFs. p62 knockdown further increased this marker of mitochondrial damage ~1.5-fold compared to Tsc2−/− shCTL MEFs (p<0.01, Fig. 5H). Together these data indicate that p62 is a key regulator of mitochondrial homeostasis in Tsc2-null cells.
Glutathione limits mitochondrial damage and promotes Tsc2-deficient growth in soft agar
To determine the importance of glutathione in mitochondrial homeostasis and Tsc2-mediated tumorigenesis, we treated cells with membrane permeable glutathione ethyl ester (GEE). We found that GEE significantly reduced the MitoSOX signal in shCTL and shp62 Tsc2−/− cells (Fig. 6A). We also found that GEE reduced PINK1 levels by 50% (p<0.05) in Tsc2−/− MEFs (Fig. 6B). Finally, treatment of shCTL and shp62 Tsc2−/− MEFs with GEE during growth in soft agar significantly enhanced colony formation by ~3-fold (p<0.001 and p<0.0001, respectively; Fig. 6C). Overexpression of Sod2, which significantly reduces mitochondrial ROS (Supplementary Fig. S2), or catalase was also sufficient to increase growth of Tsc2−/− shp62-2 cells by ~2-fold in soft agar (p<0.01, Fig. 6D). These data highlight the importance of mitochondrial ROS in limiting the growth of Tsc2-null cells following p62 knockdown in soft agar.
Figure 6.
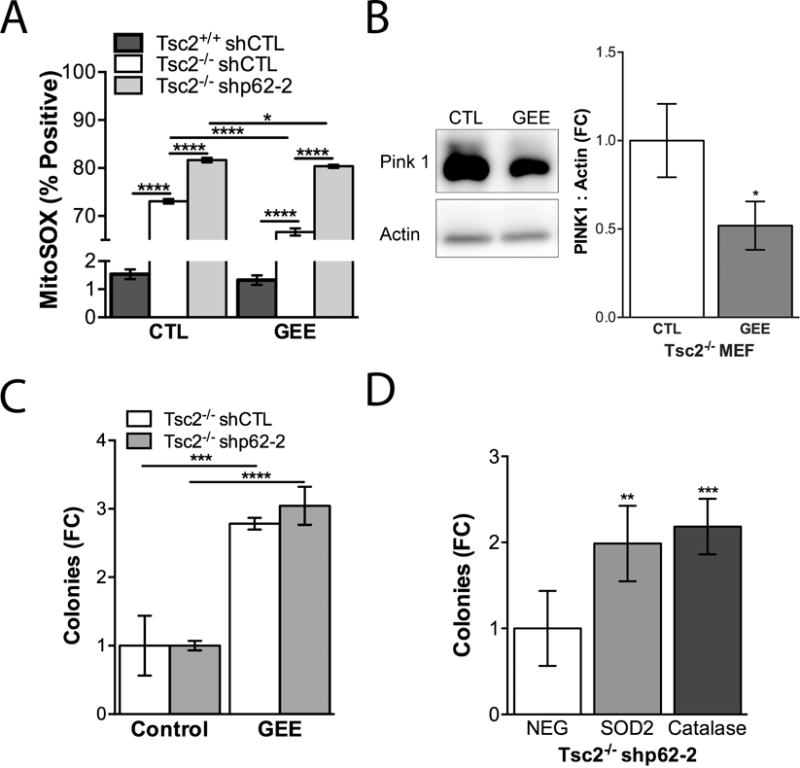
Limiting mitochondrial ROS rescues growth of Tsc2-deficient cells with p62 knockdown. A, MitoSOX was assessed by flow cytometry 24 hours after treatment with 1 mM GEE. B, Treatment of Tsc2−/− shCTL cells with 1mM GEE for 24 hours reduced Pink 1 expression. Densitometry is shown for three biological replicates. C, Cells were grown in soft agar and treated with 1 mM GEE twice per week for 4 weeks. D, Tsc2−/− shp62-2 stably overexpressing Sod2, Catalase or control empty vector (NEG) were grown in soft agar for 4 weeks. Data are presented as mean +/− SD *p<0.05, ***p<0.001, ****p<0.0001.
p62 knockdown increases ROS and sensitizes cells to GSH depletion
Since p62 knockdown reduced intracellular levels of glutathione and increased mitochondrial ROS, we hypothesized that p62 plays a critical role in global cellular redox homeostasis. Therefore, we next measured intracellular levels of ROS using H2DCFDA-CM, a compound which fluoresces as a result of oxidation (Fig. 7A). Baseline levels of ROS were higher in Tsc2−/− compared to Tsc2+/+ MEFs (25%, p<0.0001), as expected from prior studies (11), and knockdown of p62 further increased cellular ROS by ~30% (p<0.0001) in the Tsc2−/− cells. Furthermore, direct targeting of glutathione biosynthesis with the GCL inhibitor, buthionine sulfoximine (BSO; 50 μM, 48h) doubled ROS in the Tsc2+/+ MEFs and increased ROS ~40% in the Tsc2−/− MEFs. BSO treatment increased ROS in the Tsc2−/− shp62 cells by ~20% (Fig. 7A). Since the H2DCFDA signal can be affected by cellular uptake, efflux, and ester cleavage efficiency, we utilized the oxidant insensitive DCFDA and oxidant sensitive dye H2DCFDA (Supplementary Fig. S3A) as a control to determine if dye processing was equivalent in the three cell lines. The Tsc2+/+ shCTL cells had equivalent or slightly greater DCFDA fluorescence compared to Tsc2−/− shCTL; however, using H2DCFDA the Tsc2+/+ MEFs continue to exhibit decreased oxidant induced fluorescence. p62 knockdown had no effect on the oxidant insensitive DCFDA signal, while the H2DCFDA signal was increased by p62 knockdown. The H2DCFDA-SE signal was significantly repressed by treatment with pegylated superoxide dismutase (SOD) and catalase (Supplementary Fig. S3B, C), as expected.
Figure 7.
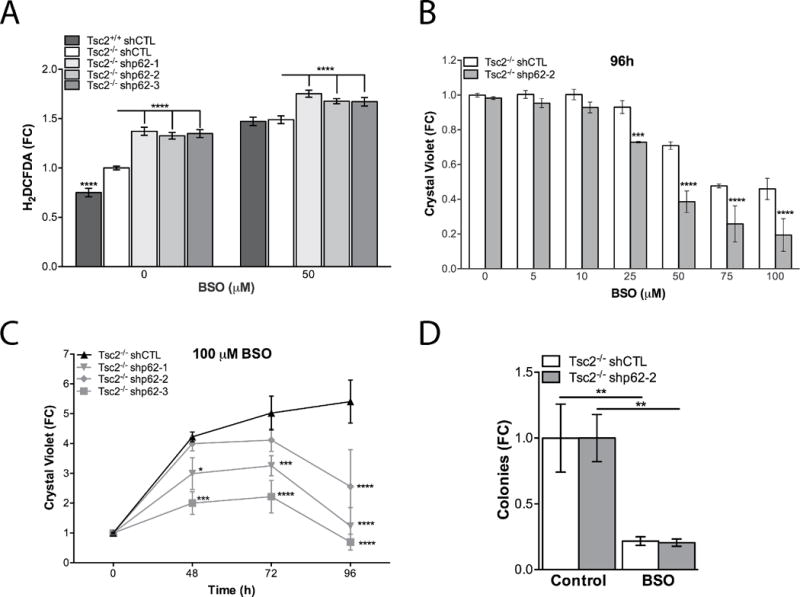
p62 promotes adaptation to oxidative stress. A, In Tsc2−/− shCTL MEFs and Tsc2−/− shp62 MEFs intracellular ROS were assessed by H2DCFDA assay in basal conditions and BSO treatment (50 μM, 48h). B, Effects of glutathione biosynthesis inhibition were assessed by crystal violet in dose response experiments at 96h. C, Effects of glutathione biosynthesis inhibition were assessed by crystal violet in time course (100 μM) experiments. D, Cells were grown in soft agar and treated with 25 μM BSO twice per week for 4 weeks. Data are mean +/− SD with *p<0.05, ***p<0.001, ****p <0.0001.
To determine the functional consequences of glutathione depletion in Tsc2−/− cells following p62 knockdown, the effects of BSO on cell proliferation were assessed. BSO was given at 6 doses (5, 10, 25, 50, 75 and 100 μM) for 96 hours and cell density was determined by crystal violet. Knockdown of p62 significantly increased sensitivity of Tsc2−/− cells to BSO at doses of 25, 50, 75 and100 μM (Fig. 7B). A time course of 100 μM BSO treatment from 48 to 96 hours also showed that p62 shRNA sensitizes Tsc2−/− cells to BSO treatment (Fig. 7C). Finally, growth of Tsc2-null cells in soft agar was ~70% reduced by treatment with BSO (p<0.01, Fig. 7D). These data suggest that p62 cooperates with mTORC1 hyperactivity to maintain intracellular glutathione, which is essential for Tsc2-null cell proliferation.
Discussion
p62 is a central signaling hub for the activation of diverse pro-survival pathways, which are frequently hijacked to support the adaptation of cancer cells during transformation (12). Herein we show that p62 is essential for kidney cyst and tumor development in two different genetic models of TSC2 deletion that have mTORC1 hyperactivation. We observed smaller and fewer renal cysts and less progression to solid tumors in both Tsc2+/− p62−/− mice and Tsc2 f/f Ksp-CreERT2+ p62−/− mice compared to littermate controls (Fig. 1 and 2).
To elucidate the mechanisms though which p62 promotes tumorigenesis in mTORC1 hyperactive cells, we performed metabolomic profiling. Metabolite set enrichment analysis highlighted pathways downregulated by loss of p62, including glutathione metabolism, nucleotide biosynthesis, urea cycle, and polyamines, all of which are dependent on glutamine as a requisite biosynthetic precursor (Fig. 4A). Previous studies have shown that mTORC1 hyperactive cells are characterized by increased glutamine uptake and dependence (5,6,41), which is a hallmark of numerous cancers (42,43). While prior studies have focused on the requirement of glutamine to generate α-ketoglutarate for the TCA cycle as the mechanism underlying glutamine “addiction” in cancer, our data highlight a role for p62 promoting glutamine uptake and glutathione synthesis in Tsc2-null cells. Implementing expression profiling of Tsc2−/− MEFs with shp62 versus shCTL, we focused on a significant oxidative stress signature of highly connected genes (Supplementary Table S2, Fig. 4E). To validate the expression data, we found that p62 directly regulates the glutamine transporter, Slc1a5 (~20%), and the rate limiting glutathione biosynthesis enzyme, GCLC and GCLM (~20%), in Tsc2-null cells, but not Slc7a11, Slc7a5 or GSS, which were previously shown to be p62 dependent in cells with wildtype Tsc2 (34). Upregulation of Slc1a5 has been implicated in numerous cancers and is target for therapeutic intervention (44,45). Interestingly, GCLC was previously implicated in the neuronal manifestations of TSC (46). Prior studies have shown that p62 activates mTORC1 resulting in Myc regulation of glutamine transporters and glutathione biosynthetic enzymes (34,41); however, we were unable to observe evidence of p62-dependent mTORC1 activity in our cells, suggesting additional mechanisms of glutathione regulation that have yet to be elucidated. Our data indicate that p62 drives glutamine uptake in Tsc2-null cells (Fig. 4G).
As an adaptor protein, p62 regulates numerous downstream pro-survival processes to support tumorigenesis. Expression profiling highlighted the role of p62 in regulating genes with known antioxidant response elements (ARE). We focused on the role of p62 in Nrf2 regulation. Immunoblot analysis did not show a strong p62 dependence of Nrf2, consistent with Zhang et al. (47), who previously demonstrated that Nrf2 is Tsc2-independent (Supplementary Fig. S4A, B). Immunohistochemistry in Tsc2+/− mice suggested that both Nrf2 and NFkB expression may be increased in tumors relative to normal adjacent kidney tissue; however, loss of p62 did not substantially suppress these pathways in vivo (Supplementary Fig. S4C, D). Furthermore, tumor burden was equivalent in the Tsc2+/− mice crossed to Nrf2−/− mice compared to Nrf2+/+ littermate controls, indicating that, unlike p62, Nrf2 is not necessary for tumor development in Tsc2+/− mice (Supplementary Fig. S4E). These findings indicate that Nrf2 may play an indirect, rather than predominant, role in Tsc2-driven renal tumors, and distinguishes TSC from models of hepatocellular carcinoma, in which p62 regulation of Nrf2 is the critical tumor driving mechanism (24,26).
Our gene expression data suggest that p62 plays a comprehensive role in limiting oxidative stress in Tsc2-null cells. Since glutathione buffers mitochondrial ROS generated during oxidative phosphorylation, we investigated effects of p62 on mitochondrial phenotype and function. It was previously shown that TSC1/2-null cells display increased numbers of mitochondria (48) and defective mitophagy (49). We observed basal and oxidative stress induced p62-dependent changes in mitochondria morphology and function. In particular, p62 knockdown reduced mitochondrial membrane polarization and increased mitochondrial superoxide production (Fig. 5E and G). Finally, maximal oxygen consumption rate induced by the electron transport uncoupler, FCCP, was reduced by ~30% in p62 knockdown, suggesting that Tsc2−/− cells depend on p62 to maintain the bioenergetic flexibility to respond to cellular stress. Importantly, the mitophagy marker PINK1 is significantly upregulated in a Tsc2- and p62-dependent manner, suggesting that limited autophagy results in accumulation of damaged mitochondria, a process exacerbated by p62 knockdown. Addition of exogenous glutathione reduced markers of mitochondrial damage and promoted growth of Tsc2-null cells in soft agar. In aggregate, our data indicate that p62 promotes tumorigenesis in mTORC1-hyperactive cells by regulating redox homeostasis. Adaptation to oxidative stress is particularly critical in the setting of mTORC1 hyperactivation because bioenergetic and biosynthetic activities place a significant burden on the capacity of cells to buffer oxidative stresses (11,50). This concept is consistent with our observation that loss of p62 suppresses ~3-fold growth in soft agar (Fig. 3C), and addition of GEE, Sod2 or catalase overexpression promoted colony formation (Fig. 6C, D), which is associated with matrix detachment, transformation, and increased oxidative stress (51,52).
mTORC1-driven tumors, including tumors in TSC patients, typically shrink as a result of mTORC1 inhibitors, but regrow after treatment cessation. Furthermore, rapid development of resistance to rapalogs occurs in many cancers with mTORC1 hyperactivation, suggesting that novel therapeutic strategies for mTORC1-driven tumors could have high clinical impact (53). Redox homeostasis may represent a critical vulnerability of mTORC1 hyperactivity with therapeutic implications. Our data show that p62 plays a critical role in cellular redox homeostasis by maintaining mitochondrial integrity and glutathione pools necessary to support mTORC1 hyperactivation. We found that p62 loss sensitized cells to further glutathione depletion with the GCL inhibitor, BSO, consistent with the role of p62 in buffering oxidative stress via glutathione. Interestingly, we previously found that chelerythrine chloride depleted p62 and increased oxidative stress in Tsc2−/− cells (54). Moreover, in recent studies combination therapy of BSO and rapamycin (50) or BSO, auranofin, and RIP kinase inhibitors selectively induced apoptosis in Tsc2−/− cells (55). We speculate that activation of autophagy by rapamycin and subsequent downregulation of p62 underlies the synergy of mTORC1 inhibition with BSO in Tsc2-null cells. Our data highlight the importance of p62 in buffering oxidative stress to support tumor growth and progression in the setting of mTORC1 hyperactivation. Therapeutic strategies that keep mTORC1 high while undermining redox homeostasis may yield increased clinical responses in TSC and other neoplastic diseases.
Supplementary Material
Acknowledgments
We thank Drs. Christian Dibble, Xingbin Ai, Soumitro Pal and Murugabaskar Balan for scientific conversations related to this project. We also thank Dr. Frank Pan for Ion Torrent sample preparation and data processing.
Financial Support: This work was supported by the Engles Program in TSC and LAM Research, R01 DK096556 (E.P. Henske), R01 HL098216, R01 DK098331 (J.J. Yu), R01 HL124021, R01 HL122596 (S.Y. Chan), P01-CA120964, 5P30CA006516 (J.M. Asara) and the Tuberous Sclerosis Alliance (C. Priolo and E.P. Henske). H.C. Lam and A.A. Parkhitko are Postdoctoral Fellows of The LAM Foundation.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- 2.Kwiatkowski DJ, Manning BD. Molecular basis of giant cells in tuberous sclerosis complex. N Engl J Med. 2014;371(8):778–80. doi: 10.1056/NEJMcibr1406613. [DOI] [PubMed] [Google Scholar]
- 3.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22(2):169–76. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38(4):487–99. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153(4):840–54. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108(30):12455–60. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parkhitko AA, Priolo C, Coloff JL, Yun J, Wu JJ, Mizumura K, et al. Autophagy-dependent metabolic reprogramming sensitizes TSC2-deficient cells to the antimetabolite 6-aminonicotinamide. Mol Cancer Res. 2014;12(1):48–57. doi: 10.1158/1541-7786.MCR-13-0258-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J, Parkhitko A, Henske EP. Autophagy: an ‘Achilles’ heel of tumorigenesis in TSC and LAM. Autophagy. 2011;7(11):1400–1. doi: 10.4161/auto.7.11.17652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–58. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 11.Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. 2009;29(18):5926–37. doi: 10.1523/JNEUROSCI.0778-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37(6):230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciuffa R, Lamark T, Tarafder AK, Guesdon A, Rybina S, Hagen WJ, et al. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015;11(5):748–58. doi: 10.1016/j.celrep.2015.03.062. [DOI] [PubMed] [Google Scholar]
- 14.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19(7):1576–86. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K, et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol Cell. 2016;63(1):34–48. doi: 10.1016/j.molcel.2016.05.027. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 17.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30(13):3275–85. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linares JF, Duran A, Reina-Campos M, Aza-Blanc P, Campos A, Moscat J, et al. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015;12(8):1339–52. doi: 10.1016/j.celrep.2015.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013;24(6):738–50. doi: 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(1):105–20. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–84. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komatsu M. Potential role of p62 in tumor development. Autophagy. 2011;7(9):1088–90. doi: 10.4161/auto.7.9.16474. [DOI] [PubMed] [Google Scholar]
- 26.Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell. 2016;29(6):935–48. doi: 10.1016/j.ccell.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23(23):3251–3. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 29.Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest. 2014;124(8):3514–28. doi: 10.1172/JCI74773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee L, Sudentas P, Donohue B, Asrican K, Worku A, Walker V, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42(3):213–27. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 31.Pollizzi K, Malinowska-Kolodziej I, Stumm M, Lane H, Kwiatkowski D. Equivalent benefit of mTORC1 blockade and combined PI3K-mTOR blockade in a mouse model of tuberous sclerosis. Mol Cancer. 2009;8:38. doi: 10.1186/1476-4598-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59(6):1206–11. [PubMed] [Google Scholar]
- 33.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7(5):872–81. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26(1):121–35. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandel NS. Mitochondria and cancer. Cancer Metab. 2014;2:8. doi: 10.1186/2049-3002-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mailloux RJ, McBride SL, Harper ME. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem Sci. 2013;38(12):592–602. doi: 10.1016/j.tibs.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285(36):27879–90. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 39.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6(8):1090–106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao K, Klionsky DJ. Participation of mitochondrial fission during mitophagy. Cell Cycle. 2013;12(19):3131–2. doi: 10.4161/cc.26352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24(19):2274–80. doi: 10.1016/j.cub.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29(3):313–24. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678–84. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu H, Li X, Lu Y, Qiu S, Fan Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Lett. 2016;381(1):23–30. doi: 10.1016/j.canlet.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016;35(24):3201–8. doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malik AR, Liszewska E, Skalecka A, Urbanska M, Iyer AM, Swiech LJ, et al. Tuberous sclerosis complex neuropathology requires glutamate-cysteine ligase. Acta Neuropathol Commun. 2015;3:48. doi: 10.1186/s40478-015-0225-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513(7518):440–3. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goto J, Talos DM, Klein P, Qin W, Chekaluk YI, Anderl S, et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci U S A. 2011;108(45):E1070–9. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebrahimi-Fakhari D, Saffari A, Wahlster L, DiNardo A, Turner D, Lewis TL, Jr, et al. Impaired Mitochondrial Dynamics And Mitophagy In Neuronal Models Of Tuberous Sclerosis Complex. Cell Rep. 2016;17(8):2162. doi: 10.1016/j.celrep.2016.10.051. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Shin S, Sun Y, Yoon SO, Li C, Zhang E, et al. mTORC1-Driven Tumor Cells Are Highly Sensitive to Therapeutic Targeting by Antagonists of Oxidative Stress. Cancer Res. 2016;76(16):4816–27. doi: 10.1158/0008-5472.CAN-15-2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461(7260):109–13. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481(7381):385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medvetz D, Sun Y, Li C, Khabibullin D, Balan M, Parkhitko A, et al. High-throughput drug screen identifies chelerythrine as a selective inducer of death in a TSC2-null setting. Mol Cancer Res. 2015;13(1):50–62. doi: 10.1158/1541-7786.MCR-14-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Filipczak PT, Thomas C, Chen W, Salzman A, McDonald JD, Lin Y, et al. TSC2 Deficiency Unmasks a Novel Necrosis Pathway That Is Suppressed by the RIP1/RIP3/MLKL Signaling Cascade. Cancer Res. 2016;76(24):7130–39. doi: 10.1158/0008-5472.CAN-16-1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.