

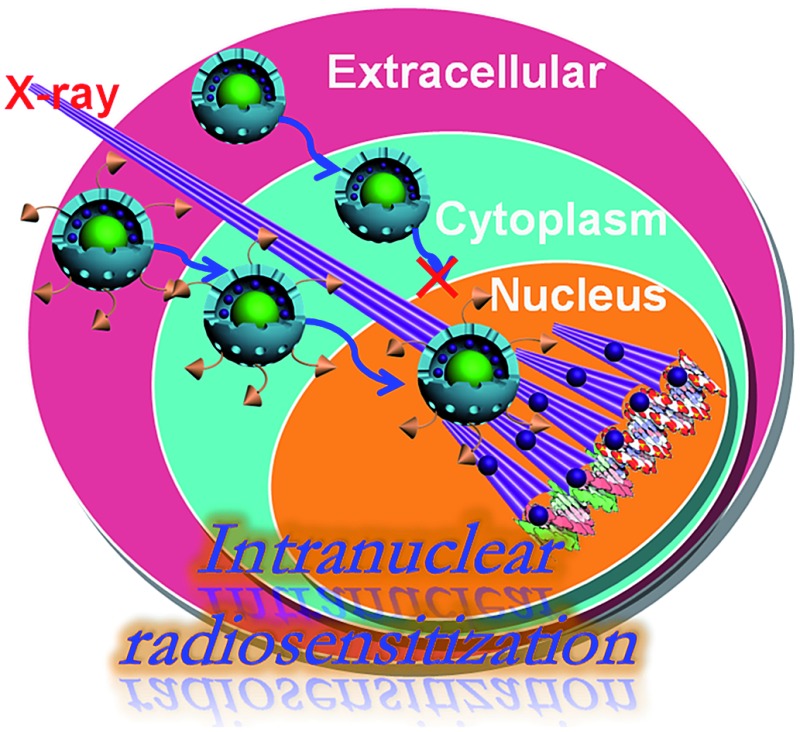
 MR/UCL bimodal imaging guided intranuclear radiosensitization was firstly achieved by an intelligent nuclear-targeting nanotheranostic system.
MR/UCL bimodal imaging guided intranuclear radiosensitization was firstly achieved by an intelligent nuclear-targeting nanotheranostic system.
Abstract
Clinically applied chemotherapy and radiotherapy is sometimes not effective due to the limited dose acting on DNA chains resident in the nuclei of cancerous cells. Herein, we develop a new theranostic technique of “intranuclear radiosensitization” aimed at directly damaging the DNA within the nucleus by a remarkable synergetic chemo-/radiotherapeutic effect based on intranuclear chemodrug-sensitized radiation enhancement. To achieve this goal, a sub-50 nm nuclear-targeting rattle-structured upconversion core/mesoporous silica nanotheranostic system was firstly constructed to directly transport the radiosensitizing drug Mitomycin C (MMC) into the nucleus for substantially enhanced synergetic chemo-/radiotherapy and simultaneous magnetic/upconversion luminescent (MR/UCL) bimodal imaging, which can lead to efficient cancer treatment as well as multi-drug resistance circumvention in vitro and in vivo. We hope the technique of intranuclear radiosensitization along with the design of nuclear-targeting nanotheranostics will contribute greatly to the development of cancer theranostics as well as to the improvement of the overall therapeutic effectiveness.
1. Introduction
During the past decades, chemotherapy has achieved great success in relieving the pain of tumor patients and extending their lifetime. However, it is difficult to further improve the overall treatment efficacy because of unspecific drug delivery. What is worse, most patients may develop multi-drug resistance (MDR) after frequent administration of free drugs.1 MDR, arising from the over-expression of the drug efflux P-glycoprotein (P-gp) pumps, has become one of the major challenges of cancer chemotherapy.2 Although various drug delivery systems (DDSs) have been reported to be capable of reversing MDR to some extent by improving the cellular drug accumulation,1b most of the released drugs within the cell cytoplasm fail to enter the nucleus, which inevitably lowers the chemotherapeutic effectiveness. Therefore, more advanced drug delivery strategies, such as intranuclear drug delivery, are in urgent need for effective anti-MDR.
In addition, radiotherapy can also efficiently kill MDR cells by focusing high-energy X-ray radiation on them to damage the DNA chains.3 However, most solid tumors are in lack of oxygen as compared to normal tissues, which often causes the failure of radiotherapy.4 Fortunately, some representative anticancer drugs, such as Mitomycin C (MMC), are selectively toxic to hypoxic solid tumors. MMC can be activated by the reducing (hypoxic) environment to inhibit DNA synthesis. More importantly, MMC can simultaneously serve as a radiosensitizer to enhance radiotherapy efficacy.5 Therefore, the combined use of MMC and X-ray radiation may contribute to the improved effectiveness of cancer therapeutics.6 Previous studies mainly focused on extracellular radiosensitization (free drugs and radiotherapy)7 or intracytoplasmic radiosensitization (intracytoplasmic delivered drugs and radiotherapy),8 which failed to achieve the optimized treatment efficacy because few drugs can passively diffuse into the cell nucleus to synergistically enhance the radiotherapy efficacy on breaking down the DNA. Therefore, the design of an active nuclear-targeting drug vehicle that can efficiently transport MMC into the nucleus9 is extremely important for the intranuclear chemodrug-sensitized radiation enhancement, which has not been reported yet.
Recent progress in nanotechnology has enabled successful syntheses of nuclear-targeting drug vehicles. For example, the small-sized (usually sub-50 nm) mesoporous nanoparticles (MSNs) conjugated with nuclear localization signal (NLS) ligands (such as TAT) may be efficiently transported into the cell nucleus.10 However, the reported intranuclear drug delivery was only achieved on the cellular level for the single mode of chemotherapy; it was not a theranostic that combined simultaneous bioimaging and multi-mode synergetic therapy. Recently, upconversion nanoparticles (UCNPs)11 have been developed as magnetic/upconversion luminescent (MR/UCL) bimodal imaging probes due to their unique physical/chemical properties.12 Therefore, the integration of UCNPs and nuclear-targeting MSNs may simultaneously achieve accurate imaging guidance and efficient intranuclear drug delivery for substantially enhanced chemo-/radiotherapy.13
In this study, a sub-50 nm nuclear-targeting rattle-structured upconversion core/mesoporous silica nanotheranostic system was firstly designed to directly deliver MMC into the nucleus for greatly enhanced damaging of the DNA with the assistance of X-ray irradiation. More importantly, we develop a new theranostic technique of “intranuclear radiosensitization”, which means that the MMC molecules released into the nucleoplasm may not only efficiently break down the intranuclear DNA, but also effectively enhance the radiotherapy efficacy due to the intranuclear chemodrug-sensitized radiation enhancement effects (Fig. 1). Therefore, our synthesized nuclear-targeting nanotheranostics are expected to achieve substantially enhanced intranuclear synergetic chemo-/radiotherapy under the monitoring of MR/UCL bimodal imaging, which may represent a significant step forward in the development of high-performance cancer theranostic upconversion nanoparticles.
Fig. 1. (a) Schematic illustration of intranuclear radiosensitization. (b) Scanning electron microscopy (SEM) image of rattle-structured upconversion core/mesoporous silica nanotheranostics (RUMSNs). (c) Scanning transmission electron microscopy (STEM) image and the corresponding element mappings of the RUMSNs: (c1) Si; (c2) O; (c3) Y; (c4) F; (c5) Yb; (c6) Gd.

2. Results and discussion
2.1. Synthesis and characterization of nuclear-targeting nanotheranostics
The sub-50 nm rattle-structured nanocomposites are usually not easy to synthesize because of the difficulties in controlling various reaction parameters (e.g., reaction time, TEOS addition amounts, etc.).14 Herein, two chemical routes were used to fabricate the sub-50 nm nuclear-targeting nanotheranostics (Fig. 2a). In the first route, uniform hexagonal phase UCNPs (NaYF4:Yb/Er/Tm/Gd) were synthesized using the traditional thermal decomposition method (Fig. S1 and 2†). Then a thickness-controlled dense silica plus mesoporous silica shell was subsequently coated onto the UCNPs (denoted as UCNPs@SiO2@mSiO2).15 In the second route, the UCNPs were transferred to a water phase by treatment in a pH = 4 aqueous solution of HCl,16 followed by depositing a thickness-controlled mesoporous silica shell onto the ligand-free UCNPs (denoted as UCNPs@mSiO2).17 Finally, by etching the intermediate dense silica or mesoporous silica with polyvinyl pyrrolidone (PVP) protecting the outer surface, the final sub-50 nm rattle-structured upconversion core/mesoporous silica nanotheranostics (RUMSNs) were successfully prepared. The transmission electron microscopy (TEM) characterizations (Fig. 2b–g) demonstrate the high dispersity of the products synthesized in each reaction, which meets the requirements of biomedical administrations.
Fig. 2. (a) Schematic diagram of the two synthetic routes to RUMSNs. (b–g) TEM images of the products: (b) UCNPs; (c) UCNPs@SiO2; (d) UCNPs@SiO2@mSiO2; (e) ligand-free UCNPs; (f) UCNPs@mSiO2; (g) RUMSNs. The core, shell and overall sizes of RUMSNs are about 19, 10 and 47 nm, respectively.

It is worth mentioning that, different from the first chemical route reported in our previous study,8b the second chemical route has unique advantages in controlling the size of the sub-50 nm RUMSNs more easily and accurately in which only the thickness of the mesoporous silica shell is necessary to regulate, as compared to the need for tuning both the thickness of the dense silica and the mesoporous silica in the first route. Secondly, by the second route, a much shorter time period is needed for synthesizing the RUMSNs as compared to the time-consuming dense silica coating by the reverse microemulsion method in the first route. Finally, the second route can be used for future scale-up production of RUMSNs. Therefore, in view of the above superiorities, the second chemical route was used to synthesize the RUMSNs for the following biological experiments.
To the best of our knowledge, this is the first report to combine UCNPs and MSNs into a sub-50 nm rattle-structured composite structure, which, as we expect, may be a significant advance in the design of multifunctional nanotheranostic systems. As we know, proper surface modification is important for the bioengineering of RUMSNs. PVP, which was covalently grafted onto the surface of the RUMSNs after hot water etching, could improve their stability and biocompatibility. Moreover, the NLS ligand TAT was covalently conjugated onto the surface of the RUMSNs by a typical esterification reaction under the catalysis of EDC and NHS, as clearly confirmed by the corresponding FT-IR characterization (Fig. S3†). The dynamic light scattering (DLS) size measurement (Fig. S4†) shows that both the RUMSNs and the RUMSNs–TAT can be well-dispersed in PBS for at least one week without any detectable aggregation, thus displaying stability over a relatively long period of time. Finally, the successful grafting of TAT onto the RUMSNs (denoted as RUMSNs–TAT) will make the RUMSNs capable of not only efficiently targeting the cell nucleus, but also capable of achieving the following intranuclear imaging/therapy by loading anticancer drugs.
2.2. Intranuclear transport observation of RUMSNs–TAT
Before conducting the in vitro biological experiments, the cytotoxicity of RUMSNs/RUMSNs–TAT should be firstly tested against both MCF-7 cells and MCF-7/ADR cells by using CCK-8 as well as lactate dehydrogenase (LDH) assays. As shown in Fig. S5 and 6,† after incubation for 24 h/48 h, the viability of these two cell lines changes very little even at a much increased concentration up to 1 mg mL–1. The negligible cytotoxicity of the RUMSNs/RUMSNs–TAT demonstrates their good in vitro biocompatibility, which is the premise for their following biomedical applications.
Then we investigated the cellular internalization of RUMSNs/RUMSNs–TAT using 2D & 3D confocal laser scanning microscopy (CLSM) imaging. As seen from Fig. 3a and b, after incubation with the RUMSNs–TAT for 24 h, the nuclei of the MCF-7 cells can be visualised by the yellow luminescence (merge of green and red luminescence) from the RUMSNs–TAT upon NIR excitation, which confirms that the RUMSNs–TAT can accumulate preferentially in the nucleus by crossing the nuclear membrane. By contrast, without the attachment of TAT, the RUMSNs could not enter the nucleus but mainly resided in the cytoplasm, as shown by the yellow luminescence surrounding the nuclei in Fig. 3e and f. In addition, as seen from the corresponding line scanning profiles of each luminescence intensity on selected MCF-7 cells, the signal of the RUMSNs is separated from that of the nucleus (Fig. 3h), as compared to the large extent of signal overlap for the RUMSNs–TAT (Fig. 3d). More importantly, the RUMSNs–TAT nanoparticles are clearly found within the nucleus from the corresponding bio-TEM images (Fig. 3c), but the RUMSNs nanoparticles can only be found in the cytoplasm (Fig. 3g). All the above results confirm that with the conjugation of the nuclear targeting ligand TAT, the efficient intranuclear localization of the RUMSNs–TAT has been achieved.
Fig. 3. (a1–6 & e1–6) Confocal laser scanning microscopy (CLSM) imaging of MCF-7 cells incubated with (a1–6) the RUMSNs–TAT and (e1–6) the RUMSNs for 24 h. Blue luminescence is from the nucleus after being stained with DAPI. Upon NIR excitation, the RUMSNs emit yellow luminescence (merge of green/red luminescence). (b1–3 & f1–3) The three-dimensional confocal luminescence reconstructions of MCF-7 cells incubated with (b1–3) the RUMSNs–TAT and (f1–3) the RUMSNs for 24 h. (c & g) Bio-TEM images of MCF-7 cells incubated with (c) the RUMSNs–TAT and (g) the RUMSNs for 24 h. Red arrow: the RUMSNs–TAT reside in the nucleus; yellow arrow: the RUMSNs reside in the cytoplasm. (d & h) Line-scanning profiles of luminescence intensity of the MCF-7 cells incubated with (d) the RUMSNs–TAT and (h) the RUMSNs for 24 h.

Next, we performed the same CLSM and bio-TEM imaging experiments on multi-drug resistant MCF-7 (MCF-7/ADR) cells. As expected, all these results from the 2D & 3D CLSM imaging/line scanning profiles/bio-TEM imaging (Fig. S7†) also show that the RUMSNs–TAT could be efficiently transported to localize within the nucleus of the MCF-7/ADR cells by the facilitation of the TAT peptide, while the RUMSNs only reach the cytoplasm.
2.3. In vitro evaluation of intranuclear radiosensitization
Due to the high BET surface area (255.92 m2 g–1) and big pores (2.5 & 3.5 nm) of the RUMSNs (Fig. S8†), MMC, an important hydrophilic antitumor drug, can be encapsulated into the cavities with a loading capacity of 7%. As seen from the release profile (Fig. S9†), MMC is released from the RUMSNs at a fast rate, which may accelerate the following drug accumulation and enhance the chemotherapeutic efficacy.18 Furthermore, the most important feature is that MMC is also a radiosensitive drug that can selectively improve the radiotherapeutic effects on hypoxic solid tumors. It is envisioned that, by encapsulation into the active nuclear-targeting DDS (RUMSNs–TAT), MMC can be directly delivered into the nucleus to play a dual role in cancer therapy based on the intranuclear chemodrug-sensitized radiation enhancement effects, which may lead to significantly enhanced synergetic chemo-/radiotherapy efficacy.
Then we tested the corresponding viabilities of MCF-7 cells after incubation with free MMC, MMC-loaded RUMSNs (RUMSNs–MMC) and MMC-loaded RUMSNs–TAT (RUMSNs–TAT–MMC). As seen from Fig. 4a and b & S10a and b,† the RUMSNs–MMC kill more cancer cells than free MMC because more MMC can be transported into the cells based on the intracytoplasmic delivery of the RUMSNs as compared to the passive diffusion of free MMC. Moreover, the RUMSNs–TAT–MMC cause higher cytotoxicity than the RUMSNs–MMC, which may be attributed to the highly efficient intranuclear drug delivery by the RUMSNs–TAT. Upon high energy X-ray irradiation, many more cells are killed due to the combined functioning of chemotherapy and radiotherapy (RT). As expected, the cells treated with the RUMSNs–TAT–MMC + RT demonstrate the lowest viability both at a high MMC concentration (10 μg mL–1) and a low concentration (0.5 μg mL–1), which indicates that the intranuclear radiosensitization (based on the synergetic interactions between MMC and X-ray radiation within the nucleus) may produce much better therapeutic effects than the intracytoplasmic radiosensitization (RUMSNs–MMC + RT) or extracellular radiosensitization (MMC + RT). Moreover, the generation of higher levels of reactive oxygen species (ROS) within cancer cells is also an indicator of higher cell death rates (Fig. 4c). Similar results are also obtained by the FITC–PI apoptosis experiments (Fig. S10c & S11a†), which further confirms that the highest cytotoxicity is achieved by the RUMSNs–TAT–MMC + RT due to the intranuclear chemodrug-sensitized radiation enhancement effects.
Fig. 4. (a & b) The viability of MCF-7 cells after different treatments for 24 h with an MMC concentration of (a) 0.5 μg mL–1 and (b) 10 μg mL–1. (c) The ROS generation monitored by 2,7-dichlorofluorescein (DCF) luminescence for MCF-7 cells after different treatments. (d1–8) Direct observation of DNA breakdown using comet assays on MCF-7 cells after different treatments: (d1) control, (d2) MMC, (d3) RUMSNs–MMC, (d4) RUMSNs–TAT–MMC, (d5) RT, (d6) MMC + RT, (d7) RUMSNs–MMC + RT, and (d8) RUMSNs–TAT–MMC + RT. [MMC] = 10 μg mL–1.

The substantially increased treatment efficiency and radiation enhancement effects are believed to be caused by the more severe DNA damage, as supported by the single-cell gel electrophoresis (comet assay) study (Fig. 4d & S11b†). The control group shows negligible DNA damage while long tails of stain can be observed in the nucleus after certain treatments, which indicates obvious DNA damage. As compared to the other treatments, the RUMSNs–TAT–MMC + RT has caused the most significant DNA damage (as evidenced by the longest tail), which further confirms the strongest radiation enhancement effects produced by the substantially enhanced synergetic chemo-/radiotherapy in the nucleus as well as the much better therapeutic effects achieved by the intranuclear radiosensitization than the intracytoplasmic or extracellular radiosensitization.
The widespread multi-drug resistance (MDR) of malignant tumors is one of the major reasons for cancer treatment failure. With single-mode chemotherapy, satisfactory treatment is usually not achievable, which highlights the demand for the combination of chemotherapy with other therapeutic modes such as radiotherapy. We performed the same in vitro experiments on the multi-drug resistant MCF-7 (MCF-7/ADR) cells to see whether the above intranuclear radiosensitization could also enhance the anticancer effects against MCF-7/ADR cells. As seen from Fig. 5a and b & S12a and b,† free MMC does not exhibit visible cytotoxicity on MCF-7/ADR cells because of the over-expressed P-gp efflux pumps on the cell membrane. Although the RUMSNs–MMC can improve the therapeutic effects due to the efficient delivery of MMC to the cytoplasm by bypassing the P-gp efflux pumps, the cell viability can be further decreased by the RUMSNs–TAT–MMC due to the direct intranuclear MMC delivery. As well, RT can also efficiently reverse MDR by suppressing the expression of P-gp (Fig. 6b). So the treatment with the RUMSNs–TAT–MMC + RT causes the most remarkable death rates of MCF-7/ADR cells, as also evidenced by the highest apoptosis rate and ROS level shown in Fig. 5c and d & S13a.† Furthermore, Fig. 6a and b show that the treatment with the RUMSNs–TAT–MMC + RT results in the most significant decrease in ATP levels and P-gp expression, which also confirms the highest MDR reversal effects caused by the intranuclear radiosensitization.
Fig. 5. (a & b) The viability of MCF-7/ADR cells after different treatments for 24 h with an MMC concentration of (a) 0.5 μg mL–1 and (b) 10 μg mL–1. (c) The ROS generation monitored by DCF luminescence for MCF-7/ADR cells after different treatments. (d1–8) Flow cytometry analysis for apoptosis of MCF-7/ADR cells after different treatments: (d1) control, (d2) MMC, (d3) RUMSNs–MMC, (d4) RUMSNs–TAT–MMC, (d5) RT, (d6) MMC + RT, (d7) RUMSNs–MMC + RT, and (d8) RUMSNs–TAT–MMC + RT. [MMC] = 10 μg mL–1.

Fig. 6. (a) The relative ATP level and (b) P-gp expression of MCF-7/ADR cells after different treatments. (c) Western blot analyses of the expression of Caspase3 and P27 protein in MCF-7/ADR cells after different treatments: (1) control, (2) MMC, (3) RUMSNs–MMC, (4) RUMSNs–TAT–MMC, (5) RT, (6) MMC + RT, (7) RUMSNs–MMC + RT, and (8) RUMSNs–TAT–MMC + RT. [MMC] = 10 μg mL–1.

Finally, we further compared these treatments from the perspective of molecular biology by testing the expressions of the apoptosis gene Caspase3 and proliferation gene P27 in the MDR cells. As well, the treatment with the RUMSNs–TAT–MMC + RT leads to the most significant up-regulation of Caspase3 and down-regulation of P27 (Fig. 6c), which demonstrates the outstanding advantages of the intranuclear radiosensitization in overcoming MDR by enhancing apoptosis and inhibiting proliferation of the MDR cells. These results are consistent with the corresponding comet assays which show that, with the synergetic interactions between MMC and X-ray irradiation directly within the nucleus, the intranuclear radiosensitization can cause the most remarkable DNA damage (Fig. S12c & S13b†). All the above results confirm that the combination of the intranuclear drug delivery and X-ray radiation may provide an excellent platform for reversing MDR based on the highly efficient intranuclear chemodrug-sensitized radiation enhancement effects.
2.4. In vivo evaluation of intranuclear radiosensitization
Encouraged by the above in vitro therapy results, we next performed the in vivo experiments to further evaluate the effectiveness of the intranuclear radiosensitization on inhibiting tumor growth. Firstly, the in vivo long-term toxicity of the RUMSNs/RUMSNs–TAT was studied by hematological and biochemical analyses. As shown by the complete blood chemistry tests in Fig. S14 and 15,† all blood parameters fall within the normal ranges even within 30 days post-injection. What is more, no noticeable tissue damage and adverse effects on the major organs (liver, spleen, etc.) can be observed from the corresponding hematoxylin and eosin (H & E) staining images (Fig. S16 and 17†), which show that the RUMSNs and RUMSNs–TAT do not cause any obvious infection/inflammation. In addition, no significant weight loss or any abnormal behaviour are observed in the mice between the RUMSNs/RUMSNs–TAT treated mice and the control mice (Fig. S18†). All these results convincingly evidence that our synthesized RUMSNs/RUMSNs–TAT are biocompatible in vivo.
Secondly, the in vivo MRI experiment was conducted to investigate the accumulation of the RUMSNs/RUMSNs–TAT (Fig. S19†) in tumors following intravenous injection. As seen from Fig. 7a, the MRI intensity of the tumor is enhanced with the increasing time period post-injection due to the successful delivery of the RUMSNs via passive targeting. More surprisingly, the MRI intensity is enhanced by about 31% 15 min post-injection of the RUMSNs–TAT in comparison to 13% of that of the RUMSNs (Fig. 7b), which may be attributed to the more significant accumulation of the RUMSNs–TAT in the tumors due to the conjugation of the nuclear-targeting ligand TAT, as also evidenced by the quantitative analysis in Fig. 7c. Therefore, the RUMSNs–TAT may be developed as magnetic imaging probes to localize to the tumor sites.
Fig. 7. (a) In vivo T1-MRI images of MCF-7 tumor-bearing mice after intravenous injection of (a1–3) the RUMSNs and (a4–6) the RUMSNs–TAT at designated time points. (b) Comparison of the MRI signal intensity of MCF-7 tumors after intravenous injection of the RUMSNs/RUMSNs–TAT at different time points. (c) Quantitative analysis of the Yttrium concentrations in tumors following the intravenous injection of the RUMSNs and RUMSNs–TAT.

Thirdly, we carried out the in vivo therapy experiments on MCF-7 tumor-bearing nude mice. As seen from the bio-distribution study in Fig. 7c & S20,† much more RUMSNs–TAT (∼4.35% ID Y g–1) can accumulate in tumors than RUMSNs (∼1.65% ID Y g–1), which may be attributed to the unique function of TAT in penetrating blood vessels/cell membranes and achieving relatively higher retention within tumor cells.19 In addition, the histological analysis of the tumors by low-resolution CLSM imaging (Fig. S21†) shows that much stronger green luminescence of RUMSNs–TAT (Fig. S21a2 †) is observed in tumors, which further evidences the greater accumulation of the RUMSNs–TAT than the RUMSNs. More importantly, the green luminescence of the RUMSNs–TAT clearly localises at the nucleus of most tumor cells (Fig. S21a3 †), which undoubtedly confirms that the RUMSNs–TAT can reach the nucleus of tumor cells with the assistance of TAT and achieve the intranuclear drug delivery in vivo following intravenous injection.
As compared to the control group, the tumors treated with the RUMSNs–TAT–MMC demonstrate more significant growth delay than those treated with the RUMSNs–MMC (Fig. 8a and b) due to the higher chemotherapeutic efficacy based on the intranuclear drug delivery rather than the intracytoplasmic delivery. With the assistance of X-ray irradiation, the synergetic chemo-/radiotherapy based on radiosensitization produces much better therapeutic effects than individual chemotherapy or radiotherapy. Similarly, the RUMSNs–TAT–MMC + RT can most effectively inhibit the tumor growth in half a month and produce much better therapeutic effects than the RUMSNs–MMC + RT, which may be attributed to the much more substantially enhanced chemo-/radiotherapy efficacy caused by the intranuclear radiosensitization than the intracytoplasmic radiosensitization, as can be clearly seen from the corresponding digital photos of tumors in Fig. S22.† Moreover, the tumors treated with the RUMSNs–TAT–MMC + RT demonstrate the most significant necrosis (as evidenced by images of hematoxylin–eosin (H & E) stained tumor sections in Fig. S23†), which further confirms the higher treatment efficiency and better radiation enhancement effects caused by the intranuclear radiosensitization in comparison to other kinds of therapies.
Fig. 8. (a) Tumor growth curves of MCF-7 tumor-bearing mice over a period of half a month after the corresponding treatments. (b) Relative MCF-7 tumor volumes of different groups after half a month of the corresponding treatments. (c) Tumor growth curves of MCF-7/ADR tumor-bearing mice over a period of half a month after treatment with the intranuclear radiosensitization. (d) Relative MCF-7/ADR tumor volumes after half a month of therapy with the intranuclear radiosensitization.

Finally, by implanting MCF-7/ADR cells into the right axilla of nude mice, we further estimated the therapeutic effects of the intranuclear radiosensitization on the MDR tumors. Surprisingly, the treatment with the RUMSNs–TAT–MMC + RT not only completely inhibits the growth of MCF-7/ADR tumors, but also leads to significant tumor regression by about 60% (Fig. 8c and d and as also clearly evidenced by the corresponding digital photos and H & E stained tumor sections in Fig. S24†), which marks one of the most significant instances in which the intranuclear radiosensitization provides a more advanced way for reversing MDR in vivo.
3. Conclusions
In summary, we have presented a new technique of “intranuclear radiosensitization” by designing a sub-50 nm multifunctional nuclear-targeting nanotheranostic system (RUMSNs–TAT) to directly deliver the radiosensitive chemotherapeutic drug MMC into the nucleus for the greatly elevated intranuclear chemodrug-sensitized radiation enhancement effects on damaging the DNA under high energy X-ray irradiation. The in vitro and in vivo therapy experiments undoubtedly demonstrate the much more enhanced treatment efficiency by the intranuclear radiosensitization than the extracellular radiosensitization and intracytoplasmic radiosensitization in killing cancer cells and inhibiting tumor growth. Moreover, as the ATP level/P-gp expression can be significantly down-regulated by the nuclear-targeting nanotheranostics to bypass efflux action, the intranuclear radiosensitization also demonstrates remarkable advantages in efficiently reversing MDR and regressing the MDR tumor growth. Finally, based on the MR/UCL bimodal imaging performance, our synthesized nanotheranostics may be developed as the next generation of accurate imaging guided nuclear-targeting nanomedicine in the future.
Acknowledgments
This work has been financially supported by the National Natural Science Foundation of China (Grant no. 51372260, 51132009, 21172043, 51102259) and the Shanghai Rising-Star Program (Grant no. 12QH1402500).
Footnotes
References
- (a) Shen J., He Q., Gao Y., Shi J., Li Y. Nanoscale. 2011;3:4314. doi: 10.1039/c1nr10580a. [DOI] [PubMed] [Google Scholar]; (b) He Q., Gao Y., Zhang L., Zhang Z., Gao F., Ji X., Li Y., Shi J. Biomaterials. 2011;32:7711. doi: 10.1016/j.biomaterials.2011.06.066. [DOI] [PubMed] [Google Scholar]; (c) Duan X., Xiao J., Yin Q., Zhang Z., Yu H., Mao S., Li Y. ACS Nano. 2013;7:5858. doi: 10.1021/nn4010796. [DOI] [PubMed] [Google Scholar]
- (a) Jabr-Milane L. S., van Vlerken L. E., Yadav S., Amiji M. M. Cancer Treat. Rev. 2008;34:592. doi: 10.1016/j.ctrv.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lage H. Cell. Mol. Life Sci. 2008;65:3145. doi: 10.1007/s00018-008-8111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ullah M. F. Asian Pac. J. Cancer Prev. 2008;9:1. [PubMed] [Google Scholar]; (d) Gao J., Feng S.-S., Guo Y. Nanomedicine. 2012;7:465. doi: 10.2217/nnm.12.11. [DOI] [PubMed] [Google Scholar]
- (a) Juzenas P., Chen W., Sun Y.-P., Coelho M. A. N., Generalov R., Generalova N., Christensen I. L. Adv. Drug Delivery Rev. 2008;60:1600. doi: 10.1016/j.addr.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hogle W. P. Semin. Oncol. Nurs. 2006;22:212. doi: 10.1016/j.soncn.2006.07.004. [DOI] [PubMed] [Google Scholar]
- (a) Hainfeld J. F., Dilmanian F. A., Slatkin D. N., Smilowitz H. M. J. Pharm. Pharmacol. 2008;60:977. doi: 10.1211/jpp.60.8.0005. [DOI] [PubMed] [Google Scholar]; (b) Wardman P. Clin. Oncol. 2007;19:397. doi: 10.1016/j.clon.2007.03.010. [DOI] [PubMed] [Google Scholar]; (c) Nakae T., Uto Y., Tanaka M., Shibata H., Nakata E., Tominaga M., Maezawa H., Hashimoto T., Kirk K. L., Nagasawa H., Hori H. Bioorg. Med. Chem. 2008;16:675. doi: 10.1016/j.bmc.2007.10.035. [DOI] [PubMed] [Google Scholar]
- (a) Kennedy K. A., Rockwell S., Sartorelli A. C. Cancer Res. 1980;40:2356. [PubMed] [Google Scholar]; (b) Tomasz M. Chem. Biol. 1995;2:575. doi: 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Grau C., Agarwal J. P., Jabeen K., Khan A. R., Abeyakoon S., Hadjieva T., Wahid I., Turkan S., Tatsuzaki H., Dinshaw K. A., Overgaard J. Radiother. Oncol. 2003;67:17. doi: 10.1016/s0167-8140(03)00020-3. [DOI] [PubMed] [Google Scholar]
- (a) Kulka U., Schaffer M., Siefert A., Schaffer P. M., Ölsner A., Kasseb K., Hofstetter A., Dühmke E., Jori G. Biochem. Biophys. Res. Commun. 2003;311:98. doi: 10.1016/j.bbrc.2003.09.170. [DOI] [PubMed] [Google Scholar]; (b) Hill B. T., Whelan R. D. H., Shellard S. A., McClean S., Hosking L. K. Invest. New Drugs. 1994;12:169. doi: 10.1007/BF00873957. [DOI] [PubMed] [Google Scholar]; (c) Rose P. G., Bundy B. N., Watkins E. B., Thigpen J. T., Deppe G., Maiman M. A., Clarke-Pearson D. L., Insalaco S. N. Engl. J. Med. 1999;340:1144. doi: 10.1056/NEJM199904153401502. [DOI] [PubMed] [Google Scholar]
- (a) Werner M. E., Copp J. A., Karve S., Cummings N. D., Sukumar R., Li C., Napier M. E., Chen R. C., Cox A. D., Wang A. Z. ACS Nano. 2011;5:8990. doi: 10.1021/nn203165z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fan W., Shen B., Bu W., Chen F., Zhao K., Zhang S., Zhou L., Peng W., Xiao Q., Xing H., Liu J., Ni D., He Q., Shi J. J. Am. Chem. Soc. 2013;135:6494. doi: 10.1021/ja312225b. [DOI] [PubMed] [Google Scholar]
- Liu J.-n., Bu W., Pan L.-m., Zhang S., Chen F., Zhou L., Zhao K.-l., Peng W., Shi J. Biomaterials. 2012;33:7282. doi: 10.1016/j.biomaterials.2012.06.035. [DOI] [PubMed] [Google Scholar]
- Pan L., He Q., Liu J., Chen Y., Ma M., Zhang L., Shi J. J. Am. Chem. Soc. 2012;134:5722. doi: 10.1021/ja211035w. [DOI] [PubMed] [Google Scholar]
- (a) Zhou J., Liu Z., Li F. Chem. Soc. Rev. 2012;41:1323. doi: 10.1039/c1cs15187h. [DOI] [PubMed] [Google Scholar]; (b) Gu Z., Yan L., Tian G., Li S., Chai Z., Zhao Y. Adv. Mater. 2013;25:3758. doi: 10.1002/adma.201301197. [DOI] [PubMed] [Google Scholar]; (c) Cao T., Yang Y., Gao Y., Zhou J., Li Z., Li F. Biomaterials. 2011;32:2959. doi: 10.1016/j.biomaterials.2010.12.050. [DOI] [PubMed] [Google Scholar]; (d) Cao T., Yang T., Gao Y., Yang Y., Hu H., Li F. Inorg. Chem. Commun. 2010;13:392. [Google Scholar]; (e) Chen G., Shen J., Ohulchanskyy T. Y., Patel N. J., Kutikov A., Li Z., Song J., Pandey R. K., Agren H., Prasad P. N., Han G. ACS Nano. 2013;6:8280. doi: 10.1021/nn302972r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shen J., Chen G., Vu A.-M., Fan W., Bilsel O. S., Chang C.-C., Han G. Adv. Opt. Mater. 2013;1:644. [Google Scholar]; (g) Shen J., Chen G., Ohulchanskyy T. Y., Kesseli S. J., Buchholz S., Li Z., Prasad P. N., Han G. Small. 2013;9:3213. doi: 10.1002/smll.201300234. [DOI] [PubMed] [Google Scholar]; (h) Punjabi A., Wu X., Tokatli-Apollon A., El-Rifai M., Lee H., Zhang Y., Wang C., Liu Z., Chan E. M., Duan C., Han G. ACS Nano. 2014;8:10621. doi: 10.1021/nn505051d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Chen F., Bu W., Zhang S., Liu X., Liu J., Xing H., Xiao Q., Zhou L., Peng W., Wang L., Shi J. Adv. Funct. Mater. 2011;21:4285. [Google Scholar]; (b) Chen F., Bu W., Zhang S., Liu J., Fan W., Zhou L., Peng W., Shi J. Adv. Funct. Mater. 2013;23:298. [Google Scholar]; (c) Park Y. I., Kim H. M., Kim J. H., Moon K. C., Yoo B., Lee K. T., Lee N., Choi Y., Park W., Ling D., Na K., Moon W. K., Choi S. H., Park H. S., Yoon S.-Y., Suh Y. D., Lee S. H., Hyeon T. Adv. Mater. 2012;24:5755. doi: 10.1002/adma.201202433. [DOI] [PubMed] [Google Scholar]; (d) Zhou J., Sun Y., Du X., Xiong L., Hu H., Li F. Biomaterials. 2010;31:3287. doi: 10.1016/j.biomaterials.2010.01.040. [DOI] [PubMed] [Google Scholar]
- (a) Li C., Hou Z., Dai Y., Yang D., Cheng Z., Ma P. a., Lin J. Biomater. Sci. 2013;1:213. doi: 10.1039/c2bm00087c. [DOI] [PubMed] [Google Scholar]; (b) Liu J., Bu W., Zhang S., Chen F., Xing H., Pan L., Zhou L., Peng W., Shi J. Chem.–Eur. J. 2012;18:2335. doi: 10.1002/chem.201102599. [DOI] [PubMed] [Google Scholar]
- Hu Y., Zhang Q., Goebl J., Zhang T., Yin Y. Phys. Chem. Chem. Phys. 2010;12:11836. doi: 10.1039/c0cp00031k. [DOI] [PubMed] [Google Scholar]
- Idris N. M., Gnanasammandhan M. K., Zhang J., Ho P. C., Mahendran R., Zhang Y. Nat. Med. 2012;18:1580. doi: 10.1038/nm.2933. [DOI] [PubMed] [Google Scholar]
- Bogdan N., Vetrone F., Ozin G. A., Capobianco J. A. Nano Lett. 2011;11:835. doi: 10.1021/nl1041929. [DOI] [PubMed] [Google Scholar]
- Liu J., Bu W., Pan L., Shi J. Angew. Chem., Int. Ed. 2013;52:4375. doi: 10.1002/anie.201300183. [DOI] [PubMed] [Google Scholar]
- Gao Y., Chen Y., Ji X., He X., Yin Q., Zhang Z., Shi J., Li Y. ACS Nano. 2011;5:9788. doi: 10.1021/nn2033105. [DOI] [PubMed] [Google Scholar]
- (a) Yuan H., Fales A. M., Vo-Dinh T. J. Am. Chem. Soc. 2012;134:11358. doi: 10.1021/ja304180y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wunderbaldinger P., Josephson L., Weissleder R. Bioconjugate Chem. 2002;13:264. doi: 10.1021/bc015563u. [DOI] [PubMed] [Google Scholar]; (c) Torchilin V. P. Adv. Drug Delivery Rev. 2008;60:548. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.