

Abstract
Three major genetic isoforms of apolipoprotein E (ApoE) exist in humans, ApoE2, ApoE3, and ApoE4 leading to differences in susceptibility to Alzheimer’s disease (AD). This study investigated the impact of human ApoE isoforms on brain metabolic pathways involved in glucose utilization and amyloid-β (Aβ) degradation, two major areas that are significantly perturbed in preclinical AD. Hippocampal RNA samples from middle-aged female mice with targeted human ApoE2, ApoE3, and ApoE4 gene replacement were comparatively analyzed with a qRT-PCR custom array for the expression of 85 genes involved in insulin/insulin-like growth factor (Igf) signaling. Consistent with its protective role against AD, ApoE2 brain exhibited the most metabolically robust profile among the three ApoE genotypes. When compared to ApoE2 brain, both ApoE3 and ApoE4 brains exhibited markedly reduced levels of Igf1, insulin receptor substrates (Irs), and facilitated glucose transporter 4 (Glut4), indicating reduced glucose uptake. Additionally, ApoE4 brain exhibited significantly decreased Pparg and insulin-degrading enzyme (Ide) indicating further compromised glucose metabolism and Aβ dysregulation associated with ApoE4. Protein analysis showed significantly decreased Igf1, Irs, and Glut4 in ApoE3 brain, and Igf1, Irs, Glut4, Pparg, and Ide in ApoE4 brain compared to ApoE2 brain. These data provide the first documented evidence that human ApoE isoforms differentially affect brain insulin/Igf signaling and downstream glucose and amyloid metabolic pathways, illustrating a potential mechanism for their differential risk in AD. A therapeutic strategy that enhances brain insulin/Igf1 signaling activity to a more robust ApoE2-like phenotype favoring both energy production and amyloid homeostasis holds promise for AD prevention and early intervention.
Keywords: Alzheimer’s disease (AD), apolipoprotein E2 (ApoE2), apolipoprotein E3 (ApoE3), apolipoprotein E4 (ApoE4), insulin-like growth factor 1 (Igf1), Insulin-degrading enzyme (Ide), glucose transporter 4 (Glut4), peroxisome proliferator-activated receptor gamma (Pparg), glucose metabolism, early intervention
Introduction
As one of the most devastating neurodegenerative disorders, Alzheimer’s disease (AD) currently affects approximately 35 million people worldwide, including 5.4 million Americans, and these numbers are predicted to triple by 2050 [1, 2]. AD disproportionally impacts females more than males; approximately two-thirds of current victims of AD are women [3]. Two classes of drugs are presently prescribed for the treatment of AD; all were FDA-approved during 1993–2003 [3]. These drugs help to regulate levels and activity of key neurotransmitters that are involved in learning, reasoning, and memory. Four of the five approved drugs are cholinesterase inhibitors that function to decrease the breakdown of acetylcholine. The other works by regulating glutamate activity. These treatments can temporarily help manage some symptoms in select patients and they do not treat the underlying causes of the disease or retard the progression of the disease [4]. In the last decade, over 100 human trials have failed in an attempt to find an effective treatment for mid to late-stage AD [5]. At present, while the cause of AD is not well understood nor is an effective treatment available, one of the key strategies to address this challenge would be to identify AD risk mechanisms that would allow institution of AD prevention and early intervention.
AD is an age-related, progressive, and extremely complex neurodegenerative disease, beginning with a long preclinical phase (10–20 years) characterized by glucose hypometabolism and increased amyloid-β (Aβ) deposition in brain regions particularly vulnerable to AD [6–8]. In addition to Aβ accumulation, which is hypothesized to be perhaps the earliest change presently detectible [8], reduced glucose utilization nearly concomitantly occurs in several brain areas typically affected in AD in preclinical, presymptomatic subjects with genetic predisposition for developing AD [6, 9]. The Alzheimer’s Association (AA) and the National Institute on Aging (NIA) have recently revised diagnostic criteria for AD and have added preclinical AD (PCAD) as a recognized stage of the disease. This PCAD stage is defined as presymptomatic but where key biological changes are underway that may begin years or even decades before memory loss, confusion, or changes in thinking or behavior occur. The AA and NIA challenge researchers to identify these early changes, measureable biomarkers, and early intervention strategies to slow or cease the progression of AD [8, 10].
Apolipoprotein E (ApoE) is primarily produced by the liver in the periphery and by astrocytes in the CNS. The primary role of ApoE is cholesterol transport, regulating lipid transport and injury repair in the brain [11]. Additionally, ApoE plays roles in glucose metabolism, mitochondrial function, neuroinflammation, neuronal signaling, and Aβ processing [11, 12]. Humans possess three major genetic isoforms of ApoE coded by three alleles, ε2, ε3, and ε4 [11, 13, 14]. Genetic variation in ApoE [15–17] has long been implicated in human risk for developing late-onset AD [18, 19]. ApoE2 is relatively rare, with only approximately 5% incidence, and it is recognized as a protective variant against AD [11, 20, 21]. ApoE3, the most common isoform, is present in approximately 75% of the population and is believed to be neutral in AD [22]. ApoE4, the strongest genetic risk factor for late-onset AD, occurs in only about 20% of the population but accounts for approximately 65% of AD cases [11, 20, 21]. ApoE4 correlates with an earlier age of onset and higher incidence of late onset AD whereas ApoE2 seems to decrease the incidence or delay disease onset [11, 23, 24]. Though conflicting evidence exists, the increased risk for AD conveyed by the ApoE4 allele appears to have a greater risk impact in females than in males [25–28]. Conversely, ApoE2 appears to afford a greater protective effect against AD in males than in females [28, 29].
In early mild cognitive impairment, the presence of the ApoE4 allele has been linked to both increased Aβ deposition in the cortex and decreased levels of Aβ in the cerebrospinal fluid (CSF) [30] demonstrating Aβ dysregulation as an early event in disease progression. In an isoform-dependent manner, ApoE isoforms differentially regulate Aβ proteolysis and Aβ clearance from brain and modulate Aβ–induced oxidative stress in synaptosomes (ApoE4<ApoE3<ApoE2) [31–34]. Moreover, the neurotoxic oligomeric form of Aβ is more abundant and even stabilized in ApoE4 over ApoE3 and ApoE2 brain, but the mechanisms by which this occurs are less clear [35–37]. Recent evidence has also shown that ApoE2 carriers have reduced AD pathology in the CSF and lower hippocampal atrophy than their non-ApoE2 counterparts [38, 39]. Despite evidence of the protective effect associated with ApoE2, remarkably few studies have been done to determine the mechanism of this protection [39]. These studies emphasize the need for further understanding of the protective effect of ApoE2.
Impaired glucose metabolism in the brain is associated with AD beginning in the earliest stages [9, 40, 41], perhaps even before amyloidogenesis and long before onset of clinical symptoms [42]. ApoE4 carriers exhibit decreased glucose metabolic rates in the brain compared to non-ApoE4 subjects [43]. As evidenced by positron emission tomography (PET), inheritance of the ApoE4 allele results in impaired cerebral glucose metabolism in subjects with a family history of AD but who do not yet have dementia [40]. Due to many overlapping abnormalities between AD and type 1 and type 2 diabetes mellitus, AD has been referred to as type 3 diabetes [44, 45]. Normally, activation of insulin or insulin-like growth factor 1 (Igf1) signaling results in an intracellular cascade leading to increased glucose uptake and utilization. Defective insulin/Igf1 signaling has been implicated in AD pathogenesis [46–49], and recent studies have shown that Igf1 polymorphism also modifies AD risk [50, 51]. However, how this signaling could be linked to different susceptibility for AD associated with human ApoE isoforms is poorly understood. In this study, using gene expression profiling analyses, we tested the hypothesis that insulin/Igf1 signaling could be differentially regulated by human ApoE isoforms, which could serve as an important mechanistic rationale for their differential risks for AD. Our results identify several areas of insulin/Igf1 signaling that are differentially regulated in brain among the three ApoE variants. These pathways could potentially serve as targets for AD prevention, risk reduction, and early intervention.
Materials and Methods
Animals
The use of animals was approved by the Institutional Animal Care and Use Committee at the University of Kansas and followed NIH guidelines for the care and use of laboratory animals. This study was carried out in human ApoE2, ApoE3, and ApoE4 gene-targeted replacement (hApoE2-TR, hApoE3-TR, and hApoE4-TR) mouse models. These mouse lines were created by gene targeting and carry one of the three human ApoE alleles in place of the endogenous murine ApoE gene while retaining the endogenous regulatory sequences required for modulating hApoE expression [52]. These mice share a C57BL/6J genetic background and express the human ApoE protein at physiological levels; thus, they provide a complete in vivo system that allows direct measurement and comparison of hApoE isoform-specific effects. The following experiments were conducted on brain tissues collected from 6-month-old hApoE2-TR, hApoE3-TR, and hApoE4-TR female mice. Our choice of using female models in this study was based on the facts that females are at a much higher risk for AD than males, and ApoE4 genotype appears to confer a greater risk for AD to females than males; therefore, we set investigating the mechanism of female susceptibility to AD as our priority.
qRT-PCR gene expression profiling
qRT-PCR gene arrays were custom manufactured at Qiagen (Valencia, CA); a list of 85 genes included on the arrays is provided in Supplemental Table 1. Total RNA was isolated from mouse hippocampal tissues using the PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA). RNA quantity and quality were analyzed using the Experion RNA StdSens Analysis Kit on an Experion Automated Electrophoresis System (Bio-Rad, Hercules, CA). RNA to cDNA synthesis was prepared using the High Capacity RNA-to-cDNA Master Mix (Life Technologies) on a MyCycler Thermal Cycler (Bio-Rad). qRT-PCR reactions were performed on 0.5 μg cDNA samples mixed with the RT2 SYBR Green qPCR Master Mix, under the thermal cycling conditions: Holding Stage: 95°C for 10 min; Cycling Stage: 40 cycles; 95°C for 15 sec; 60°C for 1 min; Melting Curve Stage: default setting from 60°C to 95°C. Fluorescence was detected on an ABI 7900HT Fast Real-Time PCR System equipped with the Sequence Detection System Software Version 2.3 (Life Technologies).
Data were analyzed using the PCR Array Data Analysis Software (Qiagen). Relative gene expression levels or fold differences (FD) relative to the comparison group were calculated by the comparative Ct (ΔΔCt) method, with Ct denoting threshold cycle [53]. Samples collected from 5 animals per ApoE genotype group were included in the analysis. For each sample, ΔCt was calculated as the difference in average Ct of the target gene and the endogenous control gene. For each ApoE genotype group, mean 2−ΔCt was calculated as the geometric mean of 2−ΔCt of the 5 samples in the group. FD was then calculated as mean 2−ΔCt (one ApoE genotype group)/mean 2−ΔCt (the other ApoE genotype group as the comparison group). FD values greater than one indicate a greater expression relative to the comparison group. FD values less than one indicate a lower expression relative to the comparison group. The 2−ΔCt values for each target gene between two ApoE genotype groups were statistically compared using Student’s t-test; p<0.05 was considered statistically significant. The heat map displays results of unsupervised hierarchical clustering; distances between target genes were calculated based on the ΔCt values using Pearson’s correlations. The volcano plot displays FD (X-axis) and p-values (Y-axis) enabling identification of genes with both large and small expression differences that are statistically significant.
Western blot protein expression analysis
Mouse cortical tissues were homogenized in T-PER Tissue Protein Extraction Buffer (Thermo Scientific, Rockford, IL) containing protease and phosphatase inhibitors using 0.5mm glass beads in a Bullet Blender homogenizer (Next Advance, Averill Park, NY). After centrifugation at 12,000 RPM for 10 min at 4°C, supernatant was transferred to fresh, labeled microfuge tubes, and saved as whole protein lysate at −80°C until analysis. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific) on a Synergy H1 Hybrid Microplate Reader equipped with Gen5 software (BioTek, Winooski, VT). Protein homogenates containing 50 ug of total protein each were mixed at 1:1 with 2x Laemmli sample buffer containing 5% β-mercaptoethanol (BME), heated for 5 min using an AccuBlock digital dry bath (Labnet, Hanover Park, IL) at 95°C, and loaded onto precast Criterion TGX (any kD) or Criterion TGX (18%) Precast Gels as appropriate for the molecular weight of the protein of interest. Precision Plus Protein All Blue Standards and samples were run using a Criterion cell and PowerPacHC at 200 V for 55–60 min in tris-glycine-SDS (TGS) running buffer. PowerPacHC, Criterion cell, gels, standards, and buffers were all from BioRad (Hercules, CA).
In-gel proteins were transferred to a nitrocellulose membrane (0.20 μm) using a Criterion Blotter and PowerPacHC at 100 V for 30 min (BioRad). After transfer, membranes were incubated in blocking solution (Blotting-Grade Blocker in phosphate buffered saline (PBS) solution with 0.2% (v/v) Tween 20) at room temperature for 1.5 h on a gentle rocking platform. Membranes were then incubated with mouse anti-Igf1 antibody, rabbit anti-Irs1 antibody, or rabbit anti-Glut4 antibody, rabbit anti-Ide antibody and either mouse anti-β-tubulin antibody or rabbit anti-β-actin antibody (Pierce) for 2 h on a gentle rocking platform, followed by three rinses for 5 min each with PBS solution with 0.2% (v/v) Tween 20. Blots were then incubated for 1 h with goat anti-mouse IgG (H+L) or goat anti-rabbit IgG (H+L), horseradish peroxidase-linked whole secondary antibody as appropriate. The resulting blots were rinsed three times for 5, 10, and 10 min each in PBS solution with 0.2% (v/v) Tween 20 and signals were detected using Clarity Western ECL Substrate (Bio-Rad) and the LI-COR scanner. Analysis was performed using the Image Studio Digits Version 4.0 software. Data are presented as mean±SEM, and statistical analysis was performed in GraphPad Prism software using ANOVA plus Bonferroni’s Multiple Comparison posttest with p<0.05 considered significant.
Results
In this study, we investigated the gene expression profiles in the brains of middle-aged female mice carrying the human ApoE2, ApoE3, or ApoE4 gene to examine the impact of ApoE genotype on brain metabolic pathways involved in glucose utilization and Aβ degradation. Hippocampal RNA samples were comparatively analyzed with qRT-PCR-based gene arrays for the expression of a focused panel of genes involved in insulin/Igf signaling (Supplemental Table 1). Distinct and consistent patterns emerged in these gene arrays demonstrating both differences and overlaps among the three ApoE brains (Fig. 1). More specifically, significant differences were found in key genes involved in Igf1/Glut4/Pparg/Ide cascades in both the neutral ApoE3 and high-risk ApoE4 groups when compared to the protective ApoE2 group (Fig. 2&3).
Fig. 1.

Gene expression profiles in the brains of middle-aged female mice carrying human ApoE2, ApoE3, or ApoE4 gene. Hippocampal RNA samples were analyzed with a qRT-PCR gene array for the expression of a focused panel of genes involved in insulin/Igf signaling. Red indicates high expression, green indicates low expression. Control Group = ApoE2 brain; Group 1 = ApoE3 brain; Group 2 = ApoE4 brain; N = 5 per ApoE genotype group. A list of all genes profiled is provided in Supplemental Table 1.
Fig. 2.

The heat map (a) includes genes that significantly differed between any two of the three ApoE genotypes. The volcano plots show fold changes (X-axis) and P-values (Y-axis) between (b) ApoE3 brain versus ApoE2 brain and (c) ApoE4 brain versus ApoE2 brain; each dot represents a gene; red indicates greater expression; green indicates lower expression; dots that fall above the horizontal blue line indicate significantly differed genes (p<0.05). Highlighted significantly altered expression - Igf1: insulin-like growth factor 1; Irs1/2: insulin receptor substrate 1/2; Slc2a4: solute carrier family 2 facilitated glucose transporter member 4 (Glut4); Pparg: peroxisome proliferator-activated receptor gamma; Ide: insulin-degrading enzyme. N = 5 per ApoE genotype group.
Fig. 3.
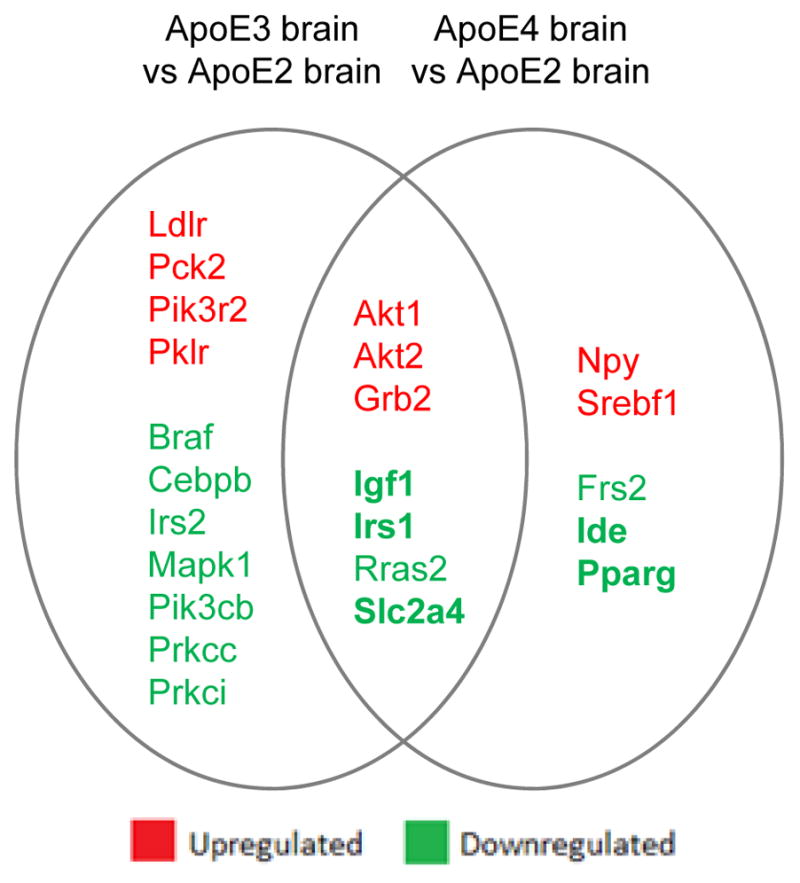
When compared to ApoE2 brain, ApoE3 and ApoE4 brain exhibit both similar and distinct expression profiles of genes involved in insulin/Igf signaling. Major genes that showed a significantly lower expression in both ApoE3 and ApoE4 brain than ApoE2 brain include Igf1, Irs1, and Glut4, involved in glucose metabolism. Ide, a key player involved in the degradation of Aβ monomers in the brain, and Pparg, involved in insulin sensitivity and mitochondrial biogenesis, were significantly low-expressed in only ApoE4 but not ApoE3 brain when compared to ApoE2 brain.
Among the 85 genes analyzed, 23 genes exhibited a significant fold difference (p<0.05) between any two of the three ApoE groups (Fig. 2). The heat map demonstrates that, among three ApoE brains, the profile associated with ApoE2 brain appeared most distinct from ApoE3 and ApoE4 brains; in comparison, ApoE3 and ApoE4 brains exhibited some degree of similarities (Fig. 2a). The volcano plots illustrate fold changes and p-values for significant differences in ApoE3 compared to ApoE2 (Fig. 2b) and ApoE4 compared to ApoE2 (Fig. 2c).
Significantly downregulated genes in ApoE3 compared to ApoE2 hippocampus included Igf1 (FD=0.79; p=0.011), insulin receptor substrate 1 (FD=0.85; p=0.048) and 2 (FD=0.82; p=0.010) (Irs1/2), and solute carrier family 2 facilitated glucose transporter member 4 (Slc2a4, Glut4) (FC=0.55; p=0.003) (Fig. 2b). Versus ApoE2 samples, ApoE4 hippocampus exhibited many of the same downregulated genes in this panel as did ApoE3, including Igf1 (FC=0.87; p=0.044), Irs1 (FC=0.86; p=0.027), and Glut4 (FC=0.61; p=0.006) (Fig. 2c). In addition, ApoE4 hippocampus was found to express significantly decreased peroxisome proliferator-activated receptor gamma (Pparg) (FD=0.82; p=0.025) versus ApoE2 and insulin-degrading enzyme (Ide) versus ApoE2 and ApoE3 (FD=0.87; p=0.013; FD=0.087; 0.043) (Fig. 2c).
Areas of discord between ApoE3 and ApoE4 brain include significantly lower in ApoE4 brain: Ide (FD=0.87, p=0.039), Ldlr (FD=0.80, p=0.039), Pck2 (FD=0.85, p=0.006), and Pklr (FD=0.82, p=0.047), and significantly higher in ApoE4 brain: Npy (FD=1.33, p=0.022), Prkcc (FD=1.16, p=0.045), and Prkci (FD=1.18, p=0.009), when compared to ApoE3 brain. The lower Ide seen here in hApoE4 mouse brain is consistent with the human Ide data found by Cook et al. in ApoE4 carriers vs. non-ApoE4 carriers [54].
Common areas of significantly altered expression between ApoE3 and ApoE4 hippocampus versus that found in ApoE2 illustrate some overlaps in risk related to Igf1 signaling deficiencies. Downregulation of Igf1, Irs1, and Glut4 present in both ApoE3 and ApoE4 brain predicts less efficient glucose metabolism in both groups compared to ApoE2 brain. Previous studies have also shown lower Irs1 in ApoE4 brain versus ApoE3 in studies in older mice [55]. Akt (protein kinase B) 1 and 2 and growth factor receptor-bound protein 2 (Grb2) were found to be upregulated in both ApoE3 and ApoE4 brains compared to the ApoE2 group. Differences in Akt gene expression is evidence of changes in regulation of glycogen synthesis and Glut4 translocation.
Further, some aspects of altered gene expression found to be unique to ApoE4 brain, i.e. deficiencies in Ide and Pparg, infer additional risk for AD associated with this genotype (Fig. 3, Table 1). Pparg also showed nearly significantly lower levels in ApoE4 brain versus ApoE3. A greater variability of Pparg in the ApoE3 group in both the mRNA and protein studies contributed to the lack of significance. A significant difference in Pparg between the ApoE3 and ApoE4 groups may be found using a larger sample size.
Table 1.
Differentially expressed genes in ApoE3 and ApoE4 brain compared to ApoE2 brain
| ApoE3 vs ApoE2 (p<0.05) | ApoE4 vs ApoE2 (p<0.05) | |||
|---|---|---|---|---|
|
| ||||
| Gene Symbol | Fold Difference | p-value | Fold Difference | p-value |
| Akt1 | 1.15 | 0.019 | 1.17 | 0.043 |
| Akt2 | 1.30 | 0.001 | 1.24 | 0.002 |
| Braf | 0.86 | 0.008 | ||
| Cebpb | 0.83 | 0.004 | ||
| Frs2 | 0.89 | 0.012 | ||
| Grb2 | 1.29 | 0.008 | 1.30 | 0.010 |
| Ide | 0.87 | 0.013 | ||
| Igf1 | 0.79 | 0.011 | 0.87 | 0.044 |
| Irs1 | 0.85 | 0.048 | 0.86 | 0.027 |
| Irs2 | 0.82 | 0.010 | ||
| Ldlr | 1.32 | 0.037 | ||
| Mapk1 | 0.93 | 0.002 | ||
| Npy | 1.27 | 0.041 | ||
| Pck2 | 1.14 | 0.048 | ||
| Pik3cb | 0.83 | 0.002 | ||
| Pik3r2 | 1.35 | 0.013 | ||
| Pklr | 1.50 | 0.007 | ||
| Pparg | 1.032 | 0.062 | 0.82 | 0.025 |
| Prkcc | 0.86 | 0.042 | ||
| Prkci | 0.80 | 0.003 | ||
| Rras2 | 0.89 | 0.013 | 0.82 | 0.009 |
| Slc2a4 | 0.55 | 0.003 | 0.61 | 0.006 |
| Srebf1 | 1.24 | 0.015 | ||
N = 5 per ApoE genotype group
To validate the results of the gene array studies (Fig. 4) and the effect of ApoE status on key proteins involved in Igf1/Ide signaling in brain, cortical protein analysis (Fig. 4) using SDS-PAGE and WB showed significantly lower levels of Igf1, Irs1, and Glut4 proteins in ApoE3 (p<0.001, p<0.01, and p<0.001, respectively) and in ApoE4 (p<0.01, p<0.01, and p<0.001, respectively) brain compared to ApoE2 brain, consistent with hippocampal gene array data. Additionally, ApoE4 brain exhibited significantly lower levels of Ide protein compared to levels found in ApoE2 and ApoE3 brain (p<0.01, p<0.05 respectively), and lower Pparg levels compared to ApoE2 brain (p<0.05), also consistent with gene array results.
Fig. 4.

ApoE isoforms differentially modulate the expression, at both the gene and protein level, of major players involved in Igf1 signaling and glucose and Aβ metabolism including Igf1, Irs1, Glut4, Pparg, and Ide. Specifically, when compared to ApoE2 brain, both ApoE3 and ApoE4 brain exhibited a significantly lower expression of Igf1, Irs1 and Glut4. Moreover, ApoE4 brain was associated with a significantly lower expression of Pparg than ApoE2 brain and significantly lower expression of Ide than both ApoE2 and ApoE3 brain, and there was not a significant difference between ApoE2 and ApoE3 brain. N = 5 per ApoE genotype group; *p<0.05, **p<0.01, and ***p<0.001.
Discussion
The brain relies almost exclusively on glucose for energy production to supply the high energy requirements, thereby directly relying on glucose availability and glucose metabolism to fill the needs. Progressive decreases in glucose metabolism begin years prior to the clinical onset of AD and continues to decline throughout the progression of the disease [6, 56, 57]. Declines in glucose metabolism are associated with decreasing cognitive test scores and correlate with the severity of dementia [58, 59]. In mouse models of AD, changes in insulin signaling and energy metabolism are seen in brain regions involved in learning and memory, and these changes overlap with patterns of Aβ deposition even in the early stages of the disease [41, 57].
Insulin resistance occurs in AD brain, and Igf1 activates similar pathways [46, 49, 60, 61]. Igf1 serves as a regulator of a variety of biological functions [60, 62] including playing essential roles in initial brain development and brain maintenance in adulthood, as well as cognition and neuroprotection [62, 63]. Pathways associated with Igf1 include roles in normal mammalian brain aging, neurogenesis and plasticity, and Aβ processing [64–66]. Disruption in insulin/Igf1 signaling could potentially lead to many of the detrimental consequences seen in AD suggesting a relationship between diabetes and AD pathogenesis [45, 61]. Markedly reduced gene expression and protein levels of key Igf1 signaling molecules, Igf1, Irs1, and Glut4, found here in ApoE3 and ApoE4 brain compared to the more metabolically robust ApoE2 brain illustrate a potential mechanism for impaired glucose metabolism lending to increased risk for developing late-onset AD. Additionally, ApoE4 brain exhibits lower levels of Pparg, a nuclear receptor involved in regulation of mitochondrial biogenesis and neuronal survival [67, 68], and Ide, a major enzyme that degrades Aβ peptide monomers in the brain [66], suggesting decreased mitochondrial efficiency and decreased ability to sustain Aβ homeostasis which could serve as additional mechanisms underlying the high risk of ApoE4 for AD. Together, these data provide the first documented evidence that human ApoE isoforms differentially affect the Igf1/Irs1/Glut4/Pparg/Ide system in the brain, which may explain, at least in part, why ApoE2 decreases whereas ApoE4 increases the risk for developing AD.
Igf1 signaling regulates multiple targets in glucose metabolism. Igf1 binds mainly to the Igf1-R, a receptor tyrosine kinase, activating multiple intracellular signaling cascades [69, 70]. Compared to healthy controls, AD brain expresses lower levels of both Igf1 and Igf1-R in brain regions responsible for learning and memory [45]. Although there are conflicting reports on changes in circulating levels of Igf1 in AD, a regression analysis shows a higher ratio of unbound to bound Igf1 in AD compared to non-AD controls, suggesting a type of progressive resistance to Igf1 in AD similar to that seen in insulin resistance [46, 71, 72]. In Igf1 deficient neurons, activation of Akt, Glut4 expression, and, consequently, glucose uptake are reduced [70]. Deficient Igf1 gene expression, reduced Igf1 protein levels, and the other concomitant changes to components of the Igf1 signaling cascade found here in ApoE3 and ApoE4 compared to ApoE2 brain indicate a potential mechanism by which glucose utilization is altered thereby conferring differential AD risk among the different ApoE genotypes.
Glut4, the primary insulin/Igf1-responsive glucose transporter, is stored in vesicles in the cytoplasm of cells [73]. These vesicles are translocated to the plasma membrane primarily in response to insulin/Igf1 binding to its receptor [73]. The insulin/Igf1 signaling pathway involves insulin/Igf1 binding to its receptor resulting in Irs phosphorylation and subsequent interaction of Irs with PI3K, leading to downstream activation of Akt. Once activated, Akt leads to the translocation of Glut4 vesicles to the plasma membrane for glucose entry into the cell as well as leading to activation of glycogen synthesis [74]. Under normal conditions, these processes respond quickly to changes in blood glucose levels and are readily reversible when insulin/Igf1 levels drop [73]. Significant downregulation of Glut4 and Irs seen here in ApoE3 and ApoE4 brain would result in lower glucose uptake into neuronal cells and less energy produced versus the more energetically robust ApoE2 brain. Upregulation of Akt1 (involved in cell survival pathways) and Akt2 (required to induce glucose transport) may be an attempt to increase glucose transport that was lost with downregulation of Glut4 and may be a compensatory response in order to rescue cells in the face of lower glucose utilization. Grb2, involved in signal transduction and cell communication, can activate the RAS/MAPK pathway in an Irs1-independent manner [74]. Grb2 upregulation may also be compensatory due to decreases in Irs1 in ApoE3 and ApoE4 brain and downregulation of Mapk1 in ApoE3 brain.
In addition to Igf1/Irs/Glut4 signaling deficiencies, downregulation of Pparg presents further challenge to the energy production problem in ApoE4 brain. Pparg is expressed in key areas of the brain that control energy balance and plays a central role in this regulation [75–78]. Pparg is a modulator of insulin sensitivity, and one consequence of ablation of Pparg is insulin resistance [79]. Synthetic Pparg agonists known to induce insulin sensitization are currently being used to treat type 2 diabetes mellitus [76, 78]. Therefore, decreased Pparg in ApoE4 brain could render neuronal cells less able to respond to changes in glucose circulating in brain capillaries [77, 78]. Pparg is also involved in the regulation of lipid uptake and storage and fatty acid oxidation [76, 80]. Under conditions of glucose deprivation or poor glucose utilization, the brain uses ketone bodies as an alternate, though less efficient, fuel source. These ketone bodies are primarily synthesized by β-oxidation of fatty acids in the liver, carried in the bloodstream, and then up-taken by the brain [81]. Limited studies have shown that astrocytes can metabolize fatty acids under starvation conditions; however, neurons do not undergo the highly oxidative process of β-oxidation [82, 83]. ApoE4 brain has been shown to be less responsive than ApoE2 or ApoE3 brain in attempts to utilize ketone bodies as alternative fuel for energy production [84]. Moreover, Pparg stimulation has been shown to promote mitochondrial biogenesis, stabilize mitochondria, reduce oxidative stress, and protect against apoptosis [67, 68]. Therefore, decreased Pparg in ApoE4 brain could be indicative of less efficient mitochondrial metabolism when compared to ApoE2 brain. Our data further show a significant increase in Npy in ApoE4 brain that favors energy storage over energy utilization [85].
Together with changes in glucose utilization, amyloid deposition in brain is thought to be among the earliest changes on the road toward AD [7, 8, 86, 87]. Mounting evidence exists that elevation of Aβ levels and subsequent Aβ-induced damage leads to neurodegeneration in AD brain [30, 88, 89]. In addition to its roles in energy regulation, Igf1 is involved in regulating Aβ levels in brain by modulating pathways involved in its degradation [48, 90, 91]. Treatment with Igf1 promotes clearance of Aβ and reverses cognitive shortcomings associated with Aβ accumulation [90, 92]. However, the potential role of Igf1 signaling in the regulation of Aβ metabolism has remained unclear and controversial.
The many functions of ApoE include inhibition of Aβ transport across the blood-brain barrier and facilitation of proteolytic degradation of Aβ by Ide [33, 35, 93]. Ide, as the name implies, functions to degrade insulin. Impairment of this ability through insufficient levels or activity results in hyperinsulinemia and glucose intolerance [94]. Ide is also known to degrade Aβ monomers, thereby potentially playing a role in preclinical development of AD [66, 95, 96]. Ide functions as a protease helping to regulate Aβ levels in the brain by degrading this neurotoxic peptide [89, 97, 98] and has been proposed as a potential junction between AD and type 2 diabetes [97, 99]. In addition to the deleterious effects on glucose utilization, downregulation of Ide found in our study in the ApoE4 group may be responsible, at least in part, for the mechanism by which ApoE status and the presence of the ApoE4 allele contributes to changes in amyloid homeostasis seen with ApoE4 in AD. Conversely, higher levels of Ide found in this study in ApoE2 brain demonstrate another potential mechanism of neuroprotection afforded by ApoE2. Aβ accumulation is a function of both increased production and decreased degradation and clearance [89, 97]. Ide levels decrease in mouse and human hippocampus as a function of age [100]. Further and earlier deficiencies in Ide levels in ApoE4 brain identified in this study would then amplify this problem leading to increased risk for AD.
Conclusions
The purpose of this study was to explore the potential differences in brain metabolic pathways, specifically insulin/Igf signaling, among the three major human ApoE genotypes using mouse models. In doing so, we were able to elucidate differences in key mechanisms involved in glucose utilization and amyloid degradation that arise from genetic differences in ApoE isoforms, which closely mimic those seen in PCAD and thus serve as potential targets for AD prevention and early intervention. Our data demonstrate that ApoE2 brain is associated with a more metabolically robust profile than both ApoE3 and ApoE4 brain, which provides a mechanistic rationale for its protective role against AD (Fig. 5). ApoE4 status and the resulting glucose and amyloid metabolic deficiencies set the stage for accelerated neurodegeneration and decrease the brain’s defense ability to respond to other AD risk stressors.
Fig. 5.

Our hypotheses and conclusion: 1) When compared to the ApoE3 and ApoE4 brain, the anti-AD ApoE2 brain is associated with a more robust Igf1 signaling and mechanisms involved in glucose uptake, glucose metabolism, and Aβ degradation, which could serve as a molecular basis underlying its protective role against the development of AD. 2) When compared to the ApoE2 and ApoE3 brain, the pro-AD ApoE4 brain is associated with a weaker Igf1 signaling and mechanisms involved in glucose uptake, glucose metabolism, and Aβ degradation, which could mechanistically contribute to its detrimental role in the etiology of AD. Therefore, targeting Igf1/Irs/Glut4/Pparg-related energy metabolism and the downstream energy production as well as Ide and associated Aβ degradation could serve as a vital strategy in order to transform a pro-AD ApoE4 brain to the anti-AD ApoE2 phenotype, and, as a result, increase the defense ability of an ApoE4 brain against the development of AD.
A major novelty of our study reported here was the comparison of ApoE2 to ApoE3 and ApoE4 genotypes. Many studies have been done comparing ApoE3 to ApoE4 or ApoE4 carriers versus ApoE4 non-carriers, but few animal models and even fewer human studies have included the ApoE2 genotype. Recent studies that have included ApoE2 have shown reduced Aβ and phospho-tau pathology and decreased hippocampal atrophy in ApoE2 carriers [102]. Those studies highlight the tremendous need for further explorations like ours that elucidate the mechanism by which ApoE2 confers this neuroprotection. Recent failures of a series of high-profile human trials aimed at treatment of mid to late-stage AD highlight the significance of AD prevention and early intervention. To that end, our findings suggest that a therapeutic strategy that converts the energy and amyloid metabolic profile of the high-risk ApoE4 and intermediate-risk ApoE3 subjects to one that more closely resembles the neuroprotective ApoE2 metabolic phenotype could hold promise for preventing, reducing the risk, or delaying the onset of AD, in particular in the high-risk ApoE4 carriers when addressed in the preclinical stages. Ongoing studies in our laboratory are aimed to further explore how human ApoE isoforms may interact with sex and age to modulate these pathways and their involvement in AD pathogenesis.
Supplementary Material
Acknowledgments
This work was supported by grants from the Alzheimer’s Association (IIRG-10-172459), the NIH-funded KU Alzheimer’s Disease Center (P30AG035982), KU general research funds and new faculty start-up funds to LZ.
References
- 1.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thies W, Bleiler L. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 3.2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10:e47–92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer’s Disease Education & Referral Center (National Institute on Aging) Alzheimer’s disease fact sheet. Alzheimer’s Disease Education & Referral (ADEAR) Center; Silver Spring, MD: 2008. [Google Scholar]
- 5.Mullard A. Sting of Alzheimer’s failures offset by upcoming prevention trials. Nat Rev Drug Discov. 2012;11:657–660. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
- 6.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging. 2005;32:486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 7.Aluise CD, Robinson RA, Beckett TL, Murphy MP, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Preclinical Alzheimer disease: brain oxidative stress, Abeta peptide and proteomics. Neurobiol Dis. 2010;39:221–228. doi: 10.1016/j.nbd.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosconi L, Sorbi S, de Leon MJ, Li Y, Nacmias B, Myoung PS, Tsui W, Ginestroni A, Bessi V, Fayyazz M, Caffarra P, Pupi A. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J Nucl Med. 2006;47:1778–1786. [PubMed] [Google Scholar]
- 10.Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, Thies B, Phelps CH. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dorey E, Chang N, Liu QY, Yang Z, Zhang W. Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci Bull. 2014;30:317–330. doi: 10.1007/s12264-013-1422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 14.Seripa D, D’Onofrio G, Panza F, Cascavilla L, Masullo C, Pilotto A. The genetics of the human APOE polymorphism. Rejuvenation Res. 2011;14:491–500. doi: 10.1089/rej.2011.1169. [DOI] [PubMed] [Google Scholar]
- 15.Lambotte C. Biochemical polymorphism in man -- its relation to disease. Anim Blood Groups Biochem Genet. 1981;12:149–166. doi: 10.1111/j.1365-2052.1981.tb01545.x. [DOI] [PubMed] [Google Scholar]
- 16.Zannis VI, Breslow JL. Human very low density lipoprotein apolipoprotein E isoprotein polymorphism is explained by genetic variation and posttranslational modification. Biochemistry. 1981;20:1033–1041. doi: 10.1021/bi00507a059. [DOI] [PubMed] [Google Scholar]
- 17.Suarez BK, Schonfeld G. Characterization of apolipoprotein E (ApoE) apoprotein levels in the various ApoE phenotypes. J Clin Endocrinol Metab. 1981;53:435–438. doi: 10.1210/jcem-53-2-435. [DOI] [PubMed] [Google Scholar]
- 18.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Roses AD, Pericak-Vance MA, Small GW, Haines JL. The apolipoprotein E E4 allele and sex-specific risk of Alzheimer’s disease. JAMA. 1995;273:373–374. [PubMed] [Google Scholar]
- 19.Lambert JC, Perez-Tur J, Dupire MJ, Galasko D, Mann D, Amouyel P, Hardy J, Delacourte A, Chartier-Harlin MC. Distortion of allelic expression of apolipoprotein E in Alzheimer’s disease. Hum Mol Genet. 1997;6:2151–2154. doi: 10.1093/hmg/6.12.2151. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 22.Frieden C, Garai K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109:8913–8918. doi: 10.1073/pnas.1207022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apostolova LG, Hwang KS, Kohannim O, Avila D, Elashoff D, Jack CR, Jr, Shaw L, Trojanowski JQ, Weiner MW, Thompson PM. ApoE4 effects on automated diagnostic classifiers for mild cognitive impairment and Alzheimer’s disease. Neuroimage Clin. 2014;4:461–472. doi: 10.1016/j.nicl.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aleshkov S, Abraham CR, Zannis VI. Interaction of nascent ApoE2, ApoE3, and ApoE4 isoforms expressed in mammalian cells with amyloid peptide beta (1–40). Relevance to Alzheimer’s disease. Biochemistry. 1997;36:10571–10580. doi: 10.1021/bi9626362. [DOI] [PubMed] [Google Scholar]
- 25.Payami H, Zareparsi S, Montee KR, Sexton GJ, Kaye JA, Bird TD, Yu CE, Wijsman EM, Heston LL, Litt M, Schellenberg GD. Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: a possible clue to the higher incidence of Alzheimer disease in women. Am J Hum Genet. 1996;58:803–811. [PMC free article] [PubMed] [Google Scholar]
- 26.Mortensen EL, Hogh P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 2001;57:89–95. doi: 10.1212/wnl.57.1.89. [DOI] [PubMed] [Google Scholar]
- 27.Anttila T, Helkala EL, Kivipelto M, Hallikainen M, Alhainen K, Heinonen H, Mannermaa A, Tuomilehto J, Soininen H, Nissinen A. Midlife income, occupation, APOE status, and dementia: a population-based study. Neurology. 2002;59:887–893. doi: 10.1212/wnl.59.6.887. [DOI] [PubMed] [Google Scholar]
- 28.Qiu C, Kivipelto M, Aguero-Torres H, Winblad B, Fratiglioni L. Risk and protective effects of the APOE gene towards Alzheimer’s disease in the Kungsholmen project: variation by age and sex. J Neurol Neurosurg Psychiatry. 2004;75:828–833. doi: 10.1136/jnnp.2003.021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson JK, McCleary R, Oshita MH, Cotman CW. Initiation and propagation stages of beta-amyloid are associated with distinctive apolipoprotein E, age, and gender profiles. Brain Res. 1998;798:18–24. doi: 10.1016/s0006-8993(98)00363-1. [DOI] [PubMed] [Google Scholar]
- 30.Risacher SL, Kim S, Shen L, Nho K, Foroud T, Green RC, Petersen RC, Jack CR, Jr, Aisen PS, Koeppe RA, Jagust WJ, Shaw LM, Trojanowski JQ, Weiner MW, Saykin AJ. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI) Front Aging Neurosci. 2013;5:11. doi: 10.3389/fnagi.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- 34.Lauderback CM, Kanski J, Hackett JM, Maeda N, Kindy MS, Butterfield DA. Apolipoprotein E modulates Alzheimer’s Abeta(1-42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res. 2002;924:90–97. doi: 10.1016/s0006-8993(01)03228-0. [DOI] [PubMed] [Google Scholar]
- 35.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerf E, Gustot A, Goormaghtigh E, Ruysschaert JM, Raussens V. High ability of apolipoprotein E4 to stabilize amyloid-beta peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J. 2011;25:1585–1595. doi: 10.1096/fj.10-175976. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, Mitani A, Joyner D, Thyssen DH, Bacskai BJ, Frosch MP, Spires-Jones TL, Finn MB, Holtzman DM, Hyman BT. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. 2012;32:15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiang GC, Insel PS, Tosun D, Schuff N, Truran-Sacrey D, Raptentsetsang ST, Jack CR, Jr, Aisen PS, Petersen RC, Weiner MW. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 2010;75:1976–1981. doi: 10.1212/WNL.0b013e3181ffe4d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caselli RJ, Dueck AC. APOE varepsilon2 and presymptomatic stage Alzheimer disease: how much is not enough? Neurology. 2010;75:1952–1953. doi: 10.1212/WNL.0b013e3181ff94f7. [DOI] [PubMed] [Google Scholar]
- 40.Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, Kaplan A, La Rue A, Adamson CF, Chang L, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273:942–947. [PubMed] [Google Scholar]
- 41.Pedros I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, Auladell C, Pallas M, Vazquez-Carrera M, Casadesus G, Folch J, Camins A. Early alterations in energy metabolism in the hippocampus of APPSwe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbadis.2014.05.025. [DOI] [PubMed] [Google Scholar]
- 42.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Yu JT, Wang HF, Han PR, Tan CC, Wang C, Meng XF, Risacher SL, Saykin AJ, Tan L. APOE genotype and neuroimaging markers of Alzheimer’s disease: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2014-307719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 46.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang B, Tang XC, Zhang HY. Alternations of central insulin-like growth factor-1 sensitivity in APP/PS1 transgenic mice and neuronal models. J Neurosci Res. 2013;91:717–725. doi: 10.1002/jnr.23201. [DOI] [PubMed] [Google Scholar]
- 48.Zhao WQ, Lacor PN, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta} J Biol Chem. 2009;284:18742–18753. doi: 10.1074/jbc.M109.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bassil F, Fernagut PO, Bezard E, Meissner WG. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog Neurobiol. 2014;118:1–18. doi: 10.1016/j.pneurobio.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Wang W, Yu JT, Tan L, Liu QY, Wang HF, Ma XY. Insulin-like growth factor 1 (IGF1) polymorphism is associated with Alzheimer’s disease in Han Chinese. Neurosci Lett. 2012;531:20–23. doi: 10.1016/j.neulet.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 51.Vargas T, Martinez-Garcia A, Antequera D, Vilella E, Clarimon J, Mateo I, Sanchez-Juan P, Rodriguez-Rodriguez E, Frank A, Rosich-Estrago M, Lleo A, Molina-Porcel L, Blesa R, Gomez-Isla T, Combarros O, Bermejo-Pareja F, Valdivieso F, Bullido MJ, Carro E. IGF-I gene variability is associated with an increased risk for AD. Neurobiol Aging. 2011;32:556e553–511. doi: 10.1016/j.neurobiolaging.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 52.Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 53.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 54.Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, Schellenberg GD, Jin LW, Kovacina KS, Craft S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003;162:313–319. doi: 10.1016/s0002-9440(10)63822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ong QR, Chan ES, Lim ML, Wong BS. Expression of human apolipoprotein E4 reduces insulin-receptor substrate 1 expression and Akt phosphorylation in the ageing liver. FEBS Open Bio. 2014;4:260–265. doi: 10.1016/j.fob.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, de Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forster S, Grimmer T, Miederer I, Henriksen G, Yousefi BH, Graner P, Wester HJ, Forstl H, Kurz A, Dickerson BC, Bartenstein P, Drzezga A. Regional expansion of hypometabolism in Alzheimer’s disease follows amyloid deposition with temporal delay. Biol Psychiatry. 2012;71:792–797. doi: 10.1016/j.biopsych.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 58.Nordberg A, Rinne JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6:78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- 59.Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Langstrom B, Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 60.McRory JE, Sherwood NM. Ancient divergence of insulin and insulin-like growth factor. DNA Cell Biol. 1997;16:939–949. doi: 10.1089/dna.1997.16.939. [DOI] [PubMed] [Google Scholar]
- 61.Cholerton B, Baker LD, Craft S. Insulin, cognition, and dementia. Eur J Pharmacol. 2013;719:170–179. doi: 10.1016/j.ejphar.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Torres-Aleman I. Toward a comprehensive neurobiology of IGF-I. Dev Neurobiol. 2010;70:384–396. doi: 10.1002/dneu.20778. [DOI] [PubMed] [Google Scholar]
- 63.Brooker GJ, Kalloniatis M, Russo VC, Murphy M, Werther GA, Bartlett PF. Endogenous IGF-1 regulates the neuronal differentiation of adult stem cells. J Neurosci Res. 2000;59:332–341. doi: 10.1002/(sici)1097-4547(20000201)59:3<332::aid-jnr6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 64.Liu W, Ye P, O’Kusky JR, D’Ercole AJ. Type 1 insulin-like growth factor receptor signaling is essential for the development of the hippocampal formation and dentate gyrus. J Neurosci Res. 2009;87:2821–2832. doi: 10.1002/jnr.22129. [DOI] [PubMed] [Google Scholar]
- 65.Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurochkin IV. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci. 2001;26:421–425. doi: 10.1016/s0968-0004(01)01876-x. [DOI] [PubMed] [Google Scholar]
- 67.Miglio G, Rosa AC, Rattazzi L, Collino M, Lombardi G, Fantozzi R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem Int. 2009;55:496–504. doi: 10.1016/j.neuint.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 68.Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, Inestrosa NC, Bronfman M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- 69.Summers SA, Birnbaum MJ. A role for the serine/threonine kinase, Akt, in insulin-stimulated glucose uptake. Biochem Soc Trans. 1997;25:981–988. doi: 10.1042/bst0250981. [DOI] [PubMed] [Google Scholar]
- 70.Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA. Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci U S A. 2000;97:10236–10241. doi: 10.1073/pnas.170008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vardy ER, Rice PJ, Bowie PC, Holmes JD, Grant PJ, Hooper NM. Increased circulating insulin-like growth factor-1 in late-onset Alzheimer’s disease. J Alzheimers Dis. 2007;12:285–290. doi: 10.3233/jad-2007-12401. [DOI] [PubMed] [Google Scholar]
- 72.Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 73.Watson RT, Pessin JE. Intracellular organization of insulin signaling and GLUT4 translocation. Recent Prog Horm Res. 2001;56:175–193. doi: 10.1210/rp.56.1.175. [DOI] [PubMed] [Google Scholar]
- 74.Bevan P. Insulin signalling. J Cell Sci. 2001;114:1429–1430. doi: 10.1242/jcs.114.8.1429. [DOI] [PubMed] [Google Scholar]
- 75.Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, Schwartz MW. Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology. 2009;150:707–712. doi: 10.1210/en.2008-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-gamma in the regulation of energy balance. Nat Med. 2011;17:623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 80.Chen YC, Wu JS, Tsai HD, Huang CY, Chen JJ, Sun GY, Lin TN. Peroxisome proliferator-activated receptor gamma (PPAR-gamma) and neurodegenerative disorders. Mol Neurobiol. 2012;46:114–124. doi: 10.1007/s12035-012-8259-8. [DOI] [PubMed] [Google Scholar]
- 81.Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005;28:109–121. doi: 10.1007/s10545-005-5518-0. [DOI] [PubMed] [Google Scholar]
- 82.Chikahisa S, Shimizu N, Shiuchi T, Sei H. Ketone body metabolism and sleep homeostasis in mice. Neuropharmacology. 2014;79:399–404. doi: 10.1016/j.neuropharm.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 83.Schonfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013;33:1493–1499. doi: 10.1038/jcbfm.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 2009;6:31. doi: 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuo LE, Kitlinska JB, Tilan JU, Li L, Baker SB, Johnson MD, Lee EW, Burnett MS, Fricke ST, Kvetnansky R, Herzog H, Zukowska Z. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med. 2007;13:803–811. doi: 10.1038/nm1611. [DOI] [PubMed] [Google Scholar]
- 86.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 87.Swomley AM, Forster S, Keeney JT, Triplett J, Zhang Z, Sultana R, Butterfield DA. Abeta, oxidative stress in Alzheimer disease: Evidence based on proteomics studies. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbadis.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Findeis MA. The role of amyloid beta peptide 42 in Alzheimer’s disease. Pharmacol Ther. 2007;116:266–286. doi: 10.1016/j.pharmthera.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 90.Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002;8:1390–1397. doi: 10.1038/nm1202-793. [DOI] [PubMed] [Google Scholar]
- 91.Sharma S, Prasanthi RPJ, Schommer E, Feist G, Ghribi O. Hypercholesterolemia-induced Abeta accumulation in rabbit brain is associated with alteration in IGF-1 signaling. Neurobiol Dis. 2008;32:426–432. doi: 10.1016/j.nbd.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 92.Carro E, Trejo JL, Gerber A, Loetscher H, Torrado J, Metzger F, Torres-Aleman I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol Aging. 2006;27:1250–1257. doi: 10.1016/j.neurobiolaging.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 93.Fan J, Donkin J, Wellington C. Greasing the wheels of Abeta clearance in Alzheimer’s disease: the role of lipids and apolipoprotein E. Biofactors. 2009;35:239–248. doi: 10.1002/biof.37. [DOI] [PubMed] [Google Scholar]
- 94.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morelli L, Llovera R, Gonzalez SA, Affranchino JL, Prelli F, Frangione B, Ghiso J, Castano EM. Differential degradation of amyloid beta genetic variants associated with hereditary dementia or stroke by insulin-degrading enzyme. J Biol Chem. 2003;278:23221–23226. doi: 10.1074/jbc.M300276200. [DOI] [PubMed] [Google Scholar]
- 96.Zhao L, Yao J, Mao Z, Chen S, Wang Y, Brinton RD. 17beta-Estradiol regulates insulin-degrading enzyme expression via an ERbeta/PI3-K pathway in hippocampus: relevance to Alzheimer’s prevention. Neurobiol Aging. 2011;32:1949–1963. doi: 10.1016/j.neurobiolaging.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eckman EA, Eckman CB. Abeta-degrading enzymes: modulators of Alzheimer’s disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans. 2005;33:1101–1105. doi: 10.1042/BST20051101. [DOI] [PubMed] [Google Scholar]
- 98.Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haque R, Nazir A. Insulin-degrading enzyme: a link between Alzheimer’s and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets. 2014;13:259–264. doi: 10.2174/18715273113126660139. [DOI] [PubMed] [Google Scholar]
- 100.Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.