

ABSTRACT
Tumor angiogenesis promotes tumor growth and metastasis. Anti-angiogenic therapy in combination with chemotherapy is used for the treatment of metastatic cancers, including breast cancer but therapeutic benefits are limited. Mobilization and accumulation of myeloid-derived suppressor cells (MDSC) during tumor progression and therapy have been implicated in metastasis formation and resistance to anti-angiogenic treatments. Here, we used the 4T1 orthotopic syngenic mouse model of mammary adenocarcinoma to investigate the effect of VEGF/VEGFR-2 axis inhibition on lung metastasis, MDSC and regulatory T cells (Tregs). We show that treatment with the anti-VEGFR-2 blocking antibody DC101 inhibits primary tumor growth, angiogenesis and lung metastasis. DC101 treatment had no effect on MDSC mobilization, but partially attenuated the inhibitory effect of mMDSC on T cell proliferation and decreased the frequency of Tregs in primary tumors and lung metastases. Strikingly, DC101 treatment induced the expression of the immune-suppressive molecule arginase I in mMDSC. Treatment with the arginase inhibitor Nω-hydroxy-nor-Arginine (Nor-NOHA) reduced the inhibitory effect of MDSC on T cell proliferation and inhibited number and size of lung metastasis but had little or no additional effects in combination with DC101.
In conclusion, DC101 treatment suppresses 4T1 tumor growth and metastasis, partially reverses the inhibitory effect of mMDSC on T cell proliferation, decreases Tregs in tumors and increases arginase I expression in mMDSC. Arginase inhibition suppresses lung metastasis independently of DC101 effects. These observations contribute to the further characterization of the immunomodulatory effect of anti-VEGF/VEGFR2 therapy and provide a rationale to pursue arginase inhibition as potential anti-metastatic therapy.
KEYWORDS: Breast cancer, 4T1, metastasis, tumor angiogenesis, MDSC, immunosuppression, Arg I, DC101, Nor-NOHA
Abbreviations
- Arg I
Arginase I
- bFGF
basic fibroblast growth factor
- COX-2
cyclooxygenase-2
- GM-CSF
granulocyte-monocyte colony stimulating factor
- IDO1–2
Indoleamine 2,3-dioxygenase
- IL
interleukin; iNOS, inducible nitric oxide synthase
- MDSC
myeloid-derived suppressor cells
- Nor-NOHA
Nω-hydroxy-nor-Arginine
- PDGF
platelet-derived growth factor
- PGE2
Prostaglandin E2
- TGFβ
transforming growth factor β
- VEGF
vascular endothelial growth factor
- VEGFR-2
VEGF receptor-2.
Introduction
Tumor progression involves reciprocal and dynamic heterotypic interactions between cancer cells and non-malignant host cells. These host cells can be either originally present in the tumor microenvironment or recruited from the surrounding environment or from distant sites.1-3 Tumor angiogenesis is a host event key to primary tumor growth and metastasis formation.4,5 Anti-angiogenic therapies, mostly targeting the VEGF/VEGFR signaling axis, are currently routinely used in the clinic to treat several advanced or metastatic cancers, including colon, kidney, liver and breast cancers.6-8 However, survival benefits are limited and experimental evidence indicates that resistance mechanisms to anti-angiogenic therapy are responsible for its weak therapeutic efficacy.9-11
The tumor stroma itself can confer resistance to anti-angiogenic therapy.9 In many human tumors and in murine models, including breast cancer, bone-marrow derived myeloid and inflammatory cells, mostly CD11b+, are mobilized and recruited at the primary and metastatic tumor sites. These cells can contribute to local tumor growth, angiogenesis and metastatic spreading.1,12-14 Correlations between inflammatory and myeloid cell infiltration in the primary tumor and negative clinical outcome have been reported in multiple preclinical models and clinical studies, including in lung, head and neck and breast cancers.1,15-18 Conversely, tumors can also recruit immune cells with potential antitumor activities, such as NK cells or T cells,17 which have been shown to suppress cancer progression and improve outcome in melanoma,19,20 colorectal21 and breast cancers.22,23
Mobilized CD11b+ cells may be arrested in their differentiation by tumor-derived factors and accumulate in blood, spleen and in the tumor microenvironment as myeloid-derived cells with immunosuppressive features (myeloid-derived suppressor cells (MDSC): CD11b+Gr1+ cells with the ability to inhibit T cell function), including in breast cancer.24,25 Mouse MDSC consist of a heterogeneous population distinct from immature myeloid cells present in tumor-free mice. Two main MDSC subsets have been described: the monocytic subset (mMDSC, CD11b+Ly6G−Ly6Chigh or CD11b+Gr1lo/intLy6ChiLy6G−) and the granulocytic subset (gMDSC, CD11b+Ly6G+Ly6Clow or CD11b+Gr1hiLy6ClowLy6G+).26 Mobilized MDSC can promote angiogenesis during natural tumor progression, in part through the release of VEGF and MMP-9.14 On the other side, MDSC can confer resistance to anti-angiogenic therapy.27,28 For instance anti-VEGF treatment promotes the recruitment of CD11b+Gr1+ MDSC at the tumor site, which mediates resistance via the IL17/GCSF/Bv8 axis.29 Elimination of MDSC with anti-Gr1 mAb treatment in combination with anti-VEGF therapy, results in a more effective response compared with anti-VEGF treatment alone.29 MDSC are now considered as candidate therapeutic targets to improve activity of anti-angiogenic therapies and boost the anti-tumoral immune response.30,31
Paradoxically to these observations, VEGF itself can induce a state of immunosuppression in the tumor microenvironment, and neutralization of VEGF abrogates it.32-35 These observations demonstrate the complexity of the interplay between tumor cells, tumor angiogenesis, the immune-inflammatory response and tumor progression and the multiple, often contrasting, effects of VEGF and anti-angiogenic treatments.17,31 It has also been speculated that combing anti-angiogenic and immunotherapies may help to improve the therapeutic efficacy of either therapy used alone.31,36,37
Here, we used the orthotopic and syngenic, highly angiogenic and metastatic 4T1 murine breast adenocarcinoma model,38 well known to effectively mobilize CD11b+ bone marrow-derived cells (BMDC),38,39 including MDSC,40,41 and to be immunogenic,42-44 to monitor the effect of anti-angiogenic therapy on MDSC mobilization and activity in relationship to tumor growth and metastasis. We show that the anti-VEGFR-2 antibody DC101,45 inhibits tumor angiogenesis, tumor growth, metastasis and partially reversed MDSC inhibitory activity on T cells, decreased the recruitment of Tregs at tumor and metastatic sites and enhanced Arg I expression. Arginase inhibition reduced lung metastasis formation and partially reversed MDSC inhibitory activity but did not enhance the anti-metastatic activity of DC101.
Results
VEGFR-2 inhibition reduces 4T1 orthotopic tumor growth and angiogenesis
Treatment with the anti-VEGFR-2 mAb DC101 was shown to inhibit angiogenesis and growth of subcutaneously implanted 4T1 tumors.45,46 We set up to test whether DC101 treatment inhibited angiogenesis, growth and metastasis of orthotopically growing 4T1 tumors. 4T1 cells were implanted in the 4th mammary gland and treatment with DC101 and IgG isotype control antibodies was started 10 d later, when tumors became palpable. The inhibitory effect of DC101 on tumor growth was evident as early as day 13 and consistently resulted in a 40–50% reduction in tumor volume at the end of the experiment (day 28) (Fig. 1A). Immunohistological analysis and microvascular density (MVD) count confirmed that DC101 treatment significantly reduced tumor vascularization (Fig. 1B and C), without causing intra-tumoral necrosis (Fig. 1D).
Figure 1.
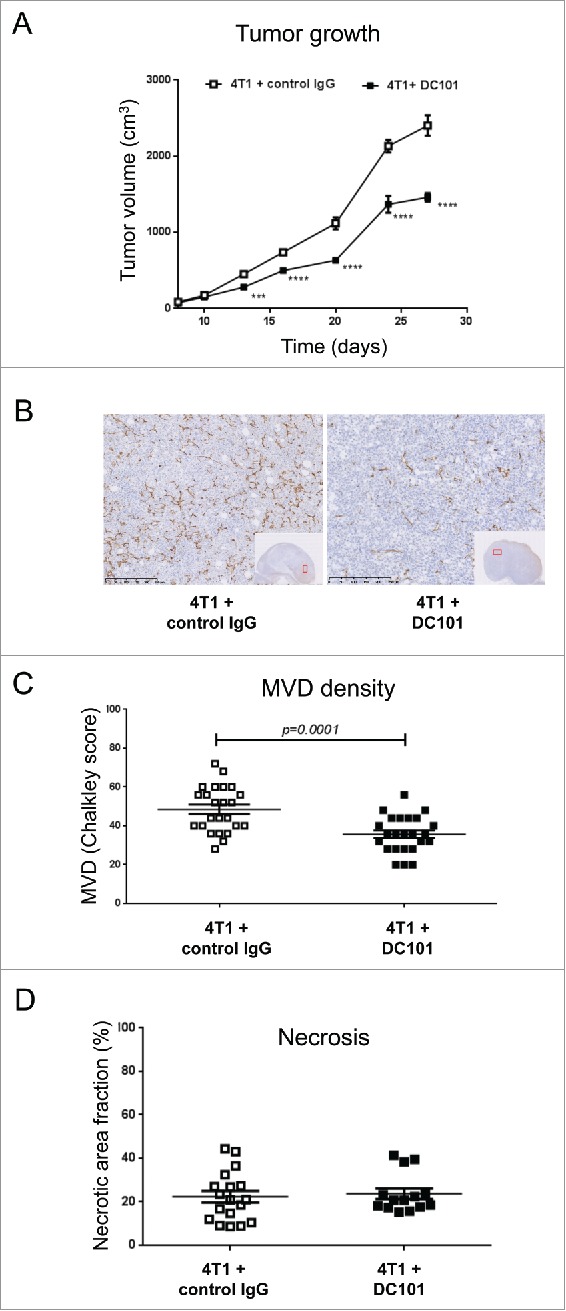
DC101 treatment delays 4T1 primary tumor growth and inhibits tumor angiogenesis. (A) Growth curves of 4T1 tumors in mice treated with the anti-VEGFR-2 antibody DC101 or a control IgG antibody. ***p < 0.001; ****p < 0.0001. (B) Representative images of CD31 staining of tumor tissues from both experimental groups. Scale bar = 250 μm. (C) Quantification of microvascular density (MVD) on multiple regions of six tumors per group. N = 3; mice per group = 7 (D) Quantification of tumor necrosis based on H&E staining and morphometric analysis with ImageJ. N = 3; mice per group = 7. Treatment conditions are indicated.
VEGFR-2 inhibition suppresses 4T1 lung metastasis from the primary tumor but not upon tail vein injection
DC101 treatment was reported to promote progression to metastasis in other tumor models in spite of primary tumor growth inhibition.47,48 We therefore, investigated its effect on 4T1 lung metastasis formation. DC101 treatment resulted in a significant reduction in the number and size of lung metastatic nodules (Fig. 2A–C). The metastatic index, which takes into account the effect on the primary tumor, was still reduced in DC101-treated mice, thereby demonstrating that VEFGR-2 inhibition has anti-metastatic effects beyond inhibition of primary tumor growth (Fig. 2D). We also observed a small reduction in MVD in the metastatic nodules of DC101-treated mice (Fig. 2E and 2F). To test whether anti-VEGFR-2 treatment affected lung metastasis formation through direct effects in the lung, we injected 4T1 cells directly in the tail vein and treated mice with DC101 and IgG isotype control. No DC101 effect on lung metastatic formation was observed (Fig. S1).
Figure 2.
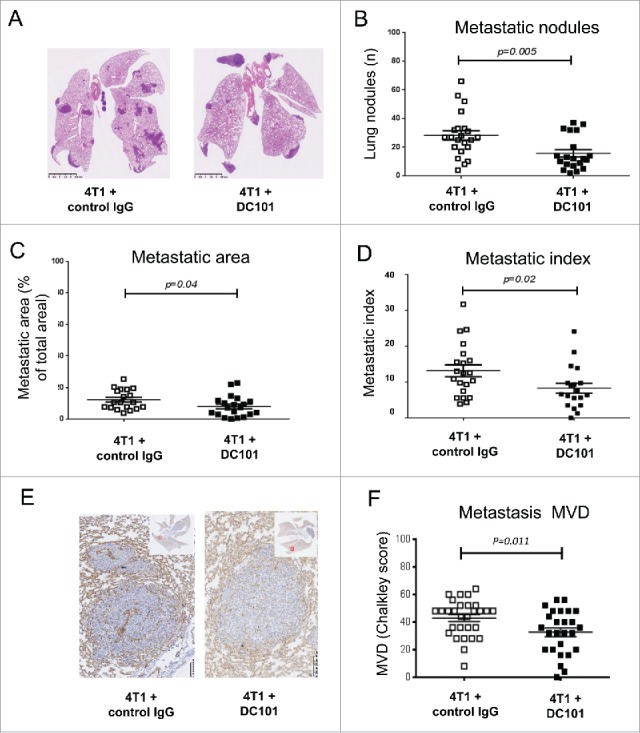
DC101 treatment inhibits 4T1 lung metastasis and associated angiogenesis. (A) Representative images of histological H&E staining of lungs from mice treated with DC101 or IgG control antibody. (B) Quantification of lung metastasis number in the two groups. (C) Quantification of lung metastasis area in the two groups. (D) Calculated metastatic index. (E) Representative images of CD31 staining of lung metastases. Scale bars = 100 μm. (F) Quantification of microvascular density (MVD) in the metastases of six representative lung images per group. N = 3; mice per group = 7. Treatment conditions are indicated.
From these experiments, we concluded that DC101 treatment reduces primary tumor growth and angiogenesis and suppresses lung metastasis formation from the primary tumor implanted orthotopically but not when tumor cells are directly injected in the venous circulation.
VEGFR-2 inhibition has no effect on the frequency of MDSC in blood, tumor and lung but decreases the frequency of regulatory T cells
To search for a cellular mechanism for the effect of DC101 treatment on tumor growth, we investigated MDSC and regulatory T cells (Tregs), two immunosuppressive cell populations present in the tumor microenvironment. MDSCs are immature myeloid cells characterized by the expression of CD11b and Gr-1 cell surface markers. MDSCs are divided into monocytic (m-MDSC, Gr-1lowLy6C+) and granulocytic (g-MDSC, Gr-1highLy6C+) subsets based on the differential expression of Ly6C and Ly6G antigens, respectively, along with the brightness of Gr-1 expression. The g-MDSC subset is the major population of MDSCs present in different tumor models.49 4T1 tumors cause expansion and accumulation of MDSCs in the blood, lungs, spleen and primary tumors (Ref. 50 and own unpublished observations). We did not observe any effect of DC101 treatment on the frequency of total CD11b+ cells or on CD11b+Gr1+ subsets in blood, tumor, spleen or lung at day 28 post injection (Fig. 3A, 3B, S2, S3). MDSC are well known to promote the conversion of T cells into Tregs.51 We therefore checked the presence of Tregs in the primary tumor and in the lung. Interestingly, DC101 treatment caused a significant decrease in the fraction of CD25+FoxP3+ T cells within CD4+ cells in tumors and lungs (Fig. 4A). Concomitantly, we observed a decrease of the fraction of CD4+ cells in the primary tumor, but not in the lung (Fig. 4B).
Figure 3.
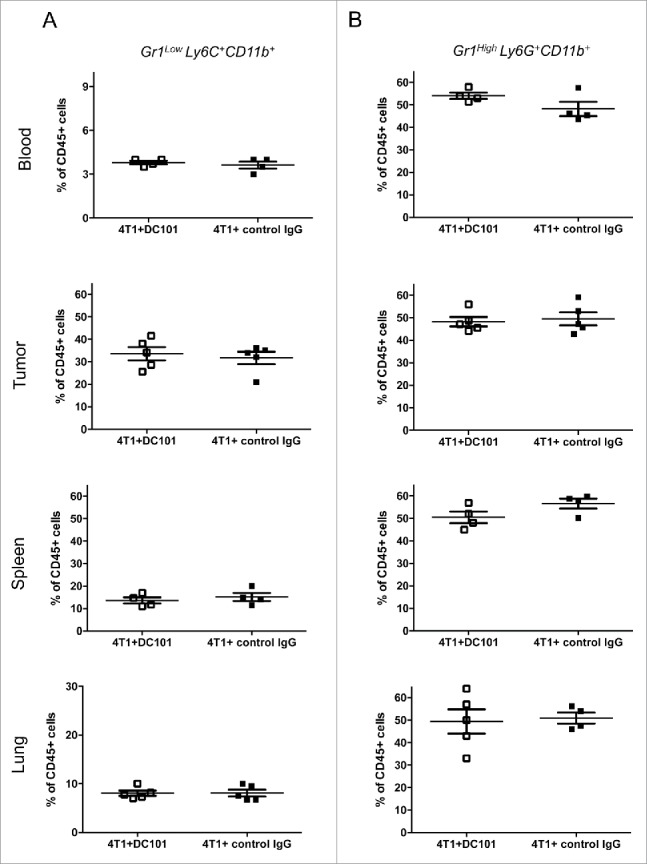
DC101 treatment has no effects on MDSC frequency. (A) Fraction of CD11b+Gr1LowLy6C+ cells (mMDSC) in the blood, primary tumors, spleens and lungs of 4T1-bearing mice treated with DC101 or IgG control antibody. (B) Frequency of CD11b+Gr1HighLy6G+ cells (gMDSC) in the same conditions as in A. Cells were isolated from DC101-treated (4T1+DC101) or control treated (4T1+control IgG) mice at the end of the experiment (d = 28). N = 3. Results from one representative experiment is shown here; mice per group = 4–5.
Figure 4.
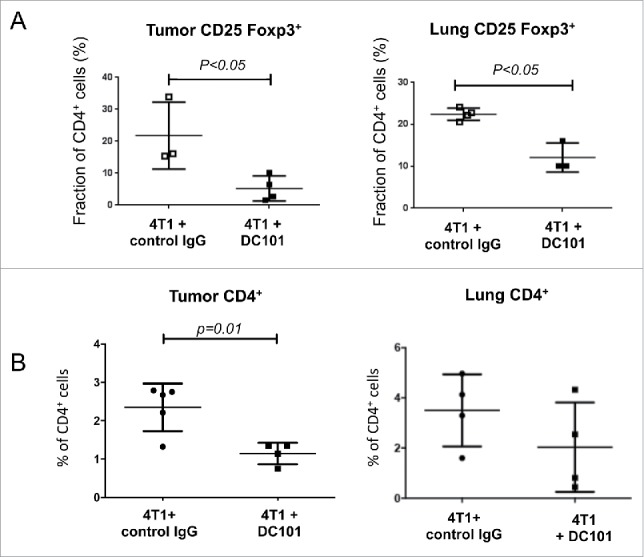
DC101 treatment reduces the frequency of tumor and lung infiltrating regulatory T cells. (A) Flow cytometry analysis of CD25+Foxp3+ cells among total CD4+ cells in tumors and lungs collected from mice treated with IgG control or DC101 antibodies. Mice analyzed per condition = 3–4. (B) Flow cytometry analysis of CD4+ cell frequency in tumor-derived from IgG control and DC101 antibodies-treated mice. DC101 treatment reduces the frequency of CD4+ cells in tumors but not lungs. Mice analyzed per group = 4–5.
From these results, we concluded that DC101 treatment has no effect on the frequency of MDSC in blood, tumor and lung, but it decreases the presence of CD25+FoxP3+ Tregs in primary tumors and metastatic lungs.
VEGFR-2 inhibition partially reverses the inhibitory effect of CD11b+Gr1lowLy6C+ cells (mMDSC) on T cell proliferation
Next, we analyzed whether DC101 treatment affected the suppressive activity of MDSC on T cell proliferation. Spleen-derived mMDSC and gMDSC were enriched by MACS using a two-step magnetic separation strategy and tested in a T cell activation assay. Interestingly, we observed that mMDSC derived from DC101-treated mice had a reduced T cell proliferation suppressive activity at all tested MDSC:T cell ratios, compared with mMDSC from the control IgG-treated mice (Fig. 5A). In contrast, DC101 treatment did not affect the immunosuppressive activity of gMDSC at equivalent MDSC:T cell ratios (Fig. 5B).
Figure 5.
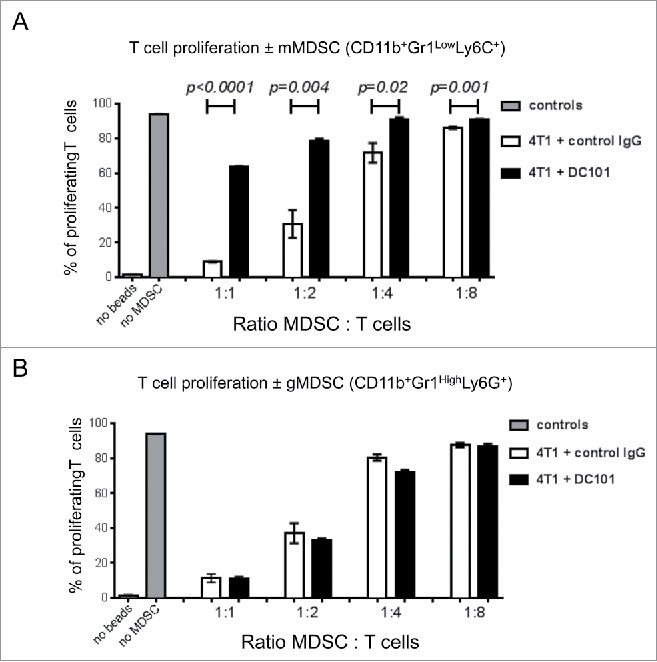
DC101 treatment reduces the inhibitory effect of mMDSC on T cell proliferation. (A) Quantification of the anti-proliferatory effect of CD11b+Gr1lowLy6G+ cells on T cells. (B) Quantification of the anti-proliferatory effect of CD11b+Gr1highLy6G+ cells on T cells. MDSC were isolated by MACS and mixed with stimulated T cells at the indicated ratios. Cells were isolated from the spleens of DC101-treated (4T1+ DC101) or control treated (4T1+control IgG) mice. Mice per group = 4–5.
From these experiments, we concluded that VEGFR-2 inhibition partially reverses the immunosuppressive activity of mMDSC (CD11b+Gr1LowLy6C+) but not of gMDSC (CD11b+Gr1HighLy6G+).
DC101 treatment induces arginase I (Arg I) expression in CD11b+ cells and MDSC
MDSC inhibit T cell function by multiple mechanisms, including depletion of L-arginine by arginase I (Arg I) and by the production of nitric oxide, reactive oxygen and nitrogen species by inducible nitric oxide synthase (iNOS).52 We therefore, monitored the expression of Arg I, iNOS and other relevant molecules (i.e., MRC-1, VEGFR-2, IL10, IL12b and TNFα) by real-time RT-PCR in blood circulating and tumor infiltrating CD11b+ cells (Fig. S4A–S4C). Strikingly, DC101 treatment induced Arg I expression in blood circulating and tumor infiltrating total CD11b+ cells (Fig. 6A, 6B, S4A, S4B) and in tumor-infiltrating CD11b+Gr1+ (total MDSC) cells (Fig. 6C). Expression of iNOS mRNA was not affected or rather downregulated by DC101 treatment (Fig. S4A–S4C). We confirmed increased Arg I protein expression by IHC (Fig. 6D and E). The total fraction of CD11b+ cells did not change as already shown by flow cytometry (Fig. S3A–D). Arg I mRNA, measured by real-time RT-PCR was detected in CD11b+ cells but not in CD11b− cells isolated from tumor tissue (Fig. S5A). Intracellular staining and flow cytometry analysis for Arg I protein in blood, tumor and metastatic lung reveled expression in approx. 20% of CD11b+ cells in metastatic nodules (Fig. S5B) and in 100% of CD11b+Gr1lowLy6C+ and CD11b+Gr1highLy6G+ cells in the blood, tumor and lungs, albeit at different levels (Fig. S5C). We also demonstrated increased Arg I enzymatic activity in CD11b+Gr1+ derived from tumors of DC101-treated mice, compared with control IgG-treated ones (Fig. 6F).
Figure 6.
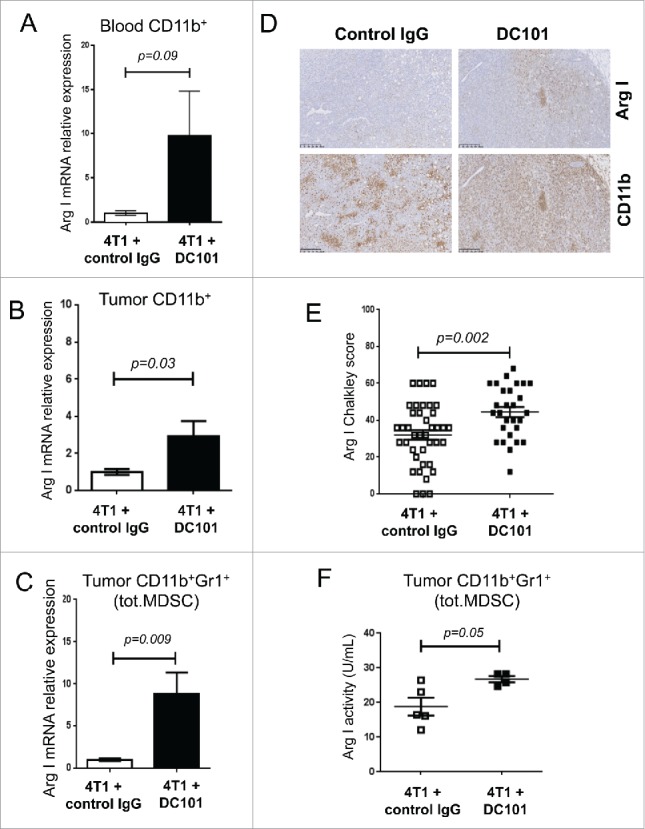
DC101 treatment induces Arg I in CD11b+ and CD11b+ Gr1+ cells. (A) Arg I mRNA expression levels in blood circulating CD11b+ cells. Mice per group = 4 and 5. (B) Arg I mRNA expression levels in tumor-derived CD11b+ cells, N = 2, mice per group = 9–10. (C) Arg I mRNA expression levels in tumor-derived CD11b+Gr1+ cells (total MDSC) mice per group = 4 and 5. (D) Representative IHC images showing expression of Arg I and CD11b in the tumor tissue in mice treated as indicated. Scale bar = 250 μm. (E) Quantification of intratumoral Arg I expression (Chalkley score). Mice per group = 7 and 10. (F) Measurement of Arg I enzymatic activity in tumor-derived CD11b+Gr1+ cells (total MDSC). Cells were isolated by MACS were taken from DC101-treated (4T1+DC101) or control treated (4T1+control IgG) mice. Mice per group = 4–5.
Further, we stained tumors and lungs from control IgG- and DC101-treated mice for Arg I, CD31 and CD11b to gain information on their localization. In tumors, Arg I was mainly localized in the periphery, in the stroma, and in CD11b+ cells rich regions surrounding necrotic areas. DC101 treatment decreased CD31+ vessel density and increased Arg I+ cell density, especially inside the tumor (Fig. S6A). In lungs, Arg I+ cells were exclusively localized in the metastatic nodules and absent in the lung parenchyma. DC101 decreased CD31+ vessel density and increased Arg I+ cell density in a non-overlapping pattern (Fig. S6B).
Collectively these data indicated that MDSC are positive for Arg I expression and that DC101 treatment further increases expression.
Arg I inhibition suppresses metastasis but not primary tumor growth and alleviates MDSC-mediated inhibition of T cell proliferation
Arg I acts as immunosuppressive molecule and could contribute to promote tumor growth and metastasis. In our model, tumor MDSC express active Arg I (Fig. 6C–F, S6). To test whether Arg I activity contributed to 4T1 tumor progression, we treated 4T1 tumor-bearing mice with the arginase inhibitor Nω-hydroxy-nor-Arginine (Nor-NOHA) starting from day 7 after tumor cell implantation till the end of the experiment. Nor-NOHA treatment did not impinge on primary tumor growth, tumor angiogenesis or necrosis (Fig. S7A–S7C). However, Nor-NOHA treatment significantly decreased the number and size of lung metastatic nodules (Fig. 7A and 7B). CD11b+Gr1+ MDSC derived from the spleen of Nor-NOHA-treated mice were less inhibitory on T cell proliferation compared with MDSC from vehicle-treated mice (Fig. 7C).
Figure 7.
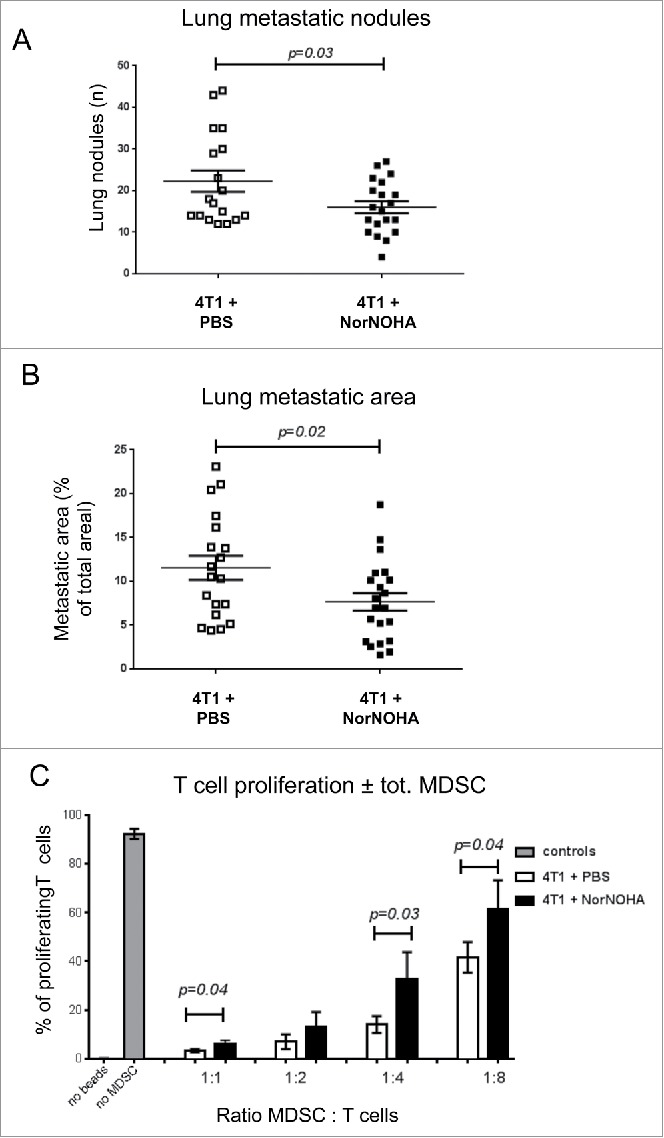
Nor-NOHA treatment reduces lung metastasis and attenuates inhibition of T cell proliferation by mMDSC. (A) Quantification of the number of lung metastatic nodules. (B) Quantification of the surface of lung metastatic nodules from DC101-treated (4T1+DC101) or IgG control antibody treated (4T1+control IgG) mice. N = 3; mice per group = 7–10. C: Inhibitory activity of CD11b+Gr1+ cells on T cell proliferation. MDSC were isolated by MACS from the spleen and co-cultured with stimulated T cells at the indicated ratios. Result from one representative experiment per condition is shown. Mice per group = 5–7, analysis done in triplicate.
Combined Nor-NOHA treatment does not further improve the anti-metastatic effect of DC101
As DC101 treatment further induced Arg I expression and activity (Fig. 6) we tested whether combined Nor-NOHA/DC101 treatment would bring additional therapeutic benefits compared with DC101 therapy alone. Combined treatment resulted in a small but significant enhancement of the inhibitory effect of DC101 on primary tumor growth (Fig. 8A), but did not provide any further benefit to the anti-metastatic effect of DC101 (Fig. 8B and C) compared with either individual treatment.
Figure 8.
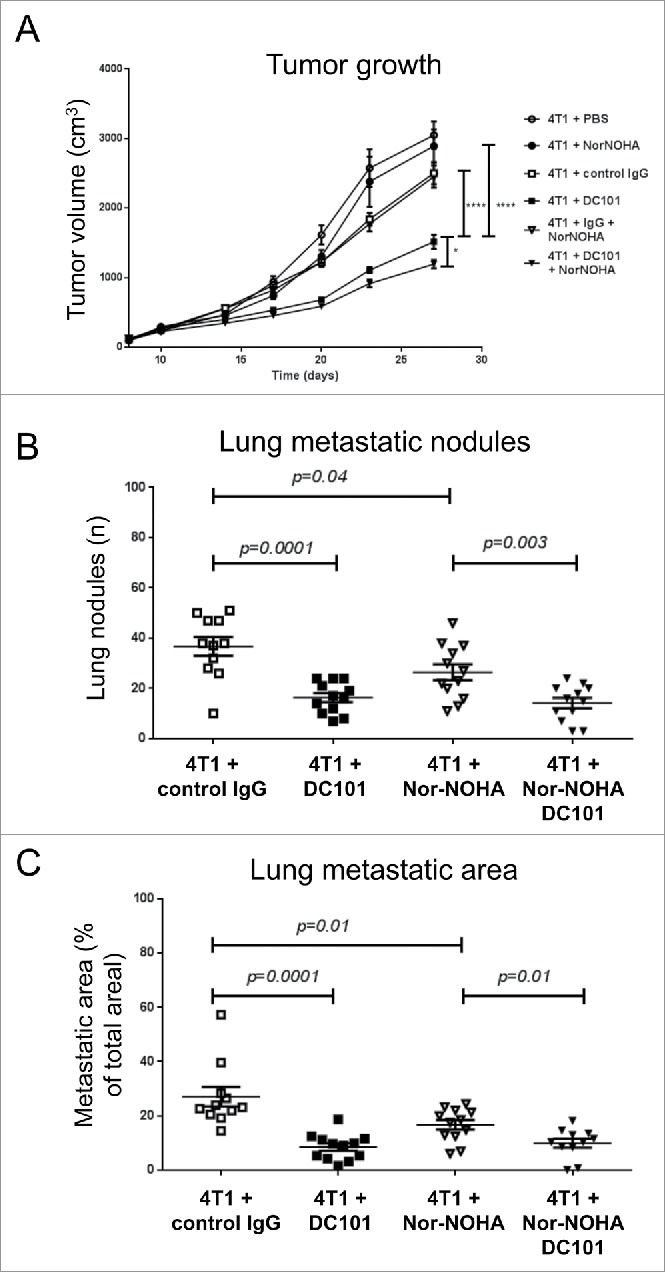
In combination with DC101, Nor-NOHA further inhibits tumor growth but not lung metastasis. (A) Growth curves of 4T1 tumors in mice treated with DC101, an IgG control antibody, Nor-NOHA, PBS, combined DC101/Nor-NOHA and combined control IgG/Nor-NOHA as indicated. (B) Quantification of the number of lung metastatic nodules in mice treated as indicated. (C) Quantification of the surface of lung metastatic nodules in mice treated as indicated. N = 2, mice per group = 10–12.
From these experiments, we concluded that Nor-NOHA treatment partially reverses the inhibitory activity of MDSC on T cell proliferation and inhibits 4T1 lung metastasis formation but has little or no additional effect in combination with DC101.
DC101 does not directly interfere with the function of MDSC generated in vitro
To investigate whether DC101 directly affects MDSC differentiation or function, bone marrow hematopoietic cells were cultured in vitro in the presence of GM-CSF and IL-6 to allow precursor cell differentiation into MDSC as described previously.53 Bone marrow cells were cultured in the presence of DC101 or IgG isotype control antibody for 3 d. Antibodies were either added to the culture during the entire process (3 d) or during the last 24 h only. In control cultures, over 90% of the cells become CD11b+Gr1+ after 3 d (not shown). Treatment with DC101, either added during 3 d or during the last 24 h, did not interfered with the generation of CD11b+Gr1+ cells or with their immunosuppressive activity relative to control IgG treatment (Fig. S8). Also, DC101 treatment did not influence Arg I expression in developing CD11b+Gr1+ cells (data not shown).
To collect evidence whether DC101 could directly affect MDSC at the primary tumor and lung metastatic sites in vivo, we analyzed VEGFR-2 expression on CD11b+, CD11b+Gr1LowLy6C+ and CD11b+Gr1highLy6G+ cells isolated form the blood, tumors and lungs. We observed that CD11b+ cells in the blood were virtually negative for VEGFR-2 expression and in tumors and lungs only about 5–6% of the cells were VEGFR-2 positive. CD11b+Gr1LowLy6C+ cells were virtually negative for VEGFR-2 expression (<1%) in all three compartments, while CD11b+Gr1highLy6G+ cells were about 3–4% positive in primary tumor and below 1% in blood and lung (Fig. 9A). In all cases, expression levels were low (Fig. 9B). RT-PCR analysis confirmed that VEGFR-2 mRNA was expressed at very low levels in tumor-derived CD11b+ cells when compared with the VEGFR-2-positive endothelioma derived cell line b.End5 (Fig. 9C, 9D). Likewise, 4T1 cells expressed very low to undetectable levels of VEGFR-2, as revealed by real-time RT-PCR and flow cytometry (Fig. 9C and 9D).
Figure 9.
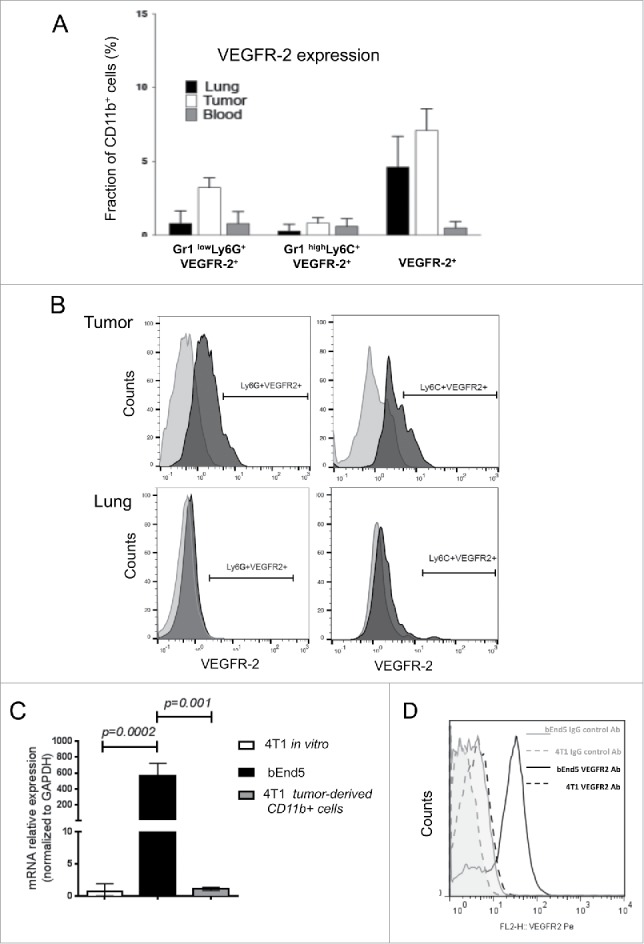
VEGFR-2 expression is very low to absent in CD11b+ and 4T1 cells. (A) VEGFR-2 expression on CD11b+ cells, CD11b+Gr1LowLy6C+ cells and CD11b+Gr1HighLy6G+ cells isolated from blood, tumor and lungs, was monitored by cell surface staining and flow cytometry analysis. N = 2, 4 mice per conditions. The percentages presented as a fraction of CD11b+ cells. (B) Representative profiles of VEGFR-2 expression from experiment in panel (A) are shown. VEGFR-2 expression is low or absent. (C) Real time RT-PCR analysis of VEGFR-2 mRNA expression in 4T1 and bEnd5 cells and CD11b+ cells recovered from 4T1 tumors. (D) Flow cytometry analysis of VEGFR-2 expression on 4T1 cells and bEnd5 cells. The bEnd5 VEGFR-2+ endothelioma cell line is used as positive control for VEGFR-2 expression. N = 2 and results from one representative experiment are shown here; mice analyzed per group = 5.
From these results, we concluded that MDSC express very low to undetectable levels of VEGFR-2 and that therefore the effect of DC101 on their activity is likely due to an indirect effect rather than a direct effect on the MDSC.
Discussion
Clinical therapeutic benefits of anti-angiogenic treatments are limited and transient. The reasons for this are not fully elucidated but may be due to the multiple and often contrasting effects of angiogenic factors on tumor cells, tumor vasculature and immune-inflammatory cells as well as to the adaptive host response to the treatment, including the mobilization and accumulation of MDSC.9-11 Here, we characterized the effects of the anti-VEGFR-2 antibody DC101 on MDSCs and Tregs in relationship to its effects on tumor angiogenesis, primary tumor growth and metastasis using the syngeneic and immunocompetent 4T1 breast adenocarcinoma model. We report three main results: First, DC101 treatment suppressed lung metastasis in addition to inhibiting primary tumor growth and angiogenesis. Second, DC101 treatment reduced significantly the inhibitory activity of CD11b+Gr1LowLy6C+ mMDSC (but not CD11b+Gr1highLy6G+ gMDSC) on T cell proliferation and also decreased the presence of Tregs in tumors and lungs but had no effects on the mobilization and recruitment of MDSC. Third, DC101 treatment enhanced Arg I mRNA and protein expression and activity in tumor-recruited MDSC. Fourth, arginase inhibition with Nor-NOHA prevented lung metastasis formation and growth but had little or no additional effect in combination with DC101.
Tumor angiogenesis has been associated with a state of immunosuppression. The VEGF/VEGFR-2 axis itself is an important mediator of immunosuppression and its inhibition was shown to restore tumor-immunity. It has been extensively described that VEGF can impair DC maturation as antigen-presenting cells, which in turn prevents T cell activation and therefore antitumor immunity. In addition, VEGF can limit T cell extravasation and induce expansion of Treg populations, thereby further enhancing the immunosuppressive state.34,54-56 It is, therefore, reasonable to expect that inhibition of the VEGF/VEGFR-2 axis would reverse tumor-mediated immunosuppression or enhance the effects of immunostimulatory antitumor therapies.31,34,36 In contrast with these observations, however, experimental data show that anti-angiogenic therapy also mobilizes MDSC, which in turn can limit the therapeutic benefit of anti-angiogenic therapy,26,29,31 and in certain tumor models may even exacerbate metastatic progression.47,48 The potential mechanisms accounting for those counteracting effects include the secretion of alternative angiogenic factors such as Bv8 and bFGF,13,29 the production of immune-suppressive molecules, such as iNOS, Arg I, IDO1–2 and PD-L1/2, the expansion of Tregs, the alteration of the IL10/IL12 balance in tumor associated macrophages (TAM) and the secretion of tumor-stimulating factors, such as IL-6, TGFβ, PDGF and GM-CSF.31,52 Recent clinical studies combining anti-angiogenic therapy and immune therapies based on checkpoint blockade inhibitors revealed promising preliminary results supporting the notion that anti-angiogenic therapy may reduce immunosuppression.57 How this is mechanistically achieved remains to be fully elucidated.
Here, we show that treatment with the VEGFR-2 blocking mAb DC101, in addition to the expected inhibitory effects on primary tumor angiogenesis and tumor growth, suppressed lung metastasis. This observation has not been previously reported and has therapeutic relevance. However, the fact that experimental metastasis (i.e., induction of lung metastasis by direct injection of tumor cells in the tail vein thereby bypassing the primary tumor) was not inhibited by DC101 treatment suggests that inhibition of angiogenesis may not be the only, or main, mechanism involved in this effect. Indeed, it has been previously speculated that lung metastasis may rather grow by vascular co-option than angiogenesis implying that anti-angiogenesis therapy per se may not be effective to control lung metastases.58-60 Although mMDSC are slightly less frequent in the primary tumors and metastases compared with gMDSC (20–40% vs. 60–80%), this population, however, was reported to be more suppressive than gMDSC. Strikingly, DC101 treatment reversed the inhibitory effect of mMDSC, but not gMDSC, on T cell proliferation. DC101 treatment also reduced the accumulation of Tregs in tumors and lungs. As MDSC have been shown to promote the differentiation of Tregs, it is possible that the reduction of Tregs is secondary to the blunting of the immunosuppressive activity of MDSC by DC101.61,62 Alternatively, decreased angiogenesis may hamper Tregs recruitment to the tumor and metastatic sites. DC101 treatment did not cause a significant mobilization and accumulation of MDSC but played a role on their suppressive function. We also calculated the ratio of CD4+ T cells over MDSC in tumors and lungs. In all conditions, the number of Gr1lowLy6C+ cells outnumbered non-Treg CD4+ T cells in tumors and lungs. Upon DC101 treatment this proportion remained virtually unchanged in the lung, while it increased about six times in the primary tumor. Taken together, our data indicate DC101 impact on MDSCs function and presence of Tregs at tumor and metastatic sites is consistent with the observed antitumor effect of DC101.
A further unexpected result was the observed increased expression of Arg I mRNA, protein and activity in MDSC upon DC101 treatment. This result is unexpected because Arg I itself is immunosuppressive, and it goes against the general notion that VEGF/VEGFR-2 inhibition reverses immunosuppression. Arg I was already expressed at high levels in control-treated mice, and treatment with the arginase inhibitor Nor-NOHA significantly reduced lung metastasis formation, but not primary tumor growth, and partially reversed MDSCs inhibition of T cell proliferation. Strikingly, Arg I expressing infiltrating CD11b+ cells in the lungs, were exclusively present within the metastatic nodules in close contact with cancer cells, while in the primary tumor they were mostly present in the tumor stroma, rather distant from cancer cells. This physical proximity is consistent the Arg I-mediated MDSC immunosuppressive activity being more effective in suppressing T cells in lung metastasis compared with primary tumors and with the more prominent effect of arginase inhibition in suppressing lung metastasis. When applied in combination with DC101, Nor-NOHA did not further enhance the anti-metastatic activity of DC101, suggesting that the global effect of DC101 is dominant over selective arginase inhibition. One outstanding question concerns the mechanism by which DC101 affects mMDSC activity. VEGFR-2 is virtually absent on MDSC and circulating CD11b+ cells, suggesting that the DC101 effect on CD11b+ cells and mMDSC is rather indirect. Consistent with this observation, DC101 was ineffective in preventing the development of immunosuppressive MDSC from bone marrow-derived progenitors in vitro. Similarly, 4T1 cells do not express VEGFR-2 and are, therefore, unlikely to be a direct target of DC101. Conversely, VEGFR-2 is highly expressed on angiogenic endothelial cells and is a key mediator of endothelial cell activation and proliferation.4 Angiogenic endothelial cells express high levels of cytokines, chemokines, growth factors or proteases modulating the immune-inflammatory response.4,31,36 DC101 may, therefore affect the production of those factors (e.g., GM-CSF, M-CSF, FLT-3 ligand, IL-3, IL-4, IL-13, IL-10, TGF-β, GM-CSF and PGE2) that could be responsible for the modulation of MDSC activity, including the stimulation of Arg I expression.4,31,36 The PI3K pathway is an attractive candidate in both endothelial and immune cells as it has been shown to induce immunostimulatory and angiostatic phenotypes in murine models of pancreatic and mammary tumors following VEGF/VEGFR blockade.31
These results have translational implications potentially relevant for therapy. First, they demonstrate that VEGFR-2 inhibition partially reverses the T-cell inhibitory activity of mMDSC and reduces the presence of Tregs and tumor and metastatic sites. Second, it highlights a dissociation between the number of MDSC and their activity. The quantitative evaluation of the frequency of MDSC currently used in clinical practice to monitor the immunosuppressive status of cancer patients may therefore not fully represent ongoing effects.63 A routine MDSC functional assay may be a useful complement to cell number. We recently analyzed cellular and biochemical parameters in the blood of patients with metastatic breast cancer treaded with chemotherapy ± bevacizumab (Avastin®), and reported that bevacizumab selectively reverses M2 polarization of CD11b+ cells and decreased IL10 levels in CD11b+ cells and plasma, consistent with a reduction of immunosuppression leveraged by bevacizumab.64 The impact of these changes on disease progression and survival, however, remains to be determined. Third, arginase inhibition has anti-metastatic activity and this effect should be further explored as a potential adjuvant therapeutic strategy to control lung metastasis progression. This may be particularly relevant for breast cancer patients at high risk for distant progression. Interestingly, COX-2 inhibitors were shown to exert antitumor activity through multiple mechanisms, including inhibition of tumor angiogenesis65 and reversion of immunosuppression via Arg I inhibition.66 Similarly, phosphodiesterase-5 inhibitors downregulate Arg I and NOS-2 expression in MDSC and boost endogenous antitumor immunity.67
In conclusion, this work has provided novel insights into the immunomodulatory effects of anti-VEGFR-2-based anti-angiogenic therapy. As DC101 blunted the inhibitory effects of mMDSC but not gMDSC on T-cell proliferation it will be important to test whether VEGF/VEGFR-2 axis inhibition may boost the antitumor immune responses in combination with immunostimulatory therapies in patients, by impinging on MDSC function or not.34,36,57 On the other side, arginase inhibition should be further explored as a potential anti-metastatic strategy in aggressive breast cancer.
Materials and methods
Cell culture and reagents
4T1 breast adenocarcinoma cells and the endothelioma-derived cell line bEnd5 were cultured in DMEM supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% non-essential amino-acids in tissue culture plates and incubated in a 37°C, 5% CO2 incubator. All cell culture reagents were purchased from Invitrogen Life Technologies (Basel, Switzerland).
Preparation of bone marrow-derived MDSC (BM-MDSC)
BM-MDSC were obtained from primary bone marrow cells as described previously.53 In short, tibias and femurs were flushed with PBS to obtain primary bone marrow cells. After red cell lysis (BD Pharm Lyse, BD Biosciences), cells were cultured in RPMI 1640 + 10% FCS, 1% sodium pyruvate, 5 × 10−5 M 2-mercaptoethanol, 1% HEPES, 50 U/mL penicillin, 50 µg/mL streptomycin, 2 mmol/L L-glutamine, supplemented with 40 ng/mL GM-CSF and 40 ng/mL IL-6 (Peprotech) for 3 d, to induce MDSC differentiation. The percentage of Gr1+CD11b+ cells was over 90%.
Mouse maintenance, tumor induction and treatments
Female BALB/c Ola/Hsd mice (4–6 weeks old) were purchased from Harlan Laboratories/Envigo, housed in single ventilated cages at 25°C and left one week for acclimation before experimentation. Then they were divided randomly into the different experimental groups. Mice in each group were given food and water ad libitum and observed daily. This study was performed according to national ethical guidelines and was approved by the veterinary service of Canton Fribourg.
For primary tumor growth, 4T1 cancer cells were orthotopically injected in the inguinal mammary fat pad as previously reported.38 Tumor growth was monitored daily and tumors measured using a microcaliper. Tumor volumes calculated with the ellipsoid volume formula: V (mm3) = L1 (major axis) × L22 (minor axis)/ π/6.
The anti-angiogenic anti-VEGFR-2 mAb (clone DC101) or the corresponding non-targeting isotype control (IgG) (BioXCell, Inc.) were administered intraperitoneally (0.8 μg per mouse), starting from day 10 after tumor cell injection in the mammary fat pad, and then every 3 d, for a total of four injections (i.e., days 10, 13, 17 and 20). The Arginase I inhibitor Nω-hydroxy-nor-Arginine (nor-NOHA) (Bachem AG) was administered intraperitoneally daily (2 mg per mouse), from day 7 until the end of the experiment. PBS injection was used as control. Combination treatments were performed using the same dosage and schedules of DC101 and Nor-NOHA used for the single treatments.
For the experimental lung metastasis model, 2.5 × 105 4T1 cells were intravenously injected in the tail vein and treatments (DC101, IgG, nor-NOHA or PBS, same dosage as above) were started from day 1 after cancer cells inoculation and followed the same schedule as for the orthotropic model, until day 9, when mice were killed for histological analysis.
Collection of peripheral blood and flow cytometry analysis
Peripheral blood BMDC for phenotype analysis were obtained from the lateral tail vein via mouse tail bleeding and analyzed at day 22 post-cancer cell injection. The following anti-mouse antibodies were used following manufacturer's instructions: anti-CD16/CD32 Fc blocking antibody (BD Biosciences), anti-CD11b-PeCy7 or Pacific Blue (clone M1/70, Biolegend) or V450 (eBiosciences), anti-Ly6C-FITC or PerCP (clone HK1.4, Biolegend), anti-Gr1-Pacific Blue (clone RB6–8C5, Biolegend), anti-Ly6G- Pe/Cy7, Alexa Fluor 647 or APC (clone 1A8, BioLegend), anti-VEGFR-2-Pe (clone Avas12a1, eBioscience), CD45 APC (clone 30-F11, BD Biosciences), CD4+-FITC (clone GK1.5, Biolegend), CD25-Pe (clone PC61.5, eBioscience), Foxp3-Alexa Fluor 647 (clone 150D, Biolegend). IgG control was from eBiosciences. Red blood cells were lysed with ACK buffer, washed and remaining leukocyte pellet was stained for FACS (fluorescence activated cell sorting) acquisition at the FACSCalibur flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Treestar Inc.). For RNA extraction, total blood was collected from heart puncture on Eppendorf tubes in presence of EDTA 0.5M and CD11b+ were isolated from the peripheral blood mononuclear fraction using Histopaque 1077 (Sigma-Aldrich) followed by CD11b-beads specific MACS Separation (Miltenyi Biotech). Gating strategy for the flow cytometry analysis is summarized in Fig. S2.
Organ digestion and flow cytometry analysis
At the conclusion of the experiment (day 28 post-cancer cell injection, unless indicated otherwise), mice were killed and tumor, spleens and lungs dissected. BMDC were isolated from sterilely harvested tumors and lungs by mechanical and enzymatic organ disruption. Organs were cut in small pieces with scissors, washed, and digested in serum free medium supplemented with Collagenase I (Worthington Biochemical Corporation), and DNAse I (Roche). The mixture was incubated at 37°C for 45 min on a shaking platform. Subsequently, serum-supplemented medium was added to neutralize the enzymatic reaction and the tissue suspensions were filtered through a 100 µm and 70 µm sterile nylon gauzes. Upon centrifugation (5 min at 1400 rpm), pellets were recovered and red blood cells lysed with ACK buffer. The staining procedure and the flow cytometry acquisition were the same as outlined above for blood.
For RNA extraction, tumor-derived CD11b+ were collected using the CD11b-beads specific MACS Separation (Miltenyi Biotech) as described above, whereas total MDSC were sorted using FACS based on CD11b+ and Ly6C/Ly6G staining (FACSARIA Fusion, Becton Dickinson), as described below for MDSC.
Spleens from tumor-bearing mice were harvested under sterile conditions. Single-cell suspensions were prepared via mechanical tissue disruption and red cells were removed using BD Pharm Lyse lysing solution (BD Biosciences). Splenocytes were incubated with the Fc block (TruStain fcX, Biolegend) for 30 min and then stained with indicated anti-mouse antibodies, accordingly to manufacturer's instructions: anti-CD11b-PeCy7 (clone M1/70, Biolegend) anti-Gr1-Pacific blue (clone RB6–8C5, Biolegend). After washing, acquisition was performed using the MACSQuant flow cytometer from Miltenyi Biotec and data analyzed by FlowJo v10.0.7 (tree Stat Inc., Ashland, USA).
Tissue histology and immunohistochemistry (IHC)
Tumors and lungs were harvested at the end of the experiment, were fixed in formalin and embedded in paraffin. 5 μm thick serial sections were cut from the tissue blocks. Hematoxylin and eosin (H&E) stained sections were used to assess and quantify intratumoral tumor necrosis and lung metastatic foci using the Image J software 1.48 v (NIH, USA). Metastasis index was calculated as ratio of metastasis number over the primary tumor volume in mm3.
Sections were stained with anti-CD31 antibody (Rabbit anti-mouse Polyclonal Antibody, NeoMarkers) using the Chalky counts method to quantify MVD,38 and with anti-Arg I (Rabbit Polyclonal to Human/Mouse, LSBio) and anti-CD11b (clone EPR1344, Abcam) antibodies to identify Arg I expression and CD11b+ cells, respectively. Quantification was performed by ImageJ software. For IHC, endogenous peroxidases were blocked by incubation with 0.6% H2O2 in methanol. HRP-conjugated secondary antibodies were used for primary antibody detection. Dako Envision+ was used with diaminobenzidine (DAB) tablets (Sigma-Aldrich) to detect HRP. Quantitative analyses of CD11b and Arg I staining were performed with the image analysis program ImageJ.68
MDSC isolation and T cell proliferation assay
At the conclusion of the experiment splenic CD11b+Gr1+ (MDSCs) cells from tumor-bearing mice in each experimental group were enriched from the spleen tissue by means of two different methods. One method consisted entirely of a MACS-based strategy, with a two-step magnetic separation via an initial depletion to collect Gr1dim/Ly-6G– myeloid cells followed by a second positive selection to collect Gr1high/Ly-6G+ cells (mouse MDSC Isolation Kit from Miltenyi Biotec). The second method consisted of an initial negative depletion system to eliminate CD3+, CD5+, CD19+, CD45R (B220)+ and Ter119+ cells using the EasySep™ Mouse Custom Enrichment Kit from Stemcell technologies. Enriched cells were further sorted by FACS based on CD11b+ and Ly6C/Ly6G staining (FACSARIA Fusion, Becton Dickinson). The following anti-mouse antibodies were used accordingly to manufacturer's instructions: anti-CD11b PeCy7 (clone M1/70, Biolegend) and Gr1-Pacific blue (clone RB6–8C5, Biolegend). The purity of cell populations was >95% as tested by flow cytometry. MDSCs were seeded in 96 well plates at the indicated ratios with 8 × 105 purified, CSFE (Biolegend) labeled T cells isolated using the Pan T cell isolation kit (Miltenyi Biotec) from the spleen of naïve BALB/c-mice. The T cells co-cultured with MDSCs were then stimulated with anti-CD3/anti-CD28 coated beads for 3 d, according to manufacturer's protocol (Dynabeads Mouse T-Activator CD3/CD28, Life Technologies AS). Proliferation of T cells was then evaluated measuring the CFSE dilution by flow cytometry using MACSQuant instrument (Miltenyi Biotec), and data analyzed with FlowJo 10.0.7.
Real-time PCR analysis
Total RNA was prepared from tumor- and blood- isolated cells obtained as outlined above using the RNAeasy Mini kit (Qiagen). cDNA was synthesized using the M-MLV reverse transcriptase, (H-) Point Mutant (Invitrogen Life Technologies) per manufacturer recommendations. Primers were designed using the NCBI tool and were purchased from Eurofins Genomics. The following primer pairs were used: GAPDH Fwd (5ʹ–CGTCCCGTAGACAAAATGGT–3ʹ) and Rev (5ʹ–TCAATGAAGGGG TCGTTGAT–3ʹ); Arg I Fwd (5ʹ–AAGCTGGTCTGCTTGAAAAA–3ʹ) and Rev (5ʹ–CCGTGGGTTCTTCACAATTT–3ʹ); iNOS Fwd (5ʹ–GCGCTCTAGTGAAGCAAAGC–3ʹ) and Rev (5ʹ–TGATGGACCCCAAGCAAGAC–3ʹ) and MRC1 Fwd (5ʹ–GATGACCTGTGCTCGAGAGG–3ʹ) and Rev (5ʹ–TCTCGCTTCCCTCAAAGTGC–3ʹ). IL10 Fwd (5ʹ–TTTGAATTCCCTGGGTGAGA–3ʹ) and Rev (5ʹ–AGACACCTTGGTCTTGGAGC–3ʹ); IL12a Fwd (5ʹ–CCTTGCATCTGGCGTCTACA–3ʹ) and Rev (5ʹ–GTCTTCAGCAGGTTTCGGGA–3ʹ); TNFa Fwd (5ʹ–CAGCCTCTTCTCATTCCTGC–3ʹ) and Rev (5ʹ–ATGAGAGGGAGGCCATTTG–3ʹ); VEGFR-2 Fwd (5ʹ–TGGGCACTCAAGTCCGAATC–3ʹ) and Rev (5ʹ–TTGGACTCAATG GGCCTTCC–3ʹ). Real-time qPCR were performed using a SYBR Green Master Mix (Applied Biosystems, Life Technologies), according to the manufacturer's recommendations and run on a StepOne SYBR System instrument (Applied Biosystems, Life Technologies). The gene expression of each individual tested gene was normalized to the corresponding GAPDH signal, as endogenous control. Relative mRNA expression levels were calculated using the 2−ΔΔCT method as reported.69
Arginase enzymatic activity
To prepare cell lysates, tumor-derived CD11b+ Gr1+ cells obtained by FACS sorting as described above, were lysed in 10 mM Tris–HCl, pH 7.4, containing 1 mM pepstatin A, 1 mM leupeptin, and 0.4% (w/v) Triton X-100 and processed accordingly to manufacturer instructions (Roche). Protein concentration was determined and equivalent amounts of total protein (10 ug) were used for all tested samples. Arginase activity was determined, using the Arginase Activity Assay Kit (Sigma Aldrich), where arginase catalyzes the conversion of arginine to urea and ornithine. One unit of Arginase is the amount of enzyme that converts 1.0 mmole of L-arginine to ornithine and urea per minute at pH 9.5 and 37°C.
Statistical analysis
All statistical analyses were performed using GraphPad PRISM 6 software (GraphPad Software, Inc. La Jolla, CA). Statistical comparisons were performed by two-way ANOVA with Bonferroni post-test by Student's t-test or by Mann–Whitney test for non-parametric distributions with small sample size. Probability values of p ≤ 0.05 were considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors' thank F.R. Miller, Michigan Cancer Foundation (Detroit, MI) for providing the 4T1 cell line, B. Engelhardt (University of Bern, Switzerland) for providing bEnd5 cells, Nathalie Duffey (University of Fribourg, for technical help), J-C. Stehle and J. Horlbeck for tissue processing and staining (mouse Pathology laboratory, University of Lausanne).
Funding
This work was supported by a Collaborative Cancer Research Project of the Swiss Cancer League (CCRP OCS-01812–12–2005), The Swiss National Science Foundation (31003A 159824), The Sassella Foundation and the MEDIC Foundation.
References
- 1.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21:309-22; PMID:22439926; https://doi.org/ 10.1016/j.ccr.2012.02.022 [DOI] [PubMed] [Google Scholar]
- 2.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; https://doi.org/ 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorusso G, Ruegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol 2008; 130:1091-103; PMID:18987874; https://doi.org/ 10.1007/s00418-008-0530-8 [DOI] [PubMed] [Google Scholar]
- 4.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011; 146:873-87; PMID:21925313; https://doi.org/ 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- 5.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 2011; 17:1359-70; PMID:22064426; https://doi.org/ 10.1038/nm.2537 [DOI] [PubMed] [Google Scholar]
- 6.Young RJ, Reed MW. Anti-angiogenic therapy: concept to clinic. Microcirculation 2012; 19:115-25; PMID:22078005; https://doi.org/ 10.1111/j.1549-8719.2011.00147.x [DOI] [PubMed] [Google Scholar]
- 7.Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med 2012; 2:a006577; PMID:23028128; https://doi.org/ 10.1101/cshperspect.a006577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldfarb SB, Traina TA, Dickler MN. Bevacizumab for advanced breast cancer. Womens Health 2010; 6:17-25; PMID:20001867; https://doi.org/ 10.2217/whe.09.71 [DOI] [PubMed] [Google Scholar]
- 9.Huijbers EJ, van Beijnum JR, Thijssen VL, Sabrkhany S, Nowak-Sliwinska P, Griffioen AW. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist Updates 2016; 25:26-37; PMID:27155374; https://doi.org/ 10.1016/j.drup.2016.02.002 [DOI] [PubMed] [Google Scholar]
- 10.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592-603; PMID:18650835; https://doi.org/ 10.1038/nrc2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, Batchelor TT, Sorensen AG. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol 2009; 6:327-38; PMID:19483739; https://doi.org/ 10.1038/nrclinonc.2009.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding Y, Song N, Luo Y. Role of bone marrow-derived cells in angiogenesis: focus on macrophages and pericytes. Cancer Microenviron 2012; 5:225-36; PMID:22528877; https://doi.org/ 10.1007/s12307-012-0106-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laurent J, Touvrey C, Botta F, Kuonen F, Ruegg C. Emerging paradigms and questions on pro-angiogenic bone marrow-derived myelomonocytic cells. Int J Dev Biol 2011; 55:527-34; PMID:21769777; https://doi.org/ 10.1387/ijdb.103228jl [DOI] [PubMed] [Google Scholar]
- 14.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008; 8:618-31; PMID:18633355; https://doi.org/ 10.1038/nrc2444 [DOI] [PubMed] [Google Scholar]
- 15.Ham M, Moon A. Inflammatory and microenvironmental factors involved in breast cancer progression. Arch Pharm Res 2013; 36:1419-31; PMID:24222504; https://doi.org/ 10.1007/s12272-013-0271-7 [DOI] [PubMed] [Google Scholar]
- 16.Rudnick JA, Kuperwasser C. Stromal biomarkers in breast cancer development and progression. Clin Exp Metastasis 2012; 29:663-72; PMID:22684404; https://doi.org/ 10.1007/s10585-012-9499-8 [DOI] [PubMed] [Google Scholar]
- 17.Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest 2015; 125:3347-55; PMID:26325032; https://doi.org/ 10.1172/JCI80007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Uden DJ, van Laarhoven HW, Westenberg AH, de Wilt JH, Blanken-Peeters CF. Inflammatory breast cancer: an overview. Crit Rev Oncol Hematol 2015; 93:116-26; PMID:25459672; https://doi.org/ 10.1016/j.critrevonc.2014.09.003 [DOI] [PubMed] [Google Scholar]
- 19.Clemente CG, Mihm MC Jr., Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996; 77:1303-10; PMID:8608507; https://doi.org/ 10.1002/(SICI)1097-0142(19960401)77:7%3c1303::AID-CNCR12%3e3.0.CO;2-5 [DOI] [PubMed] [Google Scholar]
- 20.Rao UN, Lee SJ, Luo W, Mihm MC Jr., Kirkwood JM. Presence of tumor-infiltrating lymphocytes and a dominant nodule within primary melanoma are prognostic factors for relapse-free survival of patients with thick (t4) primary melanoma: pathologic analysis of the e1690 and e1694 intergroup trials. Am J Clin Pathol 2010; 133:646-53; PMID:20231618; https://doi.org/ 10.1309/AJCPTXMEFOVYWDA6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960-4; PMID:17008531; https://doi.org/ 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 22.Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol 2014; 32:2959-66; PMID:25071121; https://doi.org/ 10.1200/JCO.2013.55.0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HJ, Kim JY, Park IA, Song IH, Yu JH, Ahn JH, Gong G. Prognostic significance of tumor-infiltrating lymphocytes and the tertiary lymphoid structures in HER2-positive breast cancer treated with adjuvant trastuzumab. Am J Clin Pathol 2015; 144:278-88; PMID:26185313; https://doi.org/ 10.1309/AJCPIXUYDVZ0RZ3G [DOI] [PubMed] [Google Scholar]
- 24.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J 2010; 16:348-53; PMID:20693846; https://doi.org/ 10.1097/PPO.0b013e3181eb3358 [DOI] [PubMed] [Google Scholar]
- 25.Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling JA. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology 2012; 1:1484-94; PMID:23264895; https://doi.org/ 10.4161/onci.21990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015; 125:3356-64; PMID:26168215; https://doi.org/ 10.1172/JCI80005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol 2011; 11:856-61; PMID:21315783; https://doi.org/ 10.1016/j.intimp.2011.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye XZ, Yu SC, Bian XW. Contribution of myeloid-derived suppressor cells to tumor-induced immune suppression, angiogenesis, invasion and metastasis. J Genet Genomics 2010; 37:423-30; PMID:20659706; https://doi.org/ 10.1016/S1673-8527(09)60061-8 [DOI] [PubMed] [Google Scholar]
- 29.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol 2007; 25:911-20; PMID:17664940; https://doi.org/ 10.1038/nbt1323 [DOI] [PubMed] [Google Scholar]
- 30.Ko JS, Bukowski RM, Fincke JH. Myeloid-derived suppressor cells: a novel therapeutic target. Curr Oncol Rep 2009; 11:87-93; PMID:19216839; https://doi.org/ 10.1007/s11912-009-0014-6 [DOI] [PubMed] [Google Scholar]
- 31.Rivera LB, Bergers G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol 2015; 36:240-9; PMID:25770923; https://doi.org/ 10.1016/j.it.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res 2001; 23:263-72; PMID:11444391; https://doi.org/ 10.1385/IR:23:2-3:263 [DOI] [PubMed] [Google Scholar]
- 33.Tartour E, Pere H, Maillere B, Terme M, Merillon N, Taieb J, Sandoval F, Quintin-Colonna F, Lacerda K, Karadimou A et al. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev 2011; 30:83-95; PMID:21249423; https://doi.org/ 10.1007/s10555-011-9281-4 [DOI] [PubMed] [Google Scholar]
- 34.Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, Terme M. Control of the immune response by pro-angiogenic factors. Front Oncol 2014; 4:70; PMID:24765614; https://doi.org/ 10.3389/fonc.2014.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Y, Yu P, Zeng D, Qian F, Lei X, Zhao Y, Tang B, Hao Y, Luo H, Chen J et al. Suppression of vascular endothelial growth factor abrogates the immunosuppressive capability of murine gastric cancer cells and elicits antitumor immunity. FEBS J 2014; 281:3882-93; PMID:25041128; https://doi.org/ 10.1111/febs.12923 [DOI] [PubMed] [Google Scholar]
- 36.Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol 2011; 11:702-11; PMID:21941296; https://doi.org/ 10.1038/nri3064 [DOI] [PubMed] [Google Scholar]
- 37.Terme M, Colussi O, Marcheteau E, Tanchot C, Tartour E, Taieb J. Modulation of immunity by antiangiogenic molecules in cancer. Clin Dev Immunol 2012; 2012:492920; PMID:23320019; https://doi.org/ 10.1155/2012/492920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuonen F, Laurent J, Secondini C, Lorusso G, Stehle JC, Rausch T, Faes-Van't Hull E, Bieler G, Alghisi GC, Schwendener R et al. Inhibition of the Kit ligand/c-Kit axis attenuates metastasis in a mouse model mimicking local breast cancer relapse after radiotherapy. Clin Cancer Res 2012; 18:4365-74; PMID:22711708; https://doi.org/ 10.1158/1078-0432.CCR-11-3028 [DOI] [PubMed] [Google Scholar]
- 39.Laurent J, Hull EF, Touvrey C, Kuonen F, Lan Q, Lorusso G, Doucey MA, Ciarloni L, Imaizumi N, Alghisi GC et al. Proangiogenic factor PlGF programs CD11b(+) myelomonocytes in breast cancer during differentiation of their hematopoietic progenitors. Cancer Res 2011; 71:3781-91; PMID:21507936; https://doi.org/ 10.1158/0008-5472.CAN-10-3684 [DOI] [PubMed] [Google Scholar]
- 40.Simpson KD, Templeton DJ, Cross JV. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol 2012; 189:5533-40; PMID:23125418; https://doi.org/ 10.4049/jimmunol.1201161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh K, Lee OY, Shon SY, Nam O, Ryu PM, Seo MW, Lee DS. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res 2013; 15:R79; PMID:24021059; https://doi.org/ 10.1186/bcr3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe H, Wada H, Baghdadi M, Nakanishi S, Usui Y, Tsuchikawa T, Shichinohe T, Hirano S, Seino K. Identification of a highly immunogenic mouse breast cancer sub cell line, 4T1-S. Hum Cell 2016; 29:58-66; PMID:26857856; https://doi.org/ 10.1007/s13577-015-0127-1 [DOI] [PubMed] [Google Scholar]
- 43.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res 2005; 11:728-34; PMID:15701862 [PubMed] [Google Scholar]
- 44.Coveney E, Wheatley GH 3rd, Lyerly HK. Active immunization using dendritic cells mixed with tumor cells inhibits the growth of primary breast cancer. Surgery 1997; 122:228-34; PMID:9288127; https://doi.org/ 10.1016/S0039-6060(97)90013-1 [DOI] [PubMed] [Google Scholar]
- 45.Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Böhlen P. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev 1998; 17:155-61; PMID:9770111; https://doi.org/ 10.1023/A:1006094117427 [DOI] [PubMed] [Google Scholar]
- 46.Franco M, Man S, Chen L, Emmenegger U, Shaked Y, Cheung AM, Brown AS, Hicklin DJ, Foster FS, Kerbel RS. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer Res 2006; 66:3639-48; PMID:16585189; https://doi.org/ 10.1158/0008-5472.CAN-05-3295 [DOI] [PubMed] [Google Scholar]
- 47.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15:220-31; PMID:19249680; https://doi.org/ 10.1016/j.ccr.2009.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005; 8:299-309; PMID:16226705; https://doi.org/ 10.1016/j.ccr.2005.09.005 [DOI] [PubMed] [Google Scholar]
- 49.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 2010; 22:238-44; PMID:20171075; https://doi.org/ 10.1016/j.coi.2010.01.021 [DOI] [PubMed] [Google Scholar]
- 50.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 2007; 67:4507-13; PMID:17483367; https://doi.org/ 10.1158/0008-5472.CAN-06-4174 [DOI] [PubMed] [Google Scholar]
- 51.Nagaraj S, Youn JI, Gabrilovich DI. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol 2013; 191:17-23; PMID:23794702; https://doi.org/ 10.4049/jimmunol.1300654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res 2007; 13:5243-8; PMID:17875751; https://doi.org/ 10.1158/1078-0432.CCR-07-0182 [DOI] [PubMed] [Google Scholar]
- 53.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010; 32:790-802; PMID:20605485; https://doi.org/ 10.1016/j.immuni.2010.05.010 [DOI] [PubMed] [Google Scholar]
- 54.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:22437938; https://doi.org/ 10.1038/nri3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terme M, Tartour E, Taieb J. VEGFA/VEGFR2-targeted therapies prevent the VEGFA-induced proliferation of regulatory T cells in cancer. Oncoimmunology 2013; 2:e25156; PMID:24083078; https://doi.org/ 10.4161/onci.25156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res 2013; 73:539-49; PMID:23108136; https://doi.org/ 10.1158/0008-5472.CAN-12-2325 [DOI] [PubMed] [Google Scholar]
- 57.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2014; 2:632-42; PMID:24838938; https://doi.org/ 10.1158/2326-6066.CIR-14-0053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, Gatter KC, Pezzella F. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med 2013; 2:427-36; PMID:24156015; https://doi.org/ 10.1002/cam4.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Welti JC, Powles T, Foo S, Gourlaouen M, Preece N, Foster J, Frentzas S, Bird D, Sharpe K, Van Weverwijk A et al. Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis 2012; 15:623-41; PMID:22843200; https://doi.org/ 10.1007/s10456-012-9291-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szabo V, Bugyik E, Dezso K, Ecker N, Nagy P, Timar J, Tovari J, Laszlo V, Bridgeman VL, Wan E et al. Mechanism of tumour vascularization in experimental lung metastases. J Pathol 2015; 235:384-96; PMID:25319725; https://doi.org/ 10.1002/path.4464 [DOI] [PubMed] [Google Scholar]
- 61.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 2008; 68:5439-49; PMID:18593947; https://doi.org/ 10.1158/0008-5472.CAN-07-6621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L, Zhao J, Ren JP, Wu XY, Morrison ZD, El Gazzar M, Ning SB, Moorman JP, Yao ZQ. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS 2016; 30:1521-31; PMID:26959508; https://doi.org/ 10.1097/QAD.0000000000001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baniyash M. Myeloid-derived suppressor cells as intruders and targets: clinical implications in cancer therapy. Cancer Immunol Immunother 2016; 65:857-67; PMID:27225641; https://doi.org/ 10.1007/s00262-016-1849-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cattin S, Fellay B, Pradervand S, Trojan A, Ruhstaller T, Ruegg C, Fürstenberger G. Bevacizumab specifically decreases elevated levels of circulating KIT+CD11b+ cells and IL-10 in metastatic breast cancer patients. Oncotarget 2016; 7:11137-50; PMID:26840567; https://doi.org/ 10.18632/oncotarget.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruegg C, Dormond O, Mariotti A. Endothelial cell integrins and COX-2: mediators and therapeutic targets of tumor angiogenesis. Biochim Biophys Acta 2004; 1654:51-67; PMID:14984767; https://doi.org/ 10.1016/j.bbcan.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 2004; 64:5839-49; PMID:15313928; https://doi.org/ 10.1158/0008-5472.CAN-04-0465 [DOI] [PubMed] [Google Scholar]
- 67.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 2006; 203:2691-702; PMID:17101732; https://doi.org/ 10.1084/jem.20061104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vrekoussis T, Chaniotis V, Navrozoglou I, Dousias V, Pavlakis K, Stathopoulos EN, Zoras O.. Image analysis of breast cancer immunohistochemistry-stained sections using ImageJ: an RGB-based model. Anticancer Res. 2009 Dec; 29(12):4995-8; PMID:20044607 [PubMed] [Google Scholar]
- 69.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101-8; PMID:18546601; https://doi.org/ 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.