

ABSTRACT
Pancreatic cancer (PaCa) has a dismal prognosis and adjuvant immunotherapy frequently is of low efficacy due to immunosuppressive features of PaCa and PaCa-stroma. We here explored, whether the efficacy of vaccination with tumor-exosome (TEX)-loaded dendritic cells (DC) can be improved by combining with drugs affecting myeloid-derived suppressor cells (MDSC). Experiments were performed with the UNKC6141 PaCa line. UNKC6141 TEX-loaded DC were weekly intravenously injected, mice additionally receiving Gemcitabine (GEM) and/or ATRA and/or Sunitinib (Sun). UNKC6141 grow aggressively after subcutaneous and orthotopic application and are consistently recovered in peripheral blood, bone marrow, lung and frequently liver. Vaccination with DC-TEX significantly prolonged the survival time, the efficacy of DC-TEX exceeding that of the cytotoxic drugs. However, ATRA, Sun and most efficiently GEM, sufficed for a pronounced reduction of MDSC including tumor-infiltrating MDSC, which was accompanied by a decrease in migrating and metastasizing tumor cells. When combined with DC-TEX vaccination, a higher number of activated T cells was recovered in the tumor and the survival time was prolonged compared with only DC-TEX vaccinated mice. As ATRA, GEM and Sun affect MDSC at distinct maturation and activation stages, a stronger support for DC-TEX vaccination was expected by the drug combination. Intrapancreatic tumor growth was prevented beyond the death of control mice. However, tumors developed after a partial breakdown of the immune system by the persisting drug application. Nonetheless, in combination with optimized drug tuning to prevent MDSC maturation and activation, vaccination with TEX-loaded DC appears a most promising option in PaCa therapy.
KEYWORDS: Cytotoxic drugs, dendritic cell vaccination, myeloid-derived suppressor cells, pancreatic cancer, tumor exosomes
Abbreviations
- AGS
ATRA+GEM+Sun
- APC
antigen-presenting cells
- ATRA
all-transretinoic acid
- BMC
bone marrow cells
- coll
collagen
- CTL
cytotoxic T cells
- DC
dendritic cells
- EpC
EpCAM
- FCS
fetal calf serum
- FN
fibronectin
- GEM
Gemcitabine
- GSH
glutathione
- Imc
immature myeloid cells
- i.v.
intravenous
- LN
laminin
- LNC
lymph node cells
- MDSC
myeloid-derived suppressor cells
- MΦ
macrophage
- o.t.
orthotopic
- OPN
osteopontin
- PaCa
pancreatic adenocarcinoma
- PBL
peripheral blood leukocytes
- PSC
pancreatic stellate cells
- ROS
reactive oxygen species
- s.c.
subcutaneous
- SC
spleen cells
- SMA
smooth muscle actin
- Sun
Sunitinib
- TAF
tumor-associated fibroblasts
- TAM
tumor-associated MΦ
- TB
tumor bearer
- TEX
tumor exosomes
- Th
helper T cells
- ThbSp
thrombospondin
- TIL
tumor-infiltrating leukocytes
- Treg
regulatory T cells
- UNKC
UNKC6141 cells.
Introduction
Pancreatic cancer (PaCa) holds the highest mortality rate,1 which is due to late diagnosis and early metastatic spread prohibiting resection, surgery being so far the only curative option.2 This is particularly alarming as PaCa incidence is raising in the Western world, expected to become the second leading cause of cancer in 2030.3 Thus, optimizing adjuvant therapies is urgent.
In the past, chemotherapy, radiation4,5 and immunotherapeutic approaches were of limited success,6,7 despite evidence for PaCa carrying immunogenic entities.8 Poor therapeutic efficacy is in part due to the strong stroma reaction in PaCa,8 which supports PaCa growth and progression, presents a barrier for cytotoxic drugs and immune effector cells and creates an immunosuppressive milieu.10-12 Major changes in the stroma of the pancreatic gland versus the dysplastic stroma in PaCa rely on upregulation of matrix proteins, MMPs, osteopontin (OPN) and thrombospondin (ThbSp),13-16 which promote survival, chemoresistance, matrix degradation-supported migration, invasion and angiogenesis. Tumor progression is assisted by the recruitment of mostly immunosuppressive inflammatory cells, like tumor associated macrophages (TAM), myeloid-derived suppressor cells (MDSC) and regulatory T cells (Treg) recruited via GM-CSF and CCR2 ligands.17 These factors are provided by fibroblasts of the PaCa stroma, a heterogeneous population, which includes myofibroblasts from the bone marrow (BM) and, as the functional dominating population, activated pancreatic stellate cells (PSC).18 PSC become activated via IL6, TGFβ, PDGF, VEGF and other cytokines provided by tumor cells and stromal elements, whereby a self-perpetuing circle between the tumor and PSC is created.18,19 Though the interplay between PaCa and the stroma is not fully elucidated, it is obvious that the stroma via recruiting immunosuppressive cells and/or by reprogramming immune cells toward a suppressive phenotype contributes to the limited success of immunotherapeutic trials including vaccination with DC.20-22 Taking this into account, most current clinical trials as well as animal models combine immunotherapy with cytotoxic drugs23-28 and are additionally concerned about attacking MDSC, the prominent immunosuppressive entity in PaCa.
MDSC are immature myeloid cells (iMC).29 In PaCa their expansion is promoted by SCF, GM-CSF, TNFα, IL1β, IL6 and VEGF provided by the tumor and stromal elements. Downstream activation of JAK-STAT3/STAT5 pathway with stimulation of cyclinD1, BclXl, survivin, c-myc and S100A8/A9 contribute to inhibition of differentiation into mature myeloid cells. The PaCa stroma adds to MDSC recruitment by provision of CCL2, CXCL12 and CXCL15, the corresponding ligands being expressed by MDSC. IL1β, IL4/IL13, TLR ligands, IFNγ and PGE2 provided by the tumor stroma and T cells account for activation of STAT1, STAT6, MyD88 with upregulation of TGFβ, IL10, iNOS, arginase and PGE2 engaged in the immunosuppressive activity of MDSC.30-32 Within the tumor environment, MDSC interfere with native and adaptive immune responses. Arginase 1 and iNOS account for depletion of L-arginine in T cells, reactive oxygen species (ROS) leads to transient loss of the TCRζ chain, TGFβ and IL10 account for downregulation of IFNγ. In addition, Treg become recruited by CXCL10, IL10 and TGFβ fostering their expansion.31
With increasing awareness of the PaCa stroma recruiting MDSC and boosting their detrimental activities, immunotherapeutic trials concomitantly take care of MDSC elimination or inactivation. One of the first drugs described to interfere with MDSC is all-transretinoic acid (ATRA), which drives iMC into differentiation toward macrophages (MΦ) and DC, upregulation of glutathione (GSH) and reduction of ROS playing an important role.33 However, ATRA also induces Treg by upregulating FoxP3 expression.34 Gemcitabine (GEM), the most frequent adjuvant drug in PaCa treatment was described to induce MDSC apoptosis and necrosis,35,36 which advised GEM for combining with immunotherapy.28 The tyrosine kinase inhibitor Sunitinib (Sun), also used in adjuvant PaCa therapy,37 provides an additional drug to interfere with MDSC by inhibiting STAT3, c-Kit and VEGFR activation and additionally decreasing Treg.36,38,39
A promising option in cancer immunotherapy is active vaccination with autologous DC, engineered or loaded to present tumor-associated peptides,40 an approach also recommended for PaCa.41,42 We recently elaborated in a murine myeloid leukemia and a renal cell cancer the superiority of loading DC with tumor-derived exosomes (TEX),43 which has several advantages: (i) there is strong evidence that TEX carry the majority of tumor-associated antigens;44-46 (ii) uptaken TEX are preferentially recruited into the MHCII-loading compartment,43,47 which implies a minor loss by degradation in lysosomes and enriched presentation in newly delivered MHCII molecules with preferential CD4+ helper T cell activation;47,48 (iii) DC express LFA1 at a high level, which is a major ligand for exosomes.49 The high therapeutic efficacy of DC-TEX vaccination demanded for a control in devasting PaCa, particularly when embedded in the tumor and host stroma, which opposes drugs and immune effector cells. We focused on coping with MDSC, elaborating whether driving into differentiation, blocking activation or attacking mature MDSC or combinations thereof may increase the therapeutic efficacy of DC-TEX vaccination. We confirmed the efficacy of DC-TEX vaccination in PaCa after s.c. application. Weaker therapeutic efficacy upon intrapancreatic growth was compensated by concomitantly attacking MDSC. Blocking both MDSC maturation and activation further strengthened the therapeutic efficacy of DC-TEX vaccination.
Results
Immunotherapy is an efficient adjuvant treatment in many tumor entities. However, in PaCa it is hampered by the strong immunosuppressive features of this tumor. Therefore, we explored in a murine PaCa model, whether vaccination with TEX-loaded DC shows any therapeutic efficacy and whether this can be strengthened by concomitantly attacking MDSC.
DC loading with PaCa-derived exosomes and DC distribution in the tumor-bearing host
UNKC6141 (UNKC) is a PaCa line derived from a Kras(G12D);Pdx1-Cre mouse.50 Aiming to vaccinate UNKC-bearing mice with TEX-loaded DC (DC-TEX), TEX and BMC-derived DC-TEX and for comparison BMC-derived MDSC were characterized. In addition, the most prominent targets and in vivo homing features were defined.
Culture supernatant-derived TEX, which were used for DC-loading, express PaCIC markers (CD44v6, c-MET, EpCAM, CD104, CD49f, CD184, Tspan8, CD133, CD24, ALDH1/2), several common tumor markers (S100A4, CD138, CD90, TGFβ1, MAGE9, Kras, ThbSp, HSP70) and constitutive exosome markers including tetraspanins (Fig. S1A). DC-TEX express CD11c, MHCI, MHCII at high, CD40, CD80 and CD86 at medium level. MDSC mostly express CD11b, Ly6C and Ly6G. DC-TEX express the homing receptors CCR6, CCR7 and CXCR4 at medium level. However, the chemokine receptors CCR6 and CXCR4 are also expressed by MDSC (Fig. S1B).
Coculture of biotinylated DC-TEX with LNC, SC and BMC from UNKC-bearing mice revealed that biotin from DC was only transferred to T cells, preferentially activated (CD69+) T cells and progenitor cells, but not NK cells, MΦ or B cells. Instead, biotin from MDSC, in vitro generated or isolated from UNKC-bearing SC, was recovered in all major leukocyte subpopulations and also in progenitor cells (Fig. S1C). CFSE-labeled DC-TEX or BM-derived MDSC (1 × 107), i.v. injected in naive, UNKC-bearing and UNKC-bearing DC-TEX vaccinated mice were recovered after 48 h in LN, spleen, BM, the intrapancreatic tumor and the lung. DC-TEX recovery in LN, spleen, tumor and lung was higher in vaccinated than non-vaccinated mice, but recovery in LN, spleen and BM was reduced compared with naive mice. In line with homing receptor expression, MDSC were also recovered in lymphoid organs, the tumor and the lung, recovery mostly exceeding that of DC-TEX. Distinct to DC-TEX, recovery of MDSC was increased in the TB spleen and BM. Despite vaccination, recovery of MDSC also increased in tumor and lung (Fig. S1D).
Taken together, the recovery of tumor markers on TEX suggests TEX to be a suited antigenic entity for DC-loading. DC-TEX preferentially interact with activated (CD69+) T cells and home into LN, BM and in vaccinated mice more efficiently in tumor and lung. However, homing of MDSC in tumor and lung is also promoted by vaccination. Thus, the question arose, whether DC vaccination surmises suppressive elements in PaCa.
Vaccination with DC-TEX prolongs the survival time and affects UNKC dissemination
In view of the immunosuppressive and tumor growth promoting features of the TB pancreatic stroma, we first controlled for the efficacy of DC-TEX vaccination after s.c. injection of 1 × 106 UNKC. Starting after 1 d, mice received weekly i.v. injections of 2 × 106 DC-TEX. Vaccination delayed the onset of tumor growth and the survival time was significantly prolonged in DC-TEX treated mice (Fig. 1A). Furthermore, vaccination affected UNKC dissemination, particularly recovery in the PB being strongly reduced (Fig. 1B).
Figure 1.
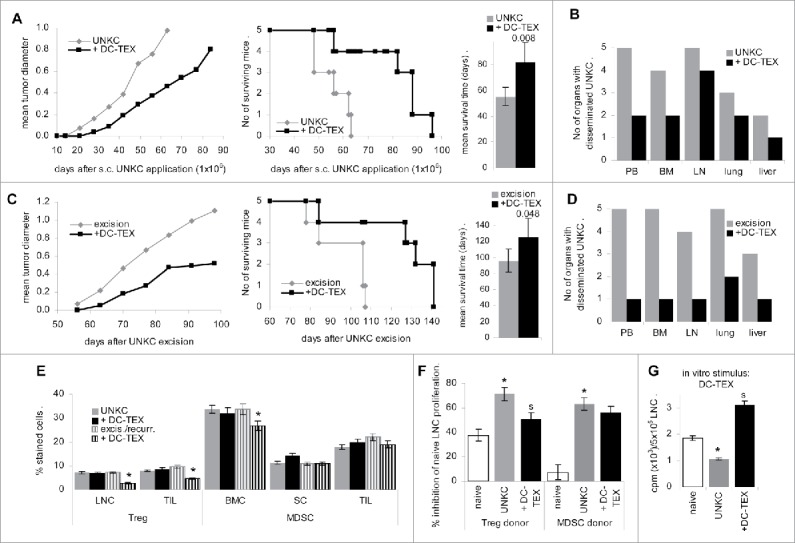
Prolonged survival time by DC-TEX vaccination after subcutaneous tumor cells application. (A, B) C57BL6 mice (5/group) received an s.c. injection of 1 × 106 UNKC and weekly i.v. injections of 2 × 106 DC-TEX. (A) Tumor growth rate, survival time and mean ± SD survival time; significant differences between untreated and vaccinated mice are shown. (B) At autopsy, dispersed cells of the indicated organs were maintained in culture for up to 6 week to survey tumor cell growth. The number of organs with disseminated tumor cells are indicated. (C, D) C57BL6 mice (5/group) received a s.c. injection of 1 × 106 UNKC. Tumors were excised reaching a mean diameter of 0.5–0.8 cm. Starting at the day of excision, mice received weekly i.v. injections of 2 × 106 DC-TEX. (C) Local recurrence, survival time and mean ± SD survival time after excision of the primary tumor; significant differences in the survival time between untreated and vaccinated mice are shown. (D) At autopsy, dispersed cells of the indicated organs were maintained in culture for up to 6 week to survey tumor cell growth. The number of organs with disseminated UNKC are indicated. (E) LNC and TIL were stained with anti-CD4+, -CD25 and -FoxP3 (Treg); BMC, SC and TIL were stained with anti-CD11b and -Gr1 (MDSC); the mean percent ± SD of Treg and MDSC in 3 mice/group is shown; significant differences between untreated and DC-TEX-treated UNKC-bearer: *. (F) Treg and MDSC were enriched by magnetic bead sorting of LNC (Treg) or SC (MDSC). LNC of naive mice were cocultured with Treg or MDSC at a 1:1 ratio for 72 h in the presence of 20 U/mL IL2, adding 3H-thymidine during the last 16 h of culture. The percent inhibition compared with LNC cultured in the absence of Treg or MDSC is shown (mean ± SD of triplicates); differences to Treg or MDSC from naive mice: *; differences between untreated vs. vaccinated UNKC-bearing mice: s. (G) LNC (5 × 105) of naive and untreated or vaccinated UNKC-bearing mice were cultured for 72 h in the presence of 1 × 105 DC-TEX. During the last 16 h, 10 µCi/mL 3H-thymidine were added; 3H-thymidine uptake (counts ± SD of triplicates/5 × 105 LNC after subtracting 3H-thymidine uptake by DC); differences to LNC from naive mice: *; differences between untreated vs. vaccinated UNKC-bearing mice: s. DC-TEX vaccination suffices for prolonged survival of UNKC-bearing mice and a reduction of tumor cell dissemination. The latter becomes stronger, when vaccination was started immediately after excision. Treg and MDSC of TB mice exhibited stronger inhibitory potential than Treg and MDSC of naive mice. Notably, Treg expansion was only reduced, when vaccination started after excision.
As DC vaccination exerted some therapeutic efficacy, the experiment was repeated starting DC-TEX treatment after excision of the primary tumor 5 week after s.c. application. All animals developed local recurrence, which was significantly delayed by DC-TEX treatment (Fig. 1C). However, whereas tumor cells were recovered in PB, BM and lung of all untreated mice, tumor cells were recovered in PB, BM, LN and liver of only 1 of 5 mice and in the lung of 2 of 5 mice (Fig. 1D), which indicated that vaccination was efficient in eradicating UNKC that had already settled in distant organs.
DC-TEX vaccination supported T cell activation. In the UNKC-bearing mice the percent of CD4+ LNC was strongly decreased. Vaccination rescued CD4+ LNC and was accompanied by upregulation of CD154+ and CD40+ LNC, but did not correct CD25 upregulation in LNC of UNKC-bearing mice. These findings also accounted for TIL (Fig. S2A and B) and fitted to a higher percent of Treg (CD4+CD25+FoxP3+) in LNC and TIL of UNKC-bearing mice irrespective of vaccination, although a decrease was noted, when mice were vaccinated after excision of the primary tumor (Fig. 1E). Increased inflammatory, immunosuppressive and immunostimulatory cytokine expression in LNC and TIL of UNKC-bearing mice was not strongly affected by DC-TEX vaccination (Fig. S2C and D) and might support MDSC activation. The latter are high in the TB BM and spleen. Only in the BM a decrease was seen in mice vaccinated after tumor excision (Fig. 1E). In addition, Treg and MDSC of TB mice exhibited a strong proliferation inhibitory potential, only Treg activity being mitigated in vaccinated mice (Fig. 1F). Nonetheless, vaccination strongly increased the proliferative activity of LNC from TB (Fig. 1G).
The findings confirm immune response induction by DC-TEX vaccination accompanied by a strong reduction of migrating and metastasizing UNKC. However, vaccination did not efficiently cope with immunosuppression, as revealed by MDSC expansion in SC and TIL. This demanded for a supportive MDSC attacking regimen.
The immunosuppressive milieu generated by orthotopic tumor growth
In advance of trying to cope with MDSC activity, the impact of intrapancreatic UNKC growth on the pancreatic stroma was evaluated, PaCa being known to induce a strong stroma reaction supporting tumor growth and immunosuppression, at least partly, via MDSC.
UNKC cells grew more rapidly in the pancreatic environment and reached a mean diameter of 0.8 cm already 21 d after application (data not shown). Compared to the naive pancreas, the matrix of the tumor-loaded pancreas showed a higher density of collagen (coll) I, coll IV, laminin (LN)111 and LN332, vessel density and VEGFR2 expression were significantly increased and lymphatic vessels (VEGFR3, Lyve) were strongly upregulated. Very few CD4+, CD8+ and GITR+ cells were recovered in the healthy pancreas. CD4+ and CD8+ cells were increased in the TB pancreas, but T cells were mostly recovered at the tumor rim and in close proximity to vessels, whereas strongly increased MDSC were spread through the tumor tissue. NK cells were recovered in the naive and TB pancreas (Fig. 2A). Flow cytometry of a small and less granulated population (leukocyte-enriched) in the dispersed naive and TB pancreas revealed no change in the relative percent of T cells, a decrease in CD154+ T cells and an increase in Treg (CD4+CD25+FoxP3+), CD11b+ and MDSC (CD11b+Gr1+) in the TB pancreas (Fig. 2B). The inflammatory cytokines TNFα, IL1β and IL6 and the immunosuppressive cytokines IL4 and IL10, but also IFNγ as well as several chemokines engaged in MDSC maturation and activation, like GM-CSF, CCL2, CCL4, CCL5, CCL25, bFGF, VEGF, OPN and LIF (Fig. 2C), the latter being involved in fibroblast activation, are upregulated in the TB pancreas. This also accounted for the death receptors CD120a, CD120b, CD253 and CD254. Accordingly, the number of apoptotic TIL slightly exceeded that of intrapancreatic leukocytes (Fig. 2D). Finally, already at 3 week after o.t. application tumor cells were recovered from the peritoneal cavity and the lung of all three mice, from liver and BM of two mice and from blood and spleen of one mouse (Fig. 2E).
Figure 2.
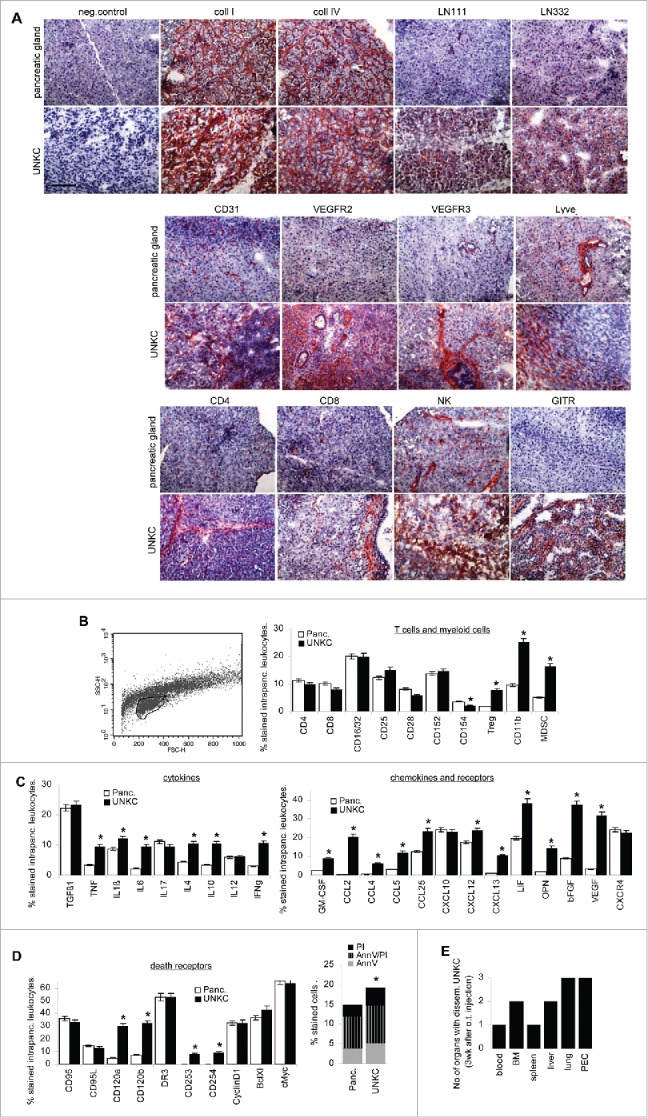
The impact of UNKC on the pancreas stroma. C57BL6 mice received an intrapancreatic (o.t.) injection of 5 × 105 UNKC. Mice were killed after 3 week with a small palpable tumor mass in the upper abdomen. (A) The cancerous pancreatic gland and the pancreatic gland of healthy mice was excised, shock frozen and sections were stained with the indicated antibodies. (B–D) Flow-cytometry analysis of pancreas resident leukocytes and TIL (encircled population in the forward/sideward scatter) after dispersion of pancreas and tumor tissue of (B) leukocyte markers, Treg (CD4+CD25+FoxP3+), MDSC (CD11b+Gr1+), (C) cytokines, chemokines, chemokine receptors, (D) death receptors and apoptotic cells. (B–D) Mean percent ± SD (3 mice/group) of stained cells; significant differences between intrapancreatic leukocytes and TIL: *. (E) Recovery of disseminated UNKC cells in tissue cultures from organs collected at autopsy. Matrix proteins, particularly LN332 are more abundant in the UNKC-bearing pancreas. Vessel density is increased and only in the cancerous pancreas lymphatic vessels are abundant. There was a slight increase in infiltrating T cells and a strong increase in Treg and MDSC. Inflammatory and immunosuppressive cytokine expression is mostly increased in TIL. Furthermore, MDSC recruiting and angiogenesis promoting chemokines are strongly upregulated. The percent of apoptotic leukocytes is slightly increased, but only TNFRI and TNFRII expression is strongly upregulated. Notably, UNKC were recovered in 3/3 mice in the peritoneal cavity and the lung already 3 week after o.t. application.
In brief, intrapancreatic UNKC growth induces a strong stroma reaction with enrichment of suppressor cells, suppressive factors and chemokines attracting MDSC, the immunosuppressive elements being stronger than in mice carrying s.c. tumors.
Coping with MDSC expansion, activation and recruitment
To cope with immunosuppression by MDSC, we evaluated the efficacy of drugs described to interfere with MDSC expansion, activation and recruitment. C57BL6 mice received an o.t. injection of UNKC and were concomitantly treated with ATRA, interfering with MDSC maturation,33 or Sun, hampering MDSC differentiation,36 or GEM, which drives MDSC into apoptosis.36 Mice concomitantly received DC-TEX, which allowed controlling for a negative impact of the cytotoxic drugs on T cells. The impact of the drugs on MDSC was evaluated in BMC, SC and TIL, the impact on T cell activation was controlled in draining LNC and TIL.
Factors known to drive myelopoiesis and to inhibit differentiation like SCF, GM-CSF, VEGF and IL6 and expression of the corresponding receptors were not strongly affected in the BM and tumor tissue of ATRA-treated mice. This also accounted for TLR2 and TLR4 and downstream signaling molecule MyD88 (data not shown). However, a reduction of NFκB was seen in BMC and of iNOS and IL10 in BMC, SC and TIL of ATRA-treated mice (Fig. S3A). It is noteworthy that ATRA did not affect chemokine expression in BMC and TIL, including CCL2 and CXCL12, particularly engaged in MDSC recruitment. Only T cell recruiting CXCL10 expression was slightly affected, indicating that ATRA hardly interfered with MDSC and T cell recruitment into the tumor (data not shown).
The impact of ATRA on T cells differed between LNC and TIL. In LNC ATRA supported CD8+ expansion and accessory molecule expression. In TIL, CD25 and CD28 expression was reduced, but rescued upon concomitant vaccination, which also boosted CD152+ and CD154+ T cells. In LNC, ATRA treatment was accompanied by a reduction in inflammatory, immunosuppressive and immunostimulatory cytokines, only IFNγ being rescued upon vaccination. The latter also accounted for TIL, the impact on cytokines delivered by TIL being less pronounced (Fig. S3B). Immunohistochemistry confirmed enrichment of CD4+ and CD8+ TIL, but free distribution into the tumor tissue was still hampered. Immunohistochemistry also indicated a strong reduction in MDSC (GITR+, Ly6C+) (Fig. S3C). Flow-cytometry reassured a significant reduction of CD11b+Gr1+ TIL, BMC and SC, which was maintained in concomitantly vaccinated mice. An increase in Treg was only seen in LNC and was waved upon concomitant vaccination (Fig. 3A).
Figure 3.
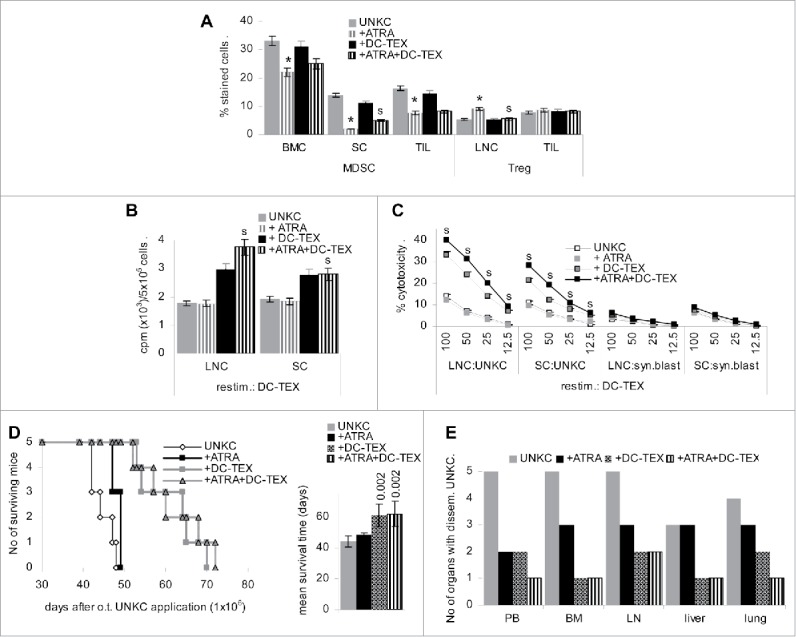
ATRA treatment strengthens the efficacy of DC-TEX vaccination: C57BL6 mice with o.t. injected (1 × 106) UNKC were treated with a s.c. depot of ATRA (5 mg), which was repeated after 3 week, and/or weekly i.v. applications of DC-TEX (2 × 106). (A) Flow-cytometry analysis of MDSC in BMC, SC and TIL and Treg in LNC and TIL of ATRA-treated and/or vaccinated mice; mean values ± SD of 3 mice/group; significant differences between untreated and ATRA-treated: *; significant differences between ATRA-treated vs. ATRA-treated plus vaccinated mice: s. (B) LNC and SC (5 × 105) were cultured for 3 d in the presence of 5 × 104 DC-TEX, adding 3H thymidine during the last 16 h of culture; mean cpm ± SD (triplicates)/5 × 105 cells after subtraction of values of DC-TEX; (C) LNC and SC were (re)stimulated in vitro for 10 d in the presence of 5 × 104 DC-TEX. Cells were harvested and seeded on 1 × 104 3H-thymidine-labeled UNKC or syngeneic blasts and incubated for 8 h; the % cytotoxicity ± SD (triplicates) is shown; (B,C) significant differences by ATRA treatment: *, significant differences between ATRA vs. ATRA plus DC-TEX treatment: s. (D) The mean survival time of the four groups of mice (5 mice/group) is shown; significant differences to untreated UNKC-bearers and the differences between ATRA vs. ATRA plus DC-TEX treated mice are indicated. (E) Recovery of disseminated tumor cells was evaluated by culturing dispersed organs collected at autopsy. The number of MDSC is reduced in ATRA-treated mice, but in LN the number of Treg is increased. ATRA does not affect proliferation or cytotoxicity, but strengthens proliferation and cytotoxic activity of concomitantly vaccinated mice. Nonetheless, ATRA and ATRA plus DC-TEX treatment does not suffice to significantly prolong survival.
ATRA treatment supported the proliferative activity of LNC and SC in response to DC-TEX, the proliferation of LNC from ATRA-treated plus DC-TEX vaccinated mice exceeding that of vaccinated mice (Fig. 3B). CTL activity of LNC and SC of ATRA-treated and concomitantly vaccinated mice, evaluated after in vitro (re)stimulation with DC-TEX, was significantly increased compared with that of LNC and SC from non-vaccinated or vaccinated, but not ATRA-treated mice (Fig. 3C). However, ATRA treatment did not suffice to prolong the survival time of UNKC-bearing mice and did not exert an additive effect on DC-TEX vaccination, the latter being less efficient than in mice bearing an s.c. tumor (Fig. 3D). It affected UNKC migration (recovery in the blood), settlement in BM, LN and weakly lung, the effect remaining below that of DC-TEX vaccination (Fig. 3E).
Taken together, ATRA drives myeloid progenitor cells into differentiation with reduced recovery of MDSC in BM, spleen and TIL. This is accompanied by an improved T cell response in DC-TEX vaccinated mice. Despite these promising effects, ATRA treatment concomitantly with vaccination did not prolong the survival time and had a weak impact on tumor cell settlement in distant organs.
The tyrosine kinase inhibitor Sun interferes with myeloid progenitor differentiation into MDSC. Flow-cytometry analysis of BMC-derived MDSC cultured in the presence of Sun revealed an increase in CD45+ cells, a decrease in CD11b+, Gr1+ and GITR+ cells and an increase in CD11c+, CD40+, CD80+, CD86+ and MHCII+ cells. These changes were accompanied by reduced GMCSF and TNFRI expression and a reduction in NFκB, cyclinD1, BclXl, cMyc, iNOS and MMP9 expressing MDSC (Fig. 4A). Despite a decrease in CD11b+Gr1+ cells, accompanied by an increase in Gr1+ cells, there was a slight increase in apoptotic cells in cultures containing Sun (Fig. 4B). When culture-derived MDSC were cocultured with SC of naive mice in the presence of IL2, the proliferation inhibitory potential of MDSC matured in the presence of Sun was reduced (Fig. 4C).
Figure 4.
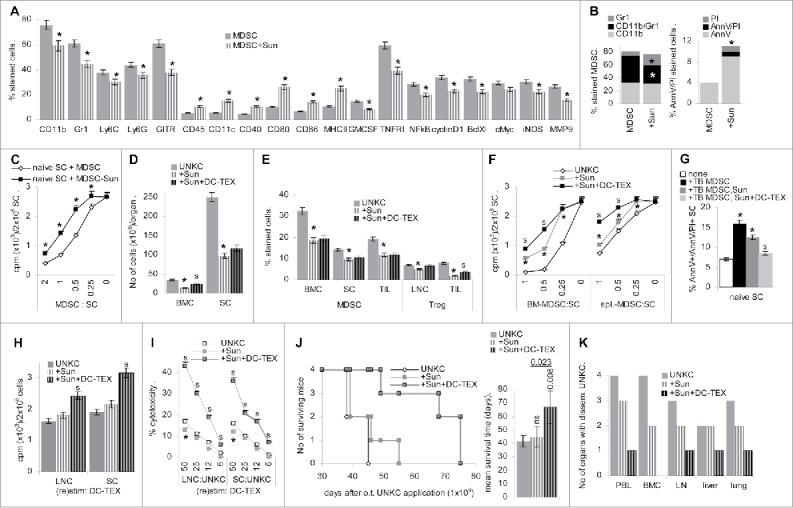
Sunitinib interferes with MDSC generation. (A–C) BMC were cultured in the presence of GM-CSF, IL6 and PGE2, where indicated cultures contained 20 µg/mL Sun. Flow-cytometry analysis of (A) MDSC marker and signaling molecules engaged in MDSC activity, (B) CD11b+/Gr1+ and apoptotic (AnnV/PtdIns stained) cells; (A,B) mean ± SD of three assays are shown, significant differences in the presence of Sun: * (C) MDSC were generated in the absence or presence of Sun as indicated above. CD11b+Gr1+ MDSC were separated and cocultured with naive SC for 3 d in the presence of 100 U/mL IL2, adding 3H-thymidine during the last 16 h of culture; mean cpm ± SD (triplicates)/2 × 105 SC after subtraction of counts of MDSC are shown; significant differences in the suppressive activity of purified MDSC generated in the presence of Sun: s. (D–K) C57BL6 mice received an o.t. injection of 1 × 106 UNKC and weekly Sun injections (20 µg/g, i.v.) and with 3 d delay i.v. injections of 2 × 106 DC-TEX. (D–I) Mice were killed after 4 week. (D) No of cells recovered in BM and spleen. (E) Flow-cytometry analysis of MDSC (CD11b+Gr1+) in BM, SC and TIL and of Treg (CD4+CD25+FoxP3+) in LNC and TIL; (D,E) mean percent cells ± SD (3 mice/group); (F) titrated number of MDSC (CD11b+Gr1+) isolated from BM and spleen were cocultured with naive SC. Cultures were maintained for 3 d in the presence of 100 U/mL IL2. Proliferation was evaluated after adding 10 µCi 3H-thymidine during the last 16 h of culture; mean cpm ± SD (triplicates); (G) MDSC isolated from SC were cocultured for 3 d in the presence of 100 U/mL IL2 with naive SC. The mean percent ± SD of apoptotic cells was evaluated by flow-cytometry; (H) proliferative activity of LNC and SC was evaluated after 3 d of culture in the presence of DC-TEX (5 × 104), adding 10 µCi 3H-thymidine during the last 16 h of culture. Mean cpm ± SD (triplicates)/2 × 105 cells; (I) LNC and SC were (re)stimulated in vitro for 10 d with 5 × 104 DC-TEX. Cells were harvested and seeded on 1 × 104 3H-thymidine labeled UNKC. Cytotoxicity was determined after 8 h incubation; the % cytotoxicity ± SD (triplicates) is shown; (D–I) significant differences by Sun-treatment: *, significant differences between Sun- vs. Sun- plus DC-TEX-treatment: s. (J) Survival time and mean ± SD survival time of the three groups of mice (4 mice/group) is shown; significant differences to untreated UNKC-bearers and between Sun- vs. Sun- plus DC-TEX-treated mice is indicated. (K) Recovery of disseminated tumor cells was evaluated by culturing dispersed organs collected at autopsy. In vitro and in vivo Sun-treatment is accompanied by a significant reduction in the generation of MDSC, which in part relies on apoptosis induction. Furthermore, MDSC generated in the presence of Sun exhibit reduced suppressive activity. Sun does not affect proliferative and cytotoxic activity of surviving lymphocytes. It does not prolong the survival time, but supports the impact of DC-TEX. This also accounts for the hindrance of metastatic settlement.
Similar effects were observed treating TB mice with Sun, which was accompanied by reduced BMC and SC recovery (Fig. 4D). BMC, SC and TIL contained fewer CD11b+Gr1+ cells and in LNC and TIL Treg were reduced, but partly regained in TIL of concomitantly vaccinated mice (Fig. 4E). Isolated CD11b+Gr1+ cells from BM and spleen of Sun-treated TB mice poorly suppressed naive LNC proliferation, the suppressive capacity being further reduced, when MDSC were isolated from vaccinated mice. Notably, the impact of Sun was stronger on BM- than spleen-derived MDSC (Fig. 4F). Finally, ∼10% of naive SC were driven into apoptosis, when cocultured with MDSC from BM of TB mice. MDSC of Sun treated mice displayed lower cytotoxic activity, the percentage of AnnV+/AnnV+/PI+ SC being further decreased with MDSC from Sun-treated, vaccinated mice (Fig. 4G).
Analyzing freshly harvested LNC and TIL of Sun-treated TB mice revealed a reduction in CD4+ cells, which was compensated in LNC, but not TIL of vaccinated mice, where CD8+ T cells became enriched. Reduced accessory molecule expression in Sun-treated mice was restored by vaccination and upregulation of CD152+ TIL by Sun was compensated (Fig. S4A). With the exception of IL12 and IFNγ, cytokine expression is reduced in LNC, but not TIL of Sun-treated mice, but is rescued in concomitantly vaccinated mice (data not shown). Immunohistology revealed large necrotic areas in the tumor. In the alive tumor tissue, despite low recovery, CD4+ cells were well distributed and CD8+ cells were enriched in the tumor tissue of Sun-treated mice. Furthermore, GITR+, but not CD11b+ cells were reduced, indicating that recruitment and/or differentiation toward MΦ was not affected by Sun (Fig. S4B).
Proliferation of LNC and SC from TB mice in response to DC-TEX was not affected by Sun-treatment, but was significantly strengthened in concomitantly vaccinated mice (Fig. 4H). Sun treatment hardly affected cytotoxic activity and Sun-treated, DC-TEX vaccinated mice displayed high cytotoxic activity toward UNKC (Fig. 4I).
Thus, Sun drives BMC into differentiation and additionally promotes apoptosis of MDSC and T cells. MDSC escaping Sun have weakened suppressive activity, but surviving lymphocytes strongly respond to DC vaccination. Accordingly, immune response induction by DC-TEX is strengthened in Sun-treated mice, which appeared promising for prolonged survival. Indeed, though Sun-treatment did not prolong the survival time of UNKC-bearing mice, the survival time of concomitantly vaccinated mice was significantly increased (Fig. 4J). Recovery of migrating UNKC was less severely affected by Sun than ATRA (Fig. 4K). We interpret these findings that Sun efficiently hampers MDSC activation. Accordingly, DC-TEX vaccination gains in efficacy. There remains the drawback of an overall strong reduction of leukocytes including T cells and, possibly as a consequence, the low efficacy of Sun in preventing metastatic spread.
GEM, frequently used in adjuvant PaCa therapy, might drive MDSC into apoptosis.
In vitro treatment of LNC and BMC with 30 nM GEM induced a minor increase in apoptotic cells. Ex vivo analysis of LNC, BMC and TIL of UNKC-bearing, GEM-treated mice indicated rather apoptosis protection in TIL and LNC and no increase in apoptotic BMC (Fig. 5A). CD4+ and CD8+ TIL and CD11b+ and CD11b+Gr1+ BMC were reduced in GEM-treated mice. However, only few GEM-treatment surviving cells were apoptotic (Fig. 5B). In vivo GEM-treatment impaired activation of STAT6 and the transcription factors HIF1α and NFκB the latter becoming activated via TLR and MyD88. Reduced activation was accompanied by impaired IL1β, IFNγ, cyclinD1, BclXl, survivin and iNOS expression in MDSC from GEM-treated mice (Fig. 5C).
Figure 5.

The impact of Gemcitabine on MDSC activation, apoptosis induction and vaccination. (A) Flow-cytometry analysis of apoptosis (AnnV/PtdIns staining) in LNC and BMC cultured for 48 h in the presence of GEM and in freshly harvested LNC, BMC and TIL of mice 4 week after o.t. UNKC (5 × 106) application. (B) Flow-cytometry analysis of leukocyte subpopulations including apoptotic cells from GEM-treated TB mice. (C) Flow-cytometry analysis of transcription factor and related gene expression in CD11b+Gr1+ SC from naive and GEM-treated mice; (A–C) the percent( ± SD) of stained cells; significant differences by GEM treatment: *. (D–F) Mice received an o.t. UNKC (1 × 106) injection, weekly i.v. injections of GEM and with 3 d delay weekly i.v. DC-TEX (2 × 106) injections. (D) Mice were killed after 4 week. LNC and SC were (re)stimulated for 7 d with DC-TEX. LNC and SC were seeded on 3H-Thymidine-labeled UNKC at the indicated ratios. Cytotoxic activity was evaluated after 8 h coculture. The mean % cytotoxicity ± SD (triplicates) is shown; significant differences by GEM treatment:*, significant differences between GEM vs. GEM plus DC-TEX treatment: s. (E) Survival time and mean ± SD survival time (5 mice/group) is shown; significant differences to untreated UNKC-bearers and between GEM- vs. GEM- plus DC-TEX-treated mice is indicated. (F) Recovery of disseminated tumor cells was evaluated by culturing dispersed organs collected at autopsy. In in vitro cultures, GEM promotes apoptosis, the effect being not seen ex vivo after 4 week GEM treatment. Instead, the impact of GEM on transcription factor and related gene expression in MDSC is more pronounced after in vivo treatment. GEM treatment strengthens the cytotoxic potential of surviving leukocytes, which is reflected by prolonged survival. In combination with DC-TEX vaccination the survival time becomes further prolonged and metastatic spread is nearly abolished.
CD4+, CD8+, CD28+ and CD154+ TIL were reduced in GEM-treated mice, but rescued in concomitantly vaccinated mice. GEM-treatment did not affect CD4+ and CD8+ LNC. The T cell activation-support by concomitant vaccination was similar to that in TIL (Fig. S5A). Neither in the draining node nor in TIL, GEM sufficed for a reduction in Treg (Fig. S5B). Inflammatory, immunosuppressive and immunostimulatory cytokine recovery in TIL of GEM-treated mice was mostly reduced, but was rescued in vaccinated mice (data not shown). GEM hardly affected MDSC- and T cell-recruiting chemokine expression in TIL, which was increased in concomitantly vaccinated mice (Fig. S5C). The impact of GEM on cytokine and chemokine expression in LNC was weak (data not shown). Immunohistology of tumor sections confirmed the increase in activated T cells, a clustered enrichment of CXCR4+ cells and a reduction in GITR+ cells in GEM-treated, vaccinated mice. High recovery of CD95L+ TIL in GEM-treated vaccinated mice could indicate pronounced cell death (Fig. S5D).
GEM treatment supported the cytotoxic activity of LNC and SC and enhanced the stimulation by DC-TEX vaccination (Fig. 5D). Distinct to ATRA and Sun, GEM treatment sufficed for a significant prolongation of the survival time and the survival time of concomitantly vaccinated mice was further prolonged, the prolongation not reaching statistical significance (Fig. 5E). GEM treatment had a minor impact on tumor cell migration and settlement in liver and lung, but strengthened the impact of vaccination (Fig. 5F).
Although we could not demonstrate direct cytotoxic activity of GEM toward MDSC, only few activated MDSC were recovered after in vitro or in vivo GEM-treatment, which sufficed for an increase in cytotoxic activity, a prolongation of the survival time and a reduction in metastatic spread in concomitantly DC-TEX vaccinated mice.
Attacking MDSC maturation and activation strengthened the efficacy of DC-TEX vaccination
ATRA, GEM and Sun (AGS) distinctly affecting MDSC, we considered the possibility that blocking MDSC maturation, activation and survival might exert an additive effect on the therapeutic efficacy of DC-TEX vaccination.
Mice received a high dose of UNKC (5 × 106, o.t.). Starting 1 d after tumor cell application mice received weekly injections of GEM and Sun (i.v.) and an s.c. depot of ATRA and, starting 4 d after UNKC application, weekly i.v. injections of 2 × 106 DC-TEX. The immune status of the mice was controlled 3 d after the second DC-TEX application. Leukocyte recovery was strongly reduced in LN, BM and spleen (Fig. 6A). MDSC were reduced in BMC, SC and TIL and Treg were decreased in LN and TIL of AGS-treated and concomitantly vaccinated mice (Fig. 6B). Activated T cells (CD25+, CD28+, CD154+) were strongly reduced, but immunosuppressive T cells (CD152+) were overpresented in AGS-treatment surviving TIL. CD40+ TIL were also diminished, but costimulatory molecules were rescued by vaccination. AGS also affected IL1β, IL6 and IL4, but not IL12 and IFNγ expression in TIL (Fig. S6A). No major changes were seen in the very few AGS-treatment surviving LNC (data not shown). Instead, chemokine expression was affected in LNC and the tumor stroma, with an increase in SCF and CXCL12 in LNC and stroma. Reduced CCL5 expression was rescued by vaccination. CXCR4 was only upregulated in LNC and CXCL10 only in the stroma of vaccinated mice (Fig. S6B). Importantly, as revealed at autopsy, AGS-treatment also affected the stroma induction by UNKC. Immunohistology revealed a reduction in collagen, LN111 (data not shown) and, particularly, LN332 deposits; SMA+ cells, though still present, were more dispersed not forming a strong shield preventing TIL infiltration. Indeed, though T cells were rare in AGS-treated mice, in concomitantly vaccinated mice, they were well distributed throughout the tumor tissue as demonstrated for CD8+ T cells. Additionally, CD31+ and VEGFR2+ cells were reduced; lymphangiogenesis (VEGFR3, Lyve) appeared less severely affected (Fig. S6C).
Figure 6.
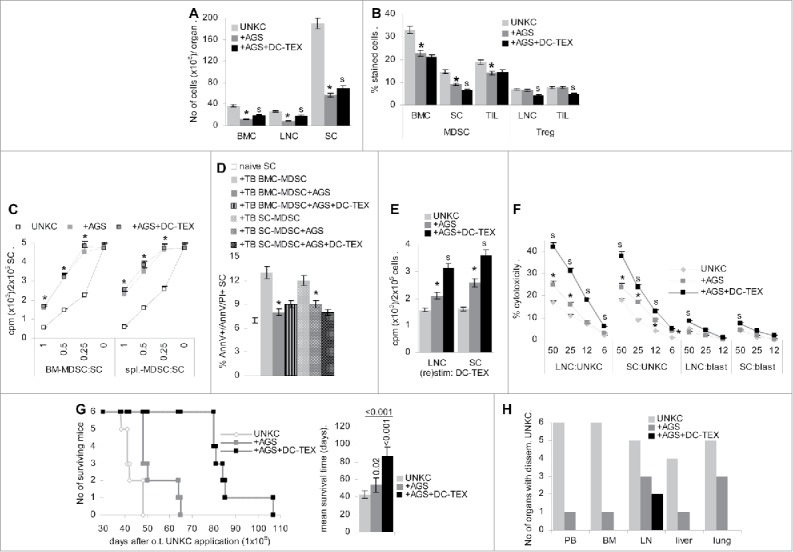
Preventing MDSC maturation and activation additively supports vaccination. (A,B) Mice received an o.t. injection of UNKC (5 × 106); after 1 d they received a s.c. pellet of ATRA and an i.v. injection of Sun and GEM. Sun and GEM treatment was repeated weekly. After 4 d and in weekly intervals mice received in i.v. injection of 2 × 106 DC-TEX. Mice were killed 3 d after the third DC-TEX vaccination. (A) No of leukocytes (mean ± SD, 3 mice/group); (B) mean percent ± SD (3 mice/group) of MDSC in BMC, SC and TIL and of Treg in LNC and TIL; (C) MDSC were isolated from BM and spleen and cocultured for 72 h with SC from naive mice in the presence of 100 U/mL IL2, adding 10 µCi 3H-thymidine during the last 16 h of culture. 3H-Thymidine incorporation (mean ± SD, triplicates) is shown; (D) MDSC isolated from BMC and SC were cocultured with naive SC for 3 d. The mean percent ± SD (triplicates) of AnnV/AnnV/PtdIns stained cells was evaluated by flow-cytometry; (E) titrated number of LN and SC were (re)stimulated in vitro with DC-TEX for 72 h, adding 10 µCi 3H-thymidine during the last 16 h of culture. 3H-Thymidine incorporation (mean ± SD, triplicates) is shown; (F) LNC and SC were (re) stimulated in vitro for 7 d with DC-TEX. Cells were seeded at the indicated ratios on 3H-thymidine-labeled UNKC or syngeneic blasts. The % cytotoxicity (mean ± SD, triplicates) was evaluated after 8 h; (A–F) significant differences compared with untreated UNKC-bearing mice: * and between AGS vs. AGS plus vaccinated mice: s. (G,H) Mice received 1 × 106 UNKC, o.t. and were treated as described above; (G) survival time and mean ± SD survival time of 6 mice/group; significant differences by AGS- or AGS- plus DC-TEX-treatment are indicated. (H) At autopsy, cells of the indicated organs were maintained in culture to survey UNKC outgrowth. The numbers of mice with disseminated UNKC are shown. AGS treatment significantly affects leukocyte survival. Nonetheless, the reduction in MDSC is dominating such that few surviving lymphocyte show increased proliferative and cytotoxic activity. However, due to a collapse of the immune system by the combined cytotoxic drugs, mice finally develop tumors despite concomitant DC-TEX vaccination, but, with the exception of draining LN, metastatic spread was prohibited.
MDSC isolated from BM and spleen of AGS-treated mice poorly suppressed proliferation of SC from naive mice, loss of suppressive activity being most pronounced in spleen-derived MDSC (Fig. 6C). Furthermore, naive SC were not driven into apoptosis in coculture with MDSC from spleen or BM of AGS-treated mice (Fig. 6D). In line with the impaired suppressive activity of MDSC, the low number of AGS-surviving LNC and SC showed increased proliferative activity toward DC-TEX and in concomitantly vaccinated mice proliferation was significantly increased (Fig. 6E). The cytotoxic activity of AGS-treatment surviving LNC and SC was slightly increased and was significantly strengthened in concomitantly DC-TEX vaccinated mice with a minor increase in autoreactivity (Fig. 6F).
AGS sufficed for a borderline significant prolongation of the survival time of mice receiving a medium high dose of UNKC (1 × 106, o.t.). Instead, no palpable tumor mass was observed in AGS-treated, DC-TEX vaccinated mice until 50 d after tumor cell application, when mice of the control group were already killed due to the tumor mass in the pancreatic gland. Nonetheless, mice additionally receiving DC-TEX started to lose weight after AGS-treatment of 60—70 d and succumbed with the tumor with a 2-fold increased mean survival time (Fig. 6G). At autopsy, BMC and SC recovery was strongly reduced and LNC were hardly recovered (data not shown), indicating exhaustion of the immune system by the persisting drug treatment as the major contributor waving the efficacy of DC vaccination. Nonetheless, tumor cells were rarely recovered in PB, BM and liver. But, in 3/6 mice UNKC grew in long-term LN and lung cultures, suggesting spread via the lymphatic route. In AGS-treated, vaccinated mice UNKC were only recovered in the draining LN of 2 mice (Fig. 6H).
Taken together driving myeloid progenitor cells into differentiation and preventing activation of remaining MDSC, though not curative by itself, most efficiently supports an active vaccination with DC-TEX. As AGS-treatment also attacks immune effector cells, a split approach between cytotoxic drugs and DC vaccination is desirable for patients with excisable PaCa, where a high cure rate becomes likely.
Discussion
Immunotherapy is a promising option in adjuvant cancer therapy as it acts systemically and is less burdened by severe side effects.51 However, immunotherapeutic trials frequently do not fulfill expectation, which counts particularly for most aggressive PaCa.6,7 Thus, life expectation prolonging combinations of adjuvant therapies become an important task. Our trial was based on the experience that TEX-loaded DC are a superior vaccine compared with peptide-loaded DC.43 In view of the immunosuppressive properties of PaCa,12 which in part are due to MDSC activation and recruitment,31 we combined DC vaccination with three chemotherapeutic drugs, ATRA, Sunitinib, Gemcitabine, described to affect different stages of MDSC activation.33,36,37 Each of the drugs hampered MDSC activity, but also affected immune cells. Only in combination with DC-TEX vaccination, survival time became prolonged and metastatic spread was strongly impaired.
Suitability of the murine model for controlling the efficacy of DC vaccination in PaCa
UNKC, spontaneously arising in a Kras(G12D) mouse,50 express human PaCIC markers,52,53 some common tumor markers, all described to contribute to PaCa progression54-58 and tumor-associated MAGE9.59 Thus, UNKC appear suited to replace human PaCa.
UNKC preferentially settle in LN, BM and lung, whereas human PaCa preferentially settle in LN, BM and liver. The metastasis profile of both, UNKC and human PaCa argues for hematogeneous and lymphatic spread with a preponderance of the latter. The high recovery rate of UNKC in the BM and the lung suggests preferential lodging and survival within a prepared premetastatic niche,60,61 where c-MET accounts for shifting hematopoiesis toward myelosuppression62,63 and the TEX tetraspanin/integrin profile accounts for preferential homing to distant organs. UNKC TEX express the Tspan8/α6β4 complex, which shows a strong affiliation to lung fibroblasts.63,64 UNKC hardly express the αV integrin (data not shown), which promotes settlement in the liver.63 This latter difference to human PaCa may account for the partly distinct metastatic organ preference. Nonetheless, early spread and the partly overlapping settlement in liver and lung confirm UNKC suited as alternative to human PaCa.
PaCa induce a strong stroma reaction that supports tumor growth and progression, shields the tumor from defense mechanisms and actively suppresses immune effector mechanisms.10-12,65 A comprehensive histological analysis of the UNKC-bearing pancreatic stroma showed abundance of ECM elements, induction of angiogenesis and lymphangiogenesis and recovery of T cells only in close vicinity to vessels. Instead, MDSC and Treg were distributed throughout the tumor and were enriched compared with the normal pancreas. Furthermore, chemokines engaged in attracting MDSC were abundantly expressed. Thus, immunotherapy of UNKC likely is burdened by an immunosuppressive environment as described for human PaCa.
Suitability of UNKC TEX loaded DC as vaccine and the likelihood of DC-TEX to reach their target
UNKC TEX express the tumor markers and several tetraspanins including CD81, which is important for antigen presentation by DC. Exosomes taken up by DC are preferentially guided to the MHCII-loading compartment with CD81 being involved in the vesicle transport.66,67 This implies that UNKC tumor markers expressed in TEX are preferentially digested in the MHCII-loading compartment and peptides are loaded into newly synthesized MHCII.43,48 Tetraspanins are also engaged in TEX binding and TEX uptake.68,69 Confirming the suitability of DC-TEX as vaccine, trogocytosis, the transfer of plasma membrane fragments70 from DC, was mostly observed in activated T cells. Some DC-TEX membrane exchange with haematopoietic progenitors was considered to be not of relevance for our vaccination protocol and was not further analyzed.
Finally, taking into account the altered stroma and the abundance of MDSC in PaCa, it became important to control for the homing of i.v. injected DC-TEX. Recovery of DC-TEX in LN is strongly reduced in the TB compared with the naive host. In the tumor tissue, recovery is low. However, recovery in LN, lung and tumor is increased after repeated DC-TEX application. The mechanisms underlying the changed homing profile remain to be defined. High level CCR6 and CXCR4 expression may support DC-TEX recruitment by CCL20 and CXCL12 in the tumor stroma.71,72 But, MDSC also preferentially home into the tumor of vaccinated mice, where CCL2, CXCL5, CXCL12 and SCF in the tumor tissue support recruitment.32,73
In brief, DC-TEX preferentially target activated T cells. Homing is impaired in the TB host, but becomes improved upon repeated vaccination possibly via high CXCR4, CCR7 and CCR6 expression. Nonetheless, in the TB pancreas MDSC exceeds DC-TEX recruitment, which demanded for dampening MDSC to assist DC-TEX vaccination.
Coping with MDSC expansion and activity in PaCa during active vaccination
From the list of drugs affecting MDSC we selected ATRA, Sun and GEM.
ATRA drives iMC into differentiation via upregulation of GSH, which reduces the level of ROS.33 From the factors known to drive iMC into activated MDSC,74 only GM-CSF expression was reduced in the BM. Nonetheless, iNOS and IL10 expression was decreased in BMC, SC and TIL of ATRA-treated TB mice. Upregulated CCL5 expression in the tumor tissue argues against ATRA affecting MDSC recruitment. Thus, lower MDSC recovery in TIL likely is a sequel of the reduced generation in the BM. There was additional evidence for pronounced T cell activation in LNC, which became stronger in DC-TEX vaccinated mice and was accompanied by higher proliferative and cytotoxic activity of LNC from ATRA-treated vaccinated mice. However, this support of immune response induction did not suffice for a prolongation of the survival time beyond that of vaccinated mice, which might be due to the overall reduced number of T cells that did not become balanced by improved activation. Nonetheless, ATRA-supported T cell activation sufficed to reduce recovery of UNKC in PB, BM and LN.
The tyrosine kinase inhibitor Sun prevents MDSC accumulation and initiates a shift toward type 1 MΦ. By targeting tyrosine kinase receptors in the tumor surrounding and tumor cells, it also directly affects tumor cells and TAM through Stat3 inhibition, which in MΦ drives IL12 and TNFα secretion, but inhibits IL10. In tumor cells, it accounts for inhibition of apoptosis; downregulation of the PDGFR drives EC in apoptosis. In addition, it strengthens the CXCR3-/CXCL9/CXCL10 axis, but downregulates CCL2, CCL17, CCL20 and CXCL12, which attract MDSC and Treg, but also effector T cells (CXCL12).75,76 Thus, Sun concomitantly affects immunosuppression and T cell activation. Our in vitro studies confirmed that Sun drives iMC toward DC and mature MΦ and revealed that the low number of CD11b+/Gr1+ cells developing in the presence of Sun exhibited reduced inhibitory potential. However, in vivo Sun treatment was not accompanied by a shift toward APC, CD4+ cells were reduced in LNC and TIL, fewer T cells expressed activation markers and only in LNC IL10 expression was mitigated. Despite the strong mitigation of in vivo T cell activation by Sun, in combination with DC vaccination the impact of Sun on MDSC maturation became dominant, which sufficed for T cell activation/expansion and pronounced recruitment into the tumor.
In brief, Sun efficiently copes with MDSC expansion and activation and the negative impact on T cell activation and recruitment into the tumor can be overcome by vaccination. Thus, Sun appears well suited to back cancer immunotherapy.77 As therapeutic weaknesses and strengths of ATRA and Sun differ, a combined treatment could provide further therapeutic advantages.
GEM is a cytidine analog. After uptake it undergoes conversion to GEM-diphosphate, which potentially hampers several steps in DNA synthesis including repair mechanisms.78 GEM is used as a cytotoxic drug in many tumors including PaCa, where it is frequently given preference.79 It is supposed to drive MDSC into apoptosis via BAX upregulation and inhibition of Bcl2 and BclXl phosphorylation.80 In patients with PaCa and in animal models GEM was repeatedly demonstrated to support DC vaccination.24,25,81 In a murine PaCa model GEM did not exert a life prolonging effect, but efficiently supported active DC vaccination due to downregulation of MDSC and Treg, which was accompanied by DC reprogramming with upregulation of IFNγ.28
We noted that in vitro, but not in vivo GEM treatment is accompanied by increased leukocyte apoptosis. However, the opposing in vivo finding does not exclude apoptotic cell death in advance of harvesting. Instead, in vitro and in vivo GEM treatment was accompanied by a blockade of MDSC activation with reduced expression of pStat6, MyD88 and downstream NFκB and HIF1α. As a consequence, IL1β, IFNγ, cyclinD1, BclXl, survivin and iNOS expression was reduced in MDSC. The impact of GEM on TIL was similar to that described for Sun, except for more pronounced reduction of immunosuppressive and immunostimulatory cytokines, a stronger reduction in MDSC, but a weaker impact on Treg. Nonetheless, the survival time of GEM treated mice is prolonged and further expanded by concomitant vaccination. In addition, GEM together with the DC vaccine reduced tumor cell spread and settlement in distant organs more efficiently than ATRA and Sun.
We had expected similar therapeutic efficacy of Sun and GEM. We suggest that the superiority of GEM is due to its activity being restricted to dividing cells such that the 3 d window between GEM and DC application suffices for an unimpaired/minimally impaired T cell activation after DC binding. In addition, there was no evidence for GEM affecting angiogenesis (data not shown). This may facilitate tumor spread, but obviously the better access of activated T cells to the tumor tissue was surmounting.
Despite the efficacy of GEM, we considered it worthwhile to combine ATRA, Sun and GEM treatment as a possible means to most efficiently eradicate MDSC. First to note, AGS treatment strongly affected leukocyte recovery, which was not efficiently coped by concomitant vaccination. Nonetheless, CD11b+Gr1+ cells were strikingly reduced and DC vaccination sufficed for a strong recruitment of CD4+ and CD8+ T cells into the tumor. Furthermore, BM-derived MDSC showed a significant loss in suppressive activity and that of spleen-derived MDSC was nearly waved. Despite the reduced leukocyte count, CD4+ and CD8+ cells were not selectively reduced and T cell activation was largely unimpaired. In LN and TIL immunostimulatory cytokines were dominating. The reduced expression of inflammatory cytokines in TIL of AGS treated mice requires further exploration. Increased CXCR4 expression in LNC could well support leukocyte egress/T cell migration with high level CXCL10 and CXCL12 expression in the tumor tissue supporting leukocyte recruitment. Furthermore, AGS did not affect the proliferative activity of surviving LNC and SC and strongly increased proliferation and cytotoxic activity in response to DC-TEX. In fact, no tumor mass was palpable in AGS plus DC-TEX vaccinated mice, when all control and AGS-treated mice had to be killed. However, after prolonged AGS application, mice developed tumors and at autopsy, only very few leukocytes were recovered from BM, LN and spleen. Immunohistology confirmed a striking stroma reduction, particularly of LN332, a disorganization of the shielding SMA+ cells, a reduction in angiogenesis and, less pronounced, lymphangiogenesis. All these features support an additive effect of ATRA, GEM and Sun, without evidence for a mutual interference. The poor recovery of migrating and metastasizing tumor cells despite the breakdown of the immune system underlines the efficacy of combining MDSC elimination with active vaccination.
Conclusion and outlook
Immunotherapy of PaCa is particularly burdened by immunosuppressive features of the tumor and the tumor-induced stroma and strong immunosuppression by MDSC and Treg. We confirmed immunogenicity of PaCa by response-induction via TEX-loaded DC, TEX-loading being well suited to present a wide panel of PaCa-associated antigens. As TEX can be recovered from the patients’ blood concomitantly with DC, this personalized vaccination approach becomes independent of the availability of tumor tissue. Confirming the strong interference of MDSC with immune response-induction toward PaCa, attacking iMC maturation, MDSC activation and driving MDSC toward apoptosis additively cope with PaCa-induced immunosuppression. The protocol requires further refinement as the repeated application of ATRA, Sun and GEM provoked a partial collapse of the immune system. The exact pausing should possibly be decided on the basis of the patient's immune status and the recovery of MDSC. Finally, though we did not take care on stroma-induced reactions besides coping with MDSC and Treg, according to the histological appearance after AGS treatment, no additional action might be required. However, this should be explicitly explored for PSC, TAM and TAF. Despite this open question, the data suggest that by attacking MDSC DC-TEX vaccination becomes a most promising approach in PaCa, where the cure rate of excisable tumors, even after metastatic spread, can be strongly uplifted.
Material and methods
Cell line: UNKC6141 (UNKC), kindly provided by SK Batra, is a pancreatic adenocarcinoma spontaneously arisen in a Kras(G12D);Pdx1-Cre mouse.50 Cells were maintained in ISCOVE'sMEM medium supplemented with 10% fetal calf serum (FCS), L-glutamine and antibiotics. Confluent cells were trypsinized and split.
Antibodies and chemicals: Suppl. Tab. 1.
Tissue preparation, cell isolation and purification: Mice were killed by cervical dislocation or were anaesthetized in CO2 for collecting heparinized PB by heart puncture. Single cell suspensions of excised organs were prepared by pressing through fine gauze; BMC were flushed from femora and tibiae; PBL were collected after FicollHypaque centrifugation. DC and MDSC were generated from BMC. For DC induction, BMC (2 × 106) were cultured in 10 cm diameter Petri-dishes in 10 mL RPMI1640/10%FCS, supplemented with recombinant mouse GMCSF (10 ng/mL) and IL4 (2 ng/mL), adding after 8 d 100µg/mL TEX for uptake, loading and peptide presentation concomitantly with 0.1 µg/mL LPS to induce DC maturation.43 MDSC were induced by culturing BMC in RPMI1640/10%FCS supplemented with 20 ng/mL GMCSF, 5 ng/mL IL6 and 1 nM PGE2 for 5 d, where indicated cultures containing 20 µg/mL Sun or 30 µM Gem. Where indicated, DC and MDSC were CFSE labeled (5 µM/106 cells). Treg, CD4+, CD8+ and CD11b+Gr1+ cells were enriched by magnetic bead sorting.
TEX preparation: UNKC6141 were cultured (48 h) in serum-free medium. Cleared supernatants (2 × 10 min, 500 g, 1 × 20 min, 2000 g, 1 × 30 min, 10,000 g) were centrifuged (120 min, 100,000 g), the pellet was washed (PBS, 90 min, 100,000 g), resuspended in 40% sucrose overlaid by a discontinuous sucrose gradient (30–5%) and centrifuged (16 h, 100,000 g). Exosomes were collected from the 10–5% sucrose interface. The protein concentration was determined by Bradford.
Flow cytometry: Cells (5 × 105) were stained according to routine procedures. TEX (10 µg) were coupled to 1 µL Latex beads. After blocking (100 mM glycine) and washing, TEX-loaded beads were stained using the same protocol as for cells. For intracellular staining, cells or bead-coated TEX were fixed and permeabilized in advance. Apoptosis was determined by AnnV/PtdIns staining. Samples were analyzed in a FACSCalibur using the CellQuest program.
Trogocytosis:82 Loaded DC and MDSC (10 × 106) were resuspended in PBS containing 1 mg/mL Sulfobiotin-X-NHS and incubated for 10 min at 25°C. After adding an equivalent volume of FCS, cells were incubated for an additional 10 min at 4°C. Washed biotinylated DC/MDSC were cocultured with LNC, SC or BMC at a ratio of 2:1 (2 h, 37°C). After washing (2 mM EDTA/PBS), LNC, SC and BMC were stained with APC-labeled antibody and counterstained with Streptavidin-FITC. Trogocytosis (transfer of biotinylated membrane particles) was evaluated by flow-cytometry.
Immunohistochemistry: Snap frozen tissue sections (8 µm) were fixed, incubated with antibodies, washed, exposed to biotinylated secondary antibodies and alkaline phosphatase conjugated avidin-biotin solution. Sections were counter-stained with hematoxilin. Digitized images were generated using a Leica DMRBE microscope.
T cell proliferation: LNC and SC were seeded in U-shaped 96 well plates (2 × 105–2.5 × 104/well) in the absence or presence of MDSC or DC at the indicated ratios. Cells were cultured for 3 d adding 3H-thymidine (10 µCi/mL) during the last 16 h. Plates were harvested and 3H-thymidine incorporation was evaluated in a β-counter. Mean values ± SD of triplicates are presented.
Cytotoxicity assay: CTL activity was evaluated after in vitro restimulation of LNC or SC by 3H-thymidine release from labeled (12 h, 10 µCi/mL 3H-thymidine) target cells (104/well), seeded on titrated numbers (1 × 106–6 × 104) of effector cells in 96 well plates. After 8 h at 37°C, cells were harvested, and radioactivity was determined in a β-counter. Cytotoxicity is presented as % cytotoxicity = 100 × (counts in control wells − counts in test wells)/(counts in control wells). The spontaneous release of tumor cells ranged between 5% and12%, and of lymphoblasts (ConA stimulated LNC) between 8% and 20%. Mean values ± SD of triplicates are presented.
In vivo assays: C57BL6 mice received a subcutaneous (s.c.) or orthotopic (o.t.) injection of 5 × 106 or 1 × 106 UNKC cells. Where indicated s.c. growing tumors were excised reaching a mean diameter of 0.5–0.8 cm. For o.t. tumor cell application and for tumor excision mice were anaesthetized with Isofluran. For immune response induction, mice received weekly 2 × 106 TEX-loaded DC (DC-TEX), i.v., starting 4 d after tumor cell application. ATRA, GEM and Sun application started 24 h after tumor cell application. An ATRA depot pellet (5 mg) was s.c. administered, application was repeated in 3 wk intervals. GEM (100 µg/g) and Sun (20 µg/g) were weekly i.v. administered. Mice receiving an i.v. injection of CFSE-labeled DC or MDSC were killed after 48 h. In other experiments, the time point of killing is indicated. Mice were killed latest, when the s.c. or o.t. tumor mass reached a mean diameter of 1 cm or after 120 d. Animal experiments were Government-approved (Baden-Wuerttemberg, Germany).
Statistics: p values <0.05 (in vitro assays: two-tailed Student's t-test, in vivo assays: Kruskal–Wallis test) were considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Angela Frank for invaluable help with flow cytometry.
Funding
This investigation was supported by the Wilhelm Sander Stiftung (MZ), the German Cancer Aid Foundation (MZ), the German Research Foundation (MZ), a grant from the Department of Oncology, Zhongshan Hospital Affiliated to Xiamen University (LX) and a PhD grant from the Chinese government (KZ).
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics 2013 CA. Cancer J Clin 2013; 63:11-30; PMID:23335087; https://doi.org/ 10.3322/caac.21166 [DOI] [PubMed] [Google Scholar]
- 2.Silvestris N, Gnoni A, Brunetti AE, Vincenti L, Santini D, Tonini G, Merchionne F, Maiello E, Lorusso V, Nardulli P et al.. Target therapies in pancreatic carcinoma. Curr Med Chem 2014; 21:948-65; PMID: 23992319; https://doi.org/ 10.2174/09298673113209990238 [DOI] [PubMed] [Google Scholar]
- 3.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74:2913-21; PMID: 24840647; https://doi.org/ 10.1158/0008-5472.CAN-14-0155 [DOI] [PubMed] [Google Scholar]
- 4.Walker EJ, Ko AH. Beyond first-line chemotherapy for advanced pancreatic cancer: an expanding array of therapeutic options?. World J Gastroenterol 2014; 20(9):2224-36; PMID: 24605022; https://doi.org/ 10.3748/wjg.v20.i9.2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyle J, Czito B, Willett C, Palta M. Adjuvant radiation therapy for pancreatic cancer: a review of the old and the new. J Gastrointest Oncol 2015; 6(4):436-44; PMID: 26261730; https://doi.org/ 10.3978/j.issn.2078-6891.2015.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Choi M. Immune therapy in pancreatic cancer: now and the future?. Rev Recent Clin Trials 2015; 10(4):317-25; PMID: 26374557; https://doi.org/ 10.2174/1574887110666150916142537 [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Zhang X. Primary analysis for clinical efficacy of immunotherapy in patients with pancreatic cancer. Immunotherapy 2016; 8(2):223-34; PMID: 26565954; https://doi.org/ 10.2217/imt.15.105 [DOI] [PubMed] [Google Scholar]
- 8.Puré E, Lo A. Can targeting stroma pave the way to enhanced antitumor immunity and Immunotherapy of solid tumors?. Cancer Immunol Res 2016; 4(4):269-78; PMID: 27036971; https://doi.org/ 10.1158/2326-6066.CIR-16-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie D, Xie K. Pancreatic cancer stromal biology and therapy. Genes Dis 2015; 2(2):133-43; PMID: 26114155; https://doi.org/ 10.1016/j.gendis.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu M, Zhou BP, Tao M, Liu J, Li W. The role of stromal components in pancreatic cancer progression. Anticancer Agents Med Chem 2016; 16(9):1117-24; PMID: 27039918; https://doi.org/ 10.2174/1871520616666160404115532 [DOI] [PubMed] [Google Scholar]
- 11.Binenbaum Y, Na'ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat 2015; 23:55-68; PMID: 26690340; https://doi.org/ 10.1016/j.drup.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 12.Schnurr M, Duewell P, Bauer C, Rothenfusser S, Lauber K, Endres S, Kobold S. Strategies to relieve immunosuppression in pancreatic cancer. Immunotherapy 2015; 7(4):363-76; PMID: 25917628; https://doi.org/ 10.2217/imt.15.9 [DOI] [PubMed] [Google Scholar]
- 13.Khan S, Ebeling MC, Chauhan N, Thompson PA, Gara RK, Ganju A, Yallapu MM, Behrman SW, Zhao H, Zafar N et al.. Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer Res 2015; 75(11):2292-304; PMID: 25840985; https://doi.org/ 10.1158/0008-5472.CAN-14-2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grünwald B, Vandooren J, Gerg M, Ahomaa K, Hunger A, Berchtold S, Akbareian S, Schaten S, Knolle P, Edwards DR et al.. Systemic ablation of MMP-9 triggers invasive growth and metastasis of pancreatic cancer via deregulation of IL6 expression in the bone marrow. Mol Cancer Res 2016; 14(11):1147-58; PMID: 27489361; https://doi.org/ 10.1158/1541-7786.MCR-16-0180 [DOI] [PubMed] [Google Scholar]
- 15.Franklin O, Öhlund D, Lundin C, Öman M, Naredi P, Wang W, Sund M. Combining conventional and stroma-derived tumour markers in pancreatic ductal adenocarcinoma. Cancer Biomark 2015; 15(1):1-10; PMID: 25524936; https://doi.org/ 10.3233/CBM-140430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okada K, Hirabayashi K, Imaizumi T, Matsuyama M, Yazawa N, Dowaki S, Tobita K, Ohtani Y, Tanaka M, Inokuchi S et al.. Stromal thrombospondin-1 expression is a prognostic indicator and a new marker of invasiveness in intraductal papillary-mucinous neoplasm of the pancreas. Biomed Res 2010; 31(1):13-19; PMID: 20203415; https://doi.org/ 10.2220/biomedres.31.13 [DOI] [PubMed] [Google Scholar]
- 17.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D et al.. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013; 73(3):1128-41; PMID: 23221383; https://doi.org/ 10.1158/0008-5472.CAN-12-2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol 2015; 31(5):416-23; PMID: 26125317; https://doi.org/ 10.1097/MOG.0000000000000196 [DOI] [PubMed] [Google Scholar]
- 19.Masamune A, Shimosegawa T. Pancreatic stellate cells: a dynamic player of the intercellular communication in pancreatic cancer. Clin Res Hepatol Gastroenterol 2015; 39 (Suppl 1):S98-103; PMID: 26189983; https://doi.org/ 10.1016/j.clinre.2015.05.018 [DOI] [PubMed] [Google Scholar]
- 20.Collignon A, Perles-Barbacaru AT, Robert S, Silvy F, Martinez E, Crenon I, Germain S, Garcia S, Viola A, Lombardo D et al.. A pancreatic tumor-specific biomarker characterized in humans and mice as an immunogenic onco-glycoprotein is efficient in dendritic cell vaccination. Oncotarget 2015; 6(27):23462-79; PMID: 26405163; https://doi.org/ 10.18632/oncotarget.4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang TH, Kim YS, Kim S, Yang B, Lee JJ, Lee HJ, Lee J, Jung ID, Han HD, Lee SH et al.. Pancreatic adenocarcinoma upregulated factor serves as adjuvant by activating dendritic cells through stimulation of TLR4. Oncotarget 2015; 6(29):27751-62; PMID: 26336989; https://doi.org/ 10.18632/oncotarget.4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Guo XZ, Li HY, Wang D, Shao XD. Comparison of cytotoxic T lymphocyte responses against pancreatic cancer induced by dendritic cells transfected with total tumor RNA and fusion hybrided with tumor cell. Exp Biol Med (Maywood) 2015; 240(10):1310-18; PMID: 25736302; https://doi.org/ 10.1177/1535370215571884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellone M, Mondino A, Corti A. Vascular targeting, chemotherapy and active immunotherapy: teaming up to attack cancer. Trends Immunol 2008; 29(5):235-41; PMID:18375183; https://doi.org/ 10.1016/j.it.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Mayanagi S, Kitago M, Sakurai T, Matsuda T, Fujita T, Higuchi H, Taguchi J, Takeuchi H, Itano O, Aiura K et al.. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci 2015; 106(4):397-406; PMID: 25614082; https://doi.org/ 10.1111/cas.12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takakura K, Koido S, Kan S, Yoshida K, Mori M, Hirano Y, Ito Z, Kobayashi H, Takami S, Matsumoto Y et al.. Prognostic markers for patient outcome following vaccination with multiple MHC Class I/II-restricted WT1 peptide-pulsed dendritic cells plus chemotherapy for pancreatic cancer. Anticancer Res 2015; 35(1):555-62; PMID: 25550602. [PubMed] [Google Scholar]
- 26.Koido S, Homma S, Okamoto M, Takakura K, Mori M, Yoshizaki S, Tsukinaga S, Odahara S, Koyama S, Imazu H et al.. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms' tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin Cancer Res 2014; 20(16):4228-39; PMID: 25056373; https://doi.org/ 10.1158/1078-0432.CCR-14-0314 [DOI] [PubMed] [Google Scholar]
- 27.Bauer C, Sterzik A, Bauernfeind F, Duewell P, Conrad C, Kiefl R, Endres S, Eigler A, Schnurr M, Dauer M. Concomitant gemcitabine therapy negatively affects DC vaccine-induced CD8(+) T-cell and B-cell responses but improves clinical efficacy in a murine pancreatic carcinoma model. Cancer Immunol Immunother 2014; 63(4):321-33; PMID: 24384835; https://doi.org/ 10.1007/s00262-013-1510-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghansah T, Vohra N, Kinney K, Weber A, Kodumudi K, Springett G, Sarnaik AA, Pilon-Thomas S. Dendritic cell immunotherapy combined with gemcitabine chemotherapy enhances survival in a murine model of pancreatic carcinoma. Cancer Immunol Immunother 2013; 62(6):1083-91; PMID: 23604104; https://doi.org/ 10.1007/s00262-013-1407-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S et al.. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7:12150; PMID: 27381735; https://doi.org/ 10.1038/ncomms12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greten TF. Myeloid-derived suppressor cells in pancreatic cancer: more than a hidden barrier for antitumour immunity?. Gut 2014; 63(11):1690-91; PMID: 24633728; https://doi.org/ 10.1136/gutjnl-2014-306790 [DOI] [PubMed] [Google Scholar]
- 31.Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther 2016. March; 24(3):100–105; PMID:27910857; https://doi.org/ 10.1038/cgt.2016.65 [DOI] [PubMed] [Google Scholar]
- 32.Motallebnezhad M, Jadidi-Niaragh F, Qamsari ES, Bagheri S, Gharibi T, Yousefi M. The immunobiology of myeloid-derived suppressor cells in cancer. Tumour Biol 2016; 37(2):1387-406; PMID: 26611648; https://doi.org/ 10.1007/s13277-015-4477-9 [DOI] [PubMed] [Google Scholar]
- 33.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 2007; 67(22):11021-28; PMID: 18006848; https://doi.org/ 10.1158/0008-5472.CAN-07-2593 [DOI] [PubMed] [Google Scholar]
- 34.Liu ZM, Wang KP, Ma J, Guo Zheng S. The role of all-trans retinoic acid in the biology of Foxp3+ regulatory T cells. Cell Mol Immunol 2015; 12(5):553-57; PMID: 25640656; https://doi.org/ 10.1038/cmi.2014.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gujar SA, Clements D, Dielschneider R, Helson E, Marcato P, Lee PW. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br J Cancer 2014; 110(1):83-93; PMID: 24281006; https://doi.org/ 10.1038/bjc.2013.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, Valle J, Smith D, Wadsley J, Cunningham D et al.. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother 2014; 63(2):175-83; PMID: 24292263; https://doi.org/ 10.1007/s00262-013-1502-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mankal P, O'Reilly E. Sunitinib malate for the treatment of pancreas malignancies—where does it fit?. Expert Opin Pharmacother 2013; 14(6):783-92; PMID: 23458511; https://doi.org/ 10.1517/14656566.2013.776540 [DOI] [PubMed] [Google Scholar]
- 38.van Hooren L, Georganaki M, Huang H, Mangsbo SM, Dimberg A. Sunitinib enhances the antitumor responses of agonistic CD40-antibody by reducing MDSCs and synergistically improving endothelial activation and T-cell recruitment. Oncotarget 2016; 7(31):50277-89; PMID: 27385210; https://doi.org/ 10.18632/oncotarget.10364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Draghiciu O, Boerma A, Hoogeboom BN, Nijman HW, Daemen T. A rationally designed combined treatment with an alphavirus-based cancer vaccine, Sunitinib and low-dose tumor irradiation completely blocks tumor development. Oncoimmunology 2015; 4(10):e1029699; PMID: 26451295; https://doi.org/ 10.1080/2162402X.2015.1029699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.H Yi D, Appel S. Current status and future perspectives of dendritic cell-based cancer immunotherapy. Scand J Immunol 2013; 78(2):167-71; PMID: 23672402; https://doi.org/ 10.1111/sji.12060 [DOI] [PubMed] [Google Scholar]
- 41.Salem ML. The use of dendritic cells for peptide-based vaccination in cancer immunotherapy. Methods Mol Biol 2014; 1139:479-503; PMID: 24619701; https://doi.org/ 10.1007/978-1-4939-0345-0_37 [DOI] [PubMed] [Google Scholar]
- 42.Amedei A, Niccolai E, Prisco D. Pancreatic cancer: role of the immune system in cancer progression and vaccine-based immunotherapy. Hum Vaccin Immunother 2014; 10(11):3354-68; PMID: 25483688; https://doi.org/ 10.4161/hv.34392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu X, Erb U, Büchler MW, Zöller M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int J Cancer 2015; 136(4):E74-84; PMID: 25066479; https://doi.org/ 10.1002/ijc.29100 [DOI] [PubMed] [Google Scholar]
- 44.Palazzolo G, Albanese NN, DI Cara G, Gygax D, Vittorelli ML, Pucci-Minafra I. Proteomic analysis of exosome-like vesicles derived from breast cancer cells. Anticancer Res 2012; 32(3):847-60; PMID: 22399603. [PubMed] [Google Scholar]
- 45.Hong CS, Muller L, Boyiadzis M, Whiteside TL. Isolation and characterization of CD34+ blast-derived exosomes in acute myeloid leukemia. PLoS One 2014; 9(8):e103310; PMID: 25093329; https://doi.org/ 10.1371/journal.pone.0103310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tu M, Wei F, Yang J, Wong D. Detection of exosomal biomarker by electric field-induced release and measurement (EFIRM). J Vis Exp 2015; 95:52439; PMID: 25650727; https://doi.org/ 10.3791/52439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skokos D, Botros HG, Demeure C, Morin J, Peronet R, Birkenmeier G, Boudaly S, Mécheri S. Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J Immunol 2003; 170(6):3037-45; PMID: 12626558; https://doi.org/ 10.4049/jimmunol.170.6.3037 [DOI] [PubMed] [Google Scholar]
- 48.Platt CD, Ma JK, Chalouni C, Ebersold M, Bou-Reslan H, Carano RA, Mellman I, Delamarre L. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc Natl Acad Sci USA 2010; 107(9):4287-92; PMID: 20142498; https://doi.org/ 10.1073/pnas.0910609107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie Y, Zhang H, Li W, Deng Y, Munegowda MA, Chibbar R, Qureshi M, Xiang J. Dendritic cells recruit T cell exosomes via exosomal LFA-1 leading to inhibition of CD8+ CTL responses through downregulation of peptide/MHC class I and Fas ligand-mediated cytotoxicity. J Immunol 2010; 185(9):5268-78; PMID: 20881190; https://doi.org/ 10.4049/jimmunol.1000386 [DOI] [PubMed] [Google Scholar]
- 50.Torres MP, Rachagani S, Souchek JJ, Mallya K, Johansson SL, Batra SK. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy. PLoS One 2013; 8(11):e80580; PMID: 24278292; https://doi.org/ 10.1371/journal.pone.0080580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamanaka R, Kajiwara K. Dendritic cell vaccines. Adv Exp Med Biol 2012; 746:187-200; PMID: 22639169; https://doi.org/ 10.1007/978-1-4614-3146-6_15 [DOI] [PubMed] [Google Scholar]
- 52.Gesierich S, Paret C, Hildebrand D, Weitz J, Zgraggen K, Schmitz-Winnenthal FH, Horejsi V, Yoshie O, Herlyn D, Ashman LK et al.. Colocalization of the tetraspanins, CO-029 and CD151, with integrins in human pancreatic adenocarcinoma: impact on cell motility. Clin Cancer Res 2005; 11(8):2840-52; PMID: 15837731; https://doi.org/ 10.1158/1078-0432.CCR-04-1935 [DOI] [PubMed] [Google Scholar]
- 53.Madhavan B, Yue S, Galli U, Rana S, Gross W, Müller M, Giese NA, Kalthoff H, Becker T, Büchler MW et al.. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Cancer 2015; 136(11):2616-27; PMID: 25388097; https://doi.org/ 10.1002/ijc.29324 [DOI] [PubMed] [Google Scholar]
- 54.Li N, Song MM, Chen XH, Liu LH, Li FS. S100A4 siRNA inhibits human pancreatic cancer cell invasion in vitro. Biomed Environ Sci 2012; 25(4):465-70; PMID: 23026527; https://doi.org/ 10.3967/0895-3988.2012.04.012 [DOI] [PubMed] [Google Scholar]
- 55.Hrabar D, Aralica G, Gomercic M, Ljubicic N, Kruslin B, Tomas D. Epithelial and stromal expression of syndecan-2 in pancreatic carcinoma. Anticancer Res 2010; 30(7):2749-53; PMID: 20683009. [PubMed] [Google Scholar]
- 56.Zhu J, Thakolwiboon S, Liu X, Zhang M, Lubman DM. Overexpression of CD90 (Thy-1) in pancreatic adenocarcinoma present in the tumor microenvironment. PLoS One 2014; 9(12):e115507; PMID: 25536077; https://doi.org/ 10.1371/journal.pone.0115507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, Menke A, Ruhland C, Adler G, Gress TM. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res 2001; 61(10):4222-28; PMID: 11358848. [PubMed] [Google Scholar]
- 58.Dutta SK, Girotra M, Singla M, Dutta A, Otis Stephen F, Nair PP, Merchant NB. Serum HSP70: a novel biomarker for early detection of pancreatic cancer. Pancreas 2012; 41(4):530-34; PMID: 22158074; https://doi.org/ 10.1097/MPA.0b013e3182374ace [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oehlrich N, Devitt G, Linnebacher M, Schwitalle Y, Grosskinski S, Stevanovic S, Zöller M. Generation of RAGE-1 and MAGE-9 peptide-specific cytotoxic T-lymphocyte lines for transfer in patients with renal cell carcinoma. Int J Cancer 2005; 117(2):256-64; PMID: 15900605; https://doi.org/ 10.1002/ijc.21200 [DOI] [PubMed] [Google Scholar]
- 60.Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol 2014; 28:39-50; PMID: 24631836; https://doi.org/ 10.1016/j.semcancer.2014.02.011 [DOI] [PubMed] [Google Scholar]
- 61.Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun 2015; 6:7164; PMID: 25968605; https://doi.org/ 10.1038/ncomms8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu GH, Huang C, Qiu ZJ, Liu J, Zhang ZH, Zhao N, Feng ZZ, Lv XH. Expression and prognostic significance of CD151, c-Met, and integrin alpha3/alpha6 in pancreatic ductal adenocarcinoma. Dig Dis Sci 2011; 56(4):1090-98; PMID: 20927591; https://doi.org/ 10.1007/s10620-010-1416-x [DOI] [PubMed] [Google Scholar]
- 63.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S et al.. Tumour exosome integrins determine organotropic metastasis. Nature 2015; 527(7578):329-35; PMID: 26524530; https://doi.org/ 10.1038/nature15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yue S, Mu W, Erb U, Zöller M. The tetraspanins CD151 and Tspan8 are essential exosome components for the crosstalk between cancer initiating cells and their surrounding. Oncotarget 2015; 6(4):2366-84; PMID: 25544774; https://doi.org/ 10.18632/oncotarget.2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maenhout SK, Thielemans K, Aerts JL. Location, location, location: functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology 2014; 3(10):e956579; PMID: 25941577; https://doi.org/ 10.4161/21624011.2014.956579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 2016; 113(8):E968-977; PMID: 26858453; https://doi.org/ 10.1073/pnas.1521230113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC et al.. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004; 104(10):3257-66; PMID: 15284116; https://doi.org/ 10.1182/blood-2004-03-0824 [DOI] [PubMed] [Google Scholar]
- 68.Zöller M. Exosomes in cancer disease. Methods Mol Biol 2016; 1381:111-49; PMID: 26667458; https://doi.org/ 10.1007/978-1-4939-3204-7_7 [DOI] [PubMed] [Google Scholar]
- 69.Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol 2014; 5:442; PMID: 25278937; https://doi.org/ 10.3389/fimmu.2014.00442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ahmed KA, Munegowda MA, Xie Y, Xiang J. Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol Immunol 2008; 5(4):261-69; PMID: 18761813; https://doi.org/ 10.1038/cmi.2008.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shih NY, Yang HY, Cheng HT, Hung YM, Yao YC, Zhu YH, Wu YC, Liu KJ. Conditioning vaccination site with irradiated MIP-3alpha-transfected tumor cells enhances efficacy of dendritic cell-based cancer vaccine. J Immunother 2009; 32(4):363-69; PMID: 19342969; https://doi.org/ 10.1097/CJI.0b013e31819d29d8 [DOI] [PubMed] [Google Scholar]
- 72.Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol Immunother 2012; 61(6):751-59; PMID: 22488275; https://doi.org/ 10.1007/s00262-012-1253-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, Shono Y, Kitabatake M, Kakimi K, Mukaida N et al.. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 2008; 111(12):5457-66; PMID: 18375791; https://doi.org/ 10.1182/blood-2008-01-136895 [DOI] [PubMed] [Google Scholar]
- 74.Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol 2015; 98(6):913-22; PMID: 26337512; https://doi.org/ 10.1189/jlb.4RI0515-204R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009; 69(6):2514-22; PMID: 19276342; https://doi.org/ 10.1158/0008-5472.CAN-08-4709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu N, Fu S, Xu Z, Liu Y, Hao J, Zhang A, Wang B. Synergistic antitumor responses by combined GITR activation and sunitinib in metastatic renal cell carcinoma. Int J Cancer 2016; 138(2):451-62; PMID: 26239999; https://doi.org/ 10.1002/ijc.29713 [DOI] [PubMed] [Google Scholar]
- 77.Kwilas AR, Donahue RN, Tsang KY, Hodge JW. Immune consequences of tyrosine kinase inhibitors that synergize with cancer immunotherapy. Cancer Cell Microenviron 2015; 2(1):pii: e677; PMID:26005708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ciccolini J, Serdjebi C, Peters GJ, Giovannetti E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: an EORTC-PAMM perspective. Cancer Chemother Pharmacol 2016; 78(1):1-12; PMID: 27007129; https://doi.org/ 10.1007/s00280-016-3003-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tu C, Zheng F, Wang JY, Li YY, Qian KQ. An updated meta-analysis and system review: is Gemcitabine+Fluoropyrimidine in combination a better therapy versus gemcitabine alone for advanced and unresectable pancreatic cancer?. Asian Pac J Cancer Prev 2015; 16(14):5681-86; PMID: 26320435; https://doi.org/ 10.7314/APJCP.2015.16.14.5681 [DOI] [PubMed] [Google Scholar]
- 80.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 2005; 11(18):6713-21; PMID: 16166452; https://doi.org/ 10.1158/1078-0432.CCR-05-0883 [DOI] [PubMed] [Google Scholar]
- 81.Pei Q, Pan J, Ding X, Wang J, Zou X, Lv Y. Gemcitabine sensitizes pancreatic cancer cells to the CTLs antitumor response induced by BCG-stimulated dendritic cells via a Fas-dependent pathway. Pancreatology 2015; 15(3):233-39; PMID: 25937078; https://doi.org/ 10.1016/j.pan.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 82.Daubeuf S, Bordier C, Hudrisier D, Joly E. Suitability of various membrane lipophilic probes for the detection of trogocytosis by flow cytometry. Cytometry A 2009; 75(5):380-89; PMID: 19051238; https://doi.org/ 10.1002/cyto.a.20679 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.