

ABSTRACT
Microenvironmental regulation in lymphoid tissues is essential for the development of chronic lymphocytic leukemia. We identified cellular and molecular factors provided by the splenic marginal zone (MZ), which alter the migratory and adhesive behavior of leukemic cells. We used the Cxcr5−/−Eµ-Tcl1 leukemia mouse model, in which tumor cells are excluded from B cell follicles and instead accumulate within the MZ. Genes involved in MZ B cell development and genes encoding for adhesion molecules were upregulated in MZ-localized Cxcr5−/−Eµ-Tcl1 cells. Likewise, surface expression of the adhesion and homing molecules, CD49d/VLA-4 and CXCR7, and of NOTCH2 was increased. In vitro, exposing Eµ-Tcl1 cells or human CLL cells to niche-specific stimuli, like B cell receptor- or Toll-like receptor ligands, caused surface expression of these molecules characteristic for a follicular or MZ-like microenvironment, respectively. In vivo, inhibition of VLA-4-mediated adhesion and CXCL13-mediated follicular homing displaced leukemic cells not only from the follicle, but also from the MZ and reduced leukemia progression. We conclude that MZ-specific factors shape the phenotype of leukemic cells and facilitate their niche-specific retention. This strong microenvironmental influence gains pathogenic significance independent from tumor-specific genetic aberrations.
KEYWORDS: Adhesion molecules, B cell leukemia, chemokine receptors, marginal zone, toll-like receptor signaling, tumor microenvironment
Introduction
Low-grade B cell malignancies, including chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), and splenic marginal zone lymphoma (SMZL), are highly dependent on the microenvironment. Tumor cells can alter their microenvironment by cell-cell contacts or by the provision of paracrine factors. Conversely, the tumor microenvironment itself can effectively support survival and proliferation of tumor cells, either by donating growth and survival factors, or by protection from immune attack.1 Much less is known about the impact of the growth and survival niche on the cellular phenotype of tumor cells itself, which is believed to depend on the tumor cells intrinsic genetic program only. Previously, the relevance of a crosstalk between neoplastic B cells and the tissue microenvironment has been recognized and critical cellular and molecular interaction partners have been identified. Malignant B cells recapitulate physiologic processes of B cells and show a conserved pattern of dissemination to anatomic niches of secondary lymphoid organs (SLOs). Surface expression of distinct chemokine receptors in cooperation with adhesion molecules are correlated with nodal homing of B cell non-Hodgkin's lymphoma (NHL) and Hodgkin's lymphoma (HL).2
In CLL, tumor cells interact with a variety of different stromal cells, such as mesenchymal stromal cells, monocyte-derived nurse-like cells, and also with T lymphocytes and myeloid cells.3,4 Employing the murine Eµ-Tcl1 transgenic model of CLL, we recently demonstrated that malignant B cells home to the B cell follicle, where they find a growth-promoting microenvironment in close proximity to the follicular dendritic cell network (FDC). FDCs secrete CXCL13, the ligand for the chemokine receptor CXCR5, and the CXCL13/CXCR5 signaling axis mediates the recruitment of leukemic cells toward follicular FDCs.5 Enhanced antigen-stimulated BCR signaling has been correlated with the clinical course of human CLL.6 In the Eµ-Tcl1 CLL model, we found enhanced expression of phosphorylated tyrosine kinases, i.e., ZAP-70 and BTK, indicating increased BCR activity. Deletion of CXCR5 blocked the entry of leukemic B cells into the B cell follicle and impaired leukemia progression. Instead, Cxcr5−/−Eµ-Tcl1 tumor cells resided in the splenic marginal zone (MZ).5 The MZ is at the border between red (RP) and white pulp (WP) and serves as a transit area for haematopoietic cells coming from the bloodstream and entering the WP. Resident cells of the MZ are involved in T cell-dependent and -independent immune responses to blood-borne pathogens. In mice, the MZ is composed of specialized macrophages, marginal reticular cells (MRC), and MZ B cells. In human SMZL, a B cell lymphoma located in the MZ of SLOs, lymphoma cells express functional toll-like receptors (TLRs) and their stimulation by microbial antigens contributes to disease pathobiology.7
Despite a denied access to the follicle, we observed expansion of Cxcr5−/−Eµ-Tcl1 leukemic cells within the MZ.5 We now asked if these tumor cells have the flexibility to adapt to their microenvironment and what factors facilitate this phenotypic diversity. We found that murine and human CLL cells acquired an inducible expression of homing and adhesion factors characteristic for a follicular or MZ-like microenvironment upon niche-specific stimuli. Finally, we identified the integrin CD49d as a crucial mediator for leukemic cell retention in the MZ and inhibiting both, the CXCR5/CXCL13-mediated migration and CD49d-mediated retention, resulted in a strongly reduced leukemia progression.
Results
Differentially expressed genes and increased surface expression of homing molecules in Cxcr5−/−Eµ-Tcl1 cells is associated with their migration and positioning within the MZ
We recently showed that Cxcr5−/−Eµ-Tcl1 leukemia cells are excluded from the B cell follicle and instead accumulate within the splenic marginal zone (MZ).5 In this study, we asked what cellular and molecular factors determine the positioning and expansion of Cxcr5−/−Eµ-Tcl cells in the MZ.
Benign MZ B cells are directed to the splenic MZ by the sphingosine 1-phosphate (S1P) receptors 1 and 38 and the chemokine receptor CXCR7.9 Hence, we addressed if S1P1 determines the positioning of Cxcr5−/−Eµ-Tcl cells in the MZ. Cxcr5−/−Eµ-Tcl1 cells showed a trend toward an enhanced S1P1 expression and an increased migratory capability in comparison to Eµ-Tcl1 cells (Figs. S1A and B). However, when we applied the S1P antagonist FTY720 13 h after adoptive transfer of SNARF-labeled Eµ-Tcl1 or Cxcr5−/−Eµ-Tcl1 cells in wt recipients, the frequency and positioning of tumor cells in the MZ, WP, and RP was not impaired (Figs. S1C and E). FTY720 treatment was confirmed by a drop in the frequency of peripheral CD3+ blood lymphocytes (Fig. S1D). Next, we analyzed CXCR7 surface expression on Eµ-Tcl1 or Cxcr5−/−Eµ-Tcl1 cells 3 d after adoptive transfer in congenic recipients. MZ-localized Cxcr5−/−Eµ-Tcl1 exhibited substantially increased CXCR7 surface expression compared with Eµ-Tcl1 cells that homed to the follicle. (Fig. S1F).
To identify additional molecules that retain Cxcr5−/−Eµ-Tcl1 cells in the MZ, we used recently generated genome-wide expression data5 and identified genes expressed differentially between Cxcr5−/−Eµ-Tcl1 and Eµ-Tcl1 cells. We found upregulation of two genes encoding for lymphocyte transcription factors associated with SMZL development in Cxcr5−/−Eµ-Tcl1 cells, Pax5 (log2 fold = 0.581, p = 0.0084) and Notch2 (log2 fold = 0.6643, p = 0.0003) (Fig. 1A). Pax5 is expressed in SMZL cells and is overexpressed in some SMZL patients due to Pax5 translocations.10 Notch2 is also frequently mutated in SMZL11 and is important in the development of MZ B cells.12
Figure 1.

Genes involved in migration and adhesion are differentially expressed between Eμ-Tcl1 and Cxcr5−/−Eµ-Tcl1 leukemia cells. (A) Genome-wide expression analysis of sorted Eμ-Tcl1 (n = 6) or Cxcr5−/−Eµ-Tcl1 (n = 5) cells was performed.5 Genes encoding lymphocyte associated transcription factors were upregulated in Cxcr5−/−Eµ-Tcl1 compared with Eµ-Tcl1 cells (black bars), genes downregulated in Cxcr5−/−Eµ-Tcl1 cells are shown with gray bars. (B) Genes that are included in gene ontology terms related to lymphocyte adhesion and migration and are differentially expressed between Cxcr5−/−Eµ-Tcl1 and Eµ-Tcl1 cells are shown. Genes implicated in MZ B cell retention and positioning are marked by a filled circle (•), genes frequently mutated in SMZL by an asterisk (*) and genes involved in integrin signaling by an open circle (°). (C) Splenic CD5±CD19± leukemia cells from Eµ-Tcl1 (n = 6) and Cxcr5−/−Eµ-Tcl1 (n = 8) mice were analyzed for surface expression of ALCAM, CD49d, CD29, and NOTCH2 in four independent experiments. Representative histograms are shown. Bar diagrams represent the gMFIs normalized against a FMO plus isotype control, means and SEMs are depicted. p values shown were determined by Mann-Whitney U test.
Genes associated with migration and adhesion were also differentially expressed in Cxcr5−/−Eµ-Tcl1 cells, i.e., genes encoding cannabinoid receptor 2 (Cnr2 log2 fold = 0.7885, p = 0.031) (Fig. S2A), integrin α8 (Itga8 log2 fold = 0.9182, p = 0.0214) and integrin α4 (Itga4 log2 fold = 0.4222, p = 0.0207) (Fig. 1B). Molecules involved in integrin activation or downstream integrin signaling (CD9 log2 fold = 0.6290, p = 0.0275; Ptk2 or FAK log2 fold = 1.668, p = 0.0033; Vav1 log2 fold = 0.3715, p = 0.0016; Pik3cb log2 fold = 0.5224, p = 0.0121) were upregulated in Cxcr5−/−Eµ-Tcl1 cells. Integrin α4 forms the heterodimer VLA-4 with integrin β1 (CD29) and is involved in retention of MZ B cells in the MZ.13 Integrin α6, an alternative binding partner of CD29,14 was downregulated in Cxcr5−/−Eµ-Tcl1 cells (Itga6 log2 fold = −1.0109, p = 0.0037), which could increase the amount of CD29 available for association with CD49d to form VLA-4.
Genes that are differentially expressed between MZ and follicular B cells may also be detectable in Cxcr5−/−Eµ-Tcl1 versus Eµ-Tcl1 cells (Figs. S2B–D). The GPI-anchored glycoprotein CD59a (log2 fold = 1.2447, p = 0.0127) was upregulated in MZ B and Cxcr5−/−Eµ-Tcl1 leukemia cells. CD59a inhibits cell lysis through complement activation by inhibiting formation of the membrane attack complex.15 Upregulation of CD59a could protect both MZ B cells and Cxcr5−/−Eµ-Tcl1 cells from complement-mediated cell lysis. Gene expression of activated leukocyte cell adhesion molecule (ALCAM), an adhesion molecule linked to migration of various leukocytes,16 was downregulated in Cxcr5−/−Eµ-Tcl1 cells and MZ B cells (log2 fold = −1.0914, p = 0.0023) (Fig. S2D).
Cell surface expression of adhesion molecules is crucial for their functionality. We found that surface expression of ALCAM was lower on Cxcr5−/−Eµ-Tcl1 cells, while CD49d, CD29, and NOTCH2 expression was higher compared with Eµ-Tcl1 cells (Fig. 1C).
In summary, genes encoding mainly for molecules involved in the positioning of MZ or follicular B cells were differentially expressed in Cxcr5−/−Eµ-Tcl1 versus Eµ-Tcl1 cells, indicating that they were linked to the specific local microenvironment tumor cells are exposed to. In line with this, MZ-positioning of Cxcr5−/−Eµ-Tcl1 leukemic cells was associated with an upregulation of CD49d, CD29, CXCR7, and NOTCH2 surface expression.
Surface expression of adhesion molecules is modulated in vitro by niche specific factors
To dissect the influence of defined stimuli in vitro, Eµ-Tcl1 cells were co-cultured on the bone marrow (BM) stromal cell line M2-10B4.17 Co-cultures were stimulated with or without different supplements and cell surface expression of ALCAM, CD49d, and NOTCH2 was analyzed after 24 h. Culturing tumor cells with stromal cells alone only modestly affected surface expression of these molecules, but increased the number of living (7-AAD−) cells (Figs. S3A and B). Next, we treated Eµ-Tcl1 cells with a BCR cross-linking anti-IgM antibody. This treatment reduced CD49d surface expression by 51 ± 15% and upregulated ALCAM expression by 2.6 ± 1.6-fold. NOTCH2 expression was reduced to some modest extent (Fig. 2A).
Figure 2.
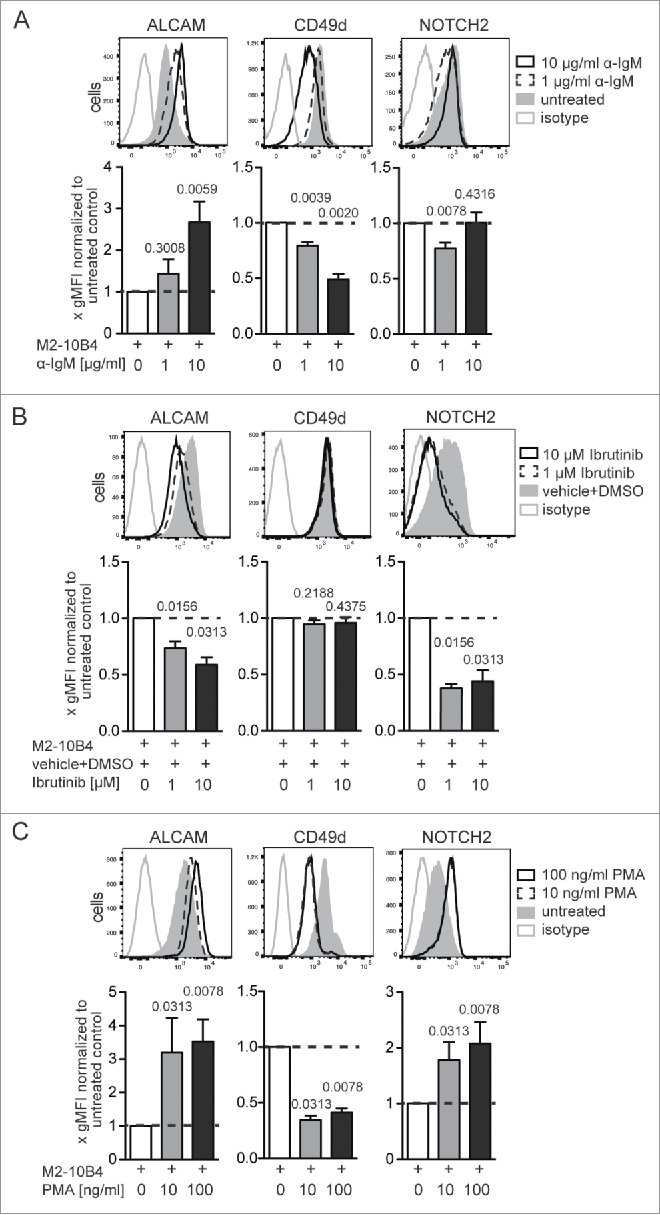
Surface expression levels of ALCAM, CD49d, and NOTCH2 on Eμ-Tcl1 tumor cells can be altered by BTK- and PKC-dependent signaling. M2-10B4 stromal cells were grown for 72 h on 24-well plates and irradiated. Four ×105 tumor cells/ml were added supplemented with (A) a BCR cross-linking α-IgM Ab, (B) the BTK inhibitor Ibrutinib, or (C) the PKC activator PMA at the indicated concentrations. Surface expression of ALCAM, CD49d, and NOTCH2 was assessed after 24 h of co-culture. Dead cells were excluded with 7-AAD. For each treatment 3–5 independent experiments were conducted with in total 6–10 different Eµ-Tcl1 cell clones. Representative histograms of one clone are shown. Clone-specific differences were normalized by dividing the gMFI of each treated sample with the gMFI of the corresponding untreated sample and bar diagrams represent means and SEMs of fold gMFIs. p values shown were calculated with the Wilcoxon signed rank test against a theoretical median of 1.
We tested if inhibition of the Bruton's tyrosine kinase (BTK), a key component of BCR signaling and function,18 had an effect on surface expression of ALCAM, CD49d, and NOTCH2. Treatment with the BTK inhibitor Ibrutinib reduced ALCAM and NOTCH2 expression levels by 40 ± 16% and 55 ± 24% on Eµ-Tcl1 cells, respectively (Fig. 2B). CD49d expression levels were unaltered 24 h after BTK inhibition. In line with our result, downregulation of VLA-4 on human CLL cells was only observed after longer treatment with Ibrutinib, while effects on adhesion were already detectable few hours after Ibrutinib treatment.19 BTK can regulate protein kinase C (PKC) activation.20 Hence, we treated leukemic cells with phorbol 12-myristate 13-acetate (PMA), which activates PKC directly. PMA treatment inversed the effect of BTK inhibition by upregulating cell surface expression of ALCAM (3.5 ± 1.9-fold) and NOTCH2 (2 ± 1.1-fold), and reduced CD49d expression levels by 58 ± 11% (Fig. 2C).
BTK is also involved in chemokine receptor-,21 IL-6-,22 CD40-,23 and TLR-mediated signaling.24,25 To inhibit chemokine receptor-mediated signals, Eµ-Tcl1 cells were treated with pertussis toxin (PTX), an inhibitor of Gαi/o-proteins. Twenty-four hours after treatment, CD49d and ALCAM expression levels were slightly downregulated. Treatment with IL-6 or with sCD40L had no influence on ALCAM and CD49d and only a minor effect on NOTCH2 expression (Fig. S3C).
Surface expression of adhesion molecules can be modulated in vitro by TLR stimulation
Within the MZ of the spleen, immune cells are strongly exposed to bacterial antigens or unmethylated DNA, which can be recognized by TLRs. Stimulation of TLR signaling activates IRAK kinases, MAPK, and NF-κB signaling pathways26 and is associated with BTK and PKC activity.25,27 Human B-CLL and SMZL cells express numerous TLRs, i.e., TLR1, TLR2, TLR6, TLR7, TLR9, and TLR10.7,28,29 Engagement of these receptors with their respective ligands leads to activation or proliferation of CLL,28,29 and SMLZ7 cells. CLL cells in LNs showed upregulation of gene sets, indicating TLR signaling, compared with CLL cells from blood and BM.30
Splenic Eµ-Tcl1 leukemic cells highly expressed TLR2 and TLR9, whereas TLR4 was only modestly expressed following LPS stimulation (Fig. 3A). Co-cultured Eµ-Tcl1 cells were stimulated with or without TLR1/2 (PAM 3CSK4), TLR6/2 (FSL-1), TLR4 (LPS), and TLR9 (CpG ODN 1826) specific agonists, and expression of ALCAM, CD49d, and NOTCH2 was analyzed after 24 h. Stimulation of TLR1/2, TLR4, or TLR9 increased ALCAM and NOTCH2 surface expression by 1.8 ± 0.5–2.7 ± 1.3-fold and 2.9 ± 1.6–4.5 ± 3.0-fold, respectively (Figs. 3B–D). CD49d expression was only significantly induced by the TLR6/2-ligand FSL-1 (Fig. 4A). FSL-1 induced strong upregulation of ALCAM (1.9 ± 0.3-fold) and NOTCH2 (3.3 ± 1.6-fold) expression but also a significant upregulation of CD49d (1.5 ± 0.3-fold). Next, we introduced an inhibitor for IRAK1/4 that inhibits the activation of IRAK kinases by TLR1/2, TLR6/2, and TLR9 stimulation.7 Three hours later, cells were stimulated with FSL-1. TLR6/2-ligand induced changes in ALCAM and CD49d surface expression could be effectively reversed by IRAK1/4 inhibition, whereas upregulation of NOTCH2 was only partially inhibited (Fig. 4B).
Figure 3.
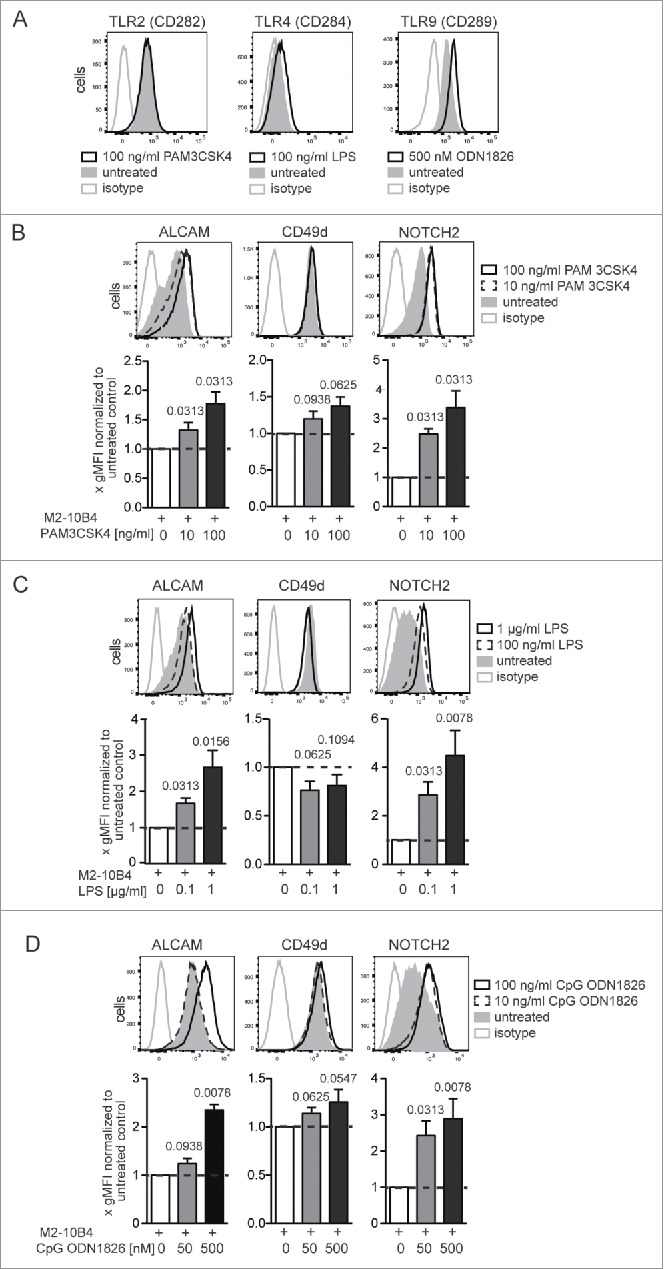
Activation of TLR-dependent signaling alters cell surface expression of ALCAM, CD49d, and NOTCH2 on Eμ-Tcl1 tumor cells. (A) 4 ×105 Eµ-Tcl1 cells/mL were co-cultured with M2-10B4 cells and supplemented with either 100 ng/mL TLR1/2 ligand PAM3CSK4, 100 ng/mL TLR4 activator LPS, 500 nM TLR9 ligand ODN1826 or without. Surface expression of TLR2 (CD282), 4 (CD284), and intracellular expression of TLR9 (CD289) was assessed after 24 h. Representative histograms from n = 7 Eµ-Tcl1 leukemia cell clones analyzed in three independent co-culture experiments are depicted. (B–D) 4 × 105 tumor cells/ml were supplemented with either (B) TLR1/2 agonist PAM 3CSK4, (C) LPS, or (D) stimulatory class B CpG ODN1826, a TLR9 agonist. Surface expression of ALCAM, CD49d, and NOTCH2 was assessed after 24 h. Per treatment 3–5 independent experiments were conducted with in total 6–10 tumor cell clones. Representative histograms of one clone are shown. Clone specific differences were normalized by dividing the gMFI of each treated sample with the gMFI of the corresponding untreated sample and bar diagrams represent means and SEMs of fold gMFIs. p values were calculated with the Wilcoxon signed rank test against a theoretical median of 1.
Figure 4.
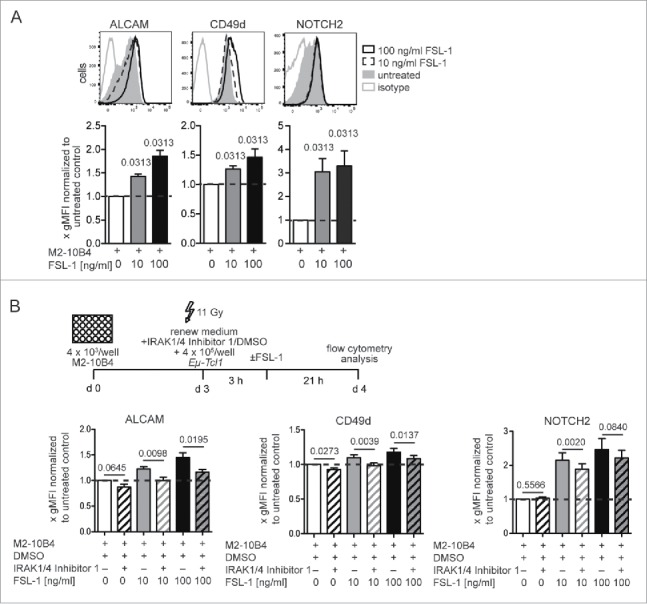
TLR6/2-ligand induced changes in cell surface expression of ALCAM and CD49d could be effectively reversed with IRAK1/4 inhibition. (A) 4 × 105 tumor cells/ml were supplemented with the TLR2/6 agonist FSL-1. Surface expression of ALCAM, CD49d, and NOTCH2 was assessed after 24 h. Per treatment 3–5 independent experiments were conducted with in total 6–10 tumor cell clones. Representative histograms of one clone are shown. Clone-specific differences were normalized by dividing the gMFI of each treated sample with the gMFI of the corresponding untreated sample and bar diagrams represent means and SEMs of fold gMFIs. (B) 4 ×105 tumor cells/mL were added in medium supplemented with 10 µM IRAK1/4-Inhibitor or DMSO. After 3 h, cultures were supplemented 1:1 with medium plus 20 µg/mL or 200 µg/mL of the TLR2/6 ligand FSL-1. Surface expression of ALCAM, CD49d, and NOTCH2 was assessed after 24 h. Means and SEMs of fold gMFIs are shown. Per treatment 3–5 independent experiments were conducted with in total 6–10 tumor cell clones. p values were calculated with the Wilcoxon signed rank test against a theoretical median of 1.
Colletively, in vitro exposure to niche-specific stimuli could recapitulate cell surface expression pattern for CD49d and NOTCH2 as observed in vivo on follicle- versus MZ-exposed tumor cells. This indicates that localization within specific splenic niches can modulate the phenotype of Eµ-Tcl1 leukemic cells.
Surface expression of ALCAM and NOTCH2 on primary human CLL cells is upregulated by BCR and TLR2/6 activation and reduced by Ibrutinib treatment
Because the provision of splenic tissue samples from untreated CLL patients is not feasible, CLL cells from the peripheral blood of six untreated CLL patients were isolated instead and co-cultured with immobilized α-IgM (Fig. 5A) or on M2-10-B4 stromal cells as described before for murine Eµ-Tcl1 cells (Fig. S3A). Culturing human CLL cells with stromal cells alone had no effect on surface expression of adhesion molecules or NOTCH2 but increased the number of living (7-AAD−) cells (Figs. S4A–C). Stimulation with a BCR cross-linking anti-IgM antibody induced only a mild upregulation of ALCAM and NOTCH2 (Fig. 5A), possibly because peripheral blood CLL cells are no longer sensitive to BCR activation through IgM-crosslinking. In line with the murine data, surface expression of ALCAM and NOTCH2 was substantially upregulated on CLL cells treated with the TLR2/6 agonist FSL-1 (Fig. 5B). Treatment with the BTK inhibitor Ibrutinib reduced expression of both molecules (Fig. 5C). CD49d expression was absent on 4 out of 6 CLL samples and expression levels were not substantially modulated by BCR or TLR activation. Culturing blood-derived CLL cells exhibiting low CD49d expression levels with stromal cells up to 96 h did not lead to further upregulation of CD49d expression (Fig. S4B). The observed discrepancy to our mouse data might be explained by this lack or downregulation of CD49d expression on peripheral blood CLL cells. CD49d/VLA-4 expression has been described as an independent negative overall survival prognosticator in CLL.31,32 Hence, the murine Eµ-Tcl1 model resembles more the agressive CD49d± subgroup of CLL patients, whereas the CLL samples made available for us displayed low or absent CD49d expression levels. This might also explain the observed discrepancies of our mouse data.
Figure 5.
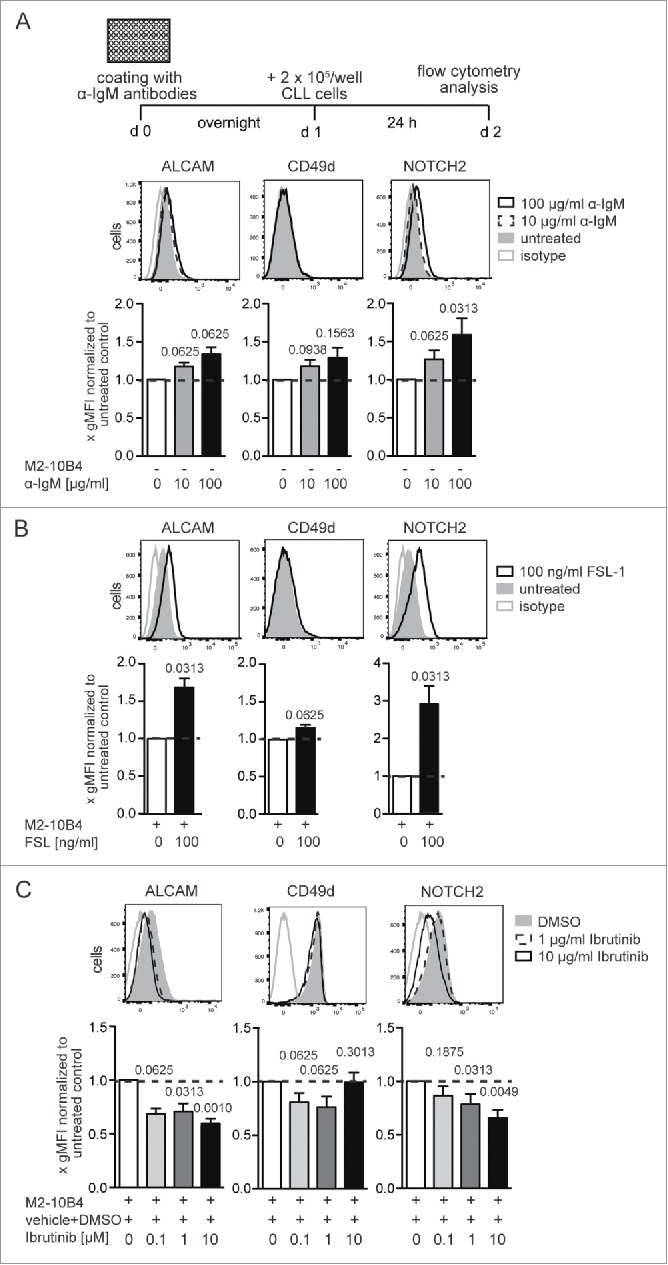
The surface expression of ALCAM and NOTCH2 on human CLL cells can be modulated by BCR and TLR activation and by Ibrutinib treatment. CLL cells were isolated from peripheral blood of six untreated CLL patients and cultured in 96-well plates with immobilized α-IgM antibodies (A) or in 24-well plates on irradiated M2-10B4 cells for 24 h supplemented with either the TLR6/2 agonist FSL-1 (B) or the BTK inhibitor Ibrutinib (C). ALCAM, CD49d, and NOTCH2 expression of CD5+CD19+7-AAD− leukemia cells was determined. Representative histograms from four independent experiments are shown. Bar diagrams represent means and SEMs of fold gMFIs. gMFI values were divided by the gMFI value of the appropriate untreated control. p values shown were calculated with Wilcoxon signed rank test against a theoretical median of 1.
Nevertheless, the modulation of expression pattern of ALCAM and NOTCH2 by niche-specific stimuli could be confirmed for human CLL cells, supporting their phenotypic adaption and flexibility.
Differential surface expression of adhesion molecules is regulated by CXCR5-dependent compartment-specific homing
Gene expression analysis and co-culture experiments were performed with splenic tumor cells of Eµ-Tcl1 and Cxcr5−/−Eµ-Tcl1 mice with a tumor burden from 20 to 40%. Hence, the splenic compartimentalization of the B, T cell zone, and MZ was no longer conserved. To reintroduce Eµ-Tcl1 cells into an intact splenic architecture, we transferred Eµ-Tcl1 and Cxcr5−/−Eµ-Tcl1 cells into wt mice (Fig. 6A). Three days after transfer, surface expression of ALCAM and NOTCH2 was assessed on re-isolated splenic tumor cells (Fig. 6A). As observed in the transgenic mice, ALCAM expression was higher in Eµ-Tcl1 cells, which homed to the follicle, whereas NOTCH2 expression was higher in MZ-localized Cxcr5−/−Eµ-Tcl1 cells. In benign B cells, interferon regulatory factor-4 (IRF4) expression is associated with NOTCH2 activity. IRF4-deficiency caused upregulation of NOTCH2 and replacement of mature B cells from the follicle into the MZ.33 Here, we isolated mRNA from Eµ-Tcl1 and Cxcr5−/−Eµ-Tcl1 cells 3 d after transfer and found that NOTCH2 expression was higher in Cxcr5−/−Eµ-Tcl1 cells, whereas IRF4 expression was reduced (Fig. 6B). This implicates that NOTCH2 expression could be negatively controlled by IRF4 signaling also in malignant B cells.
Figure 6.
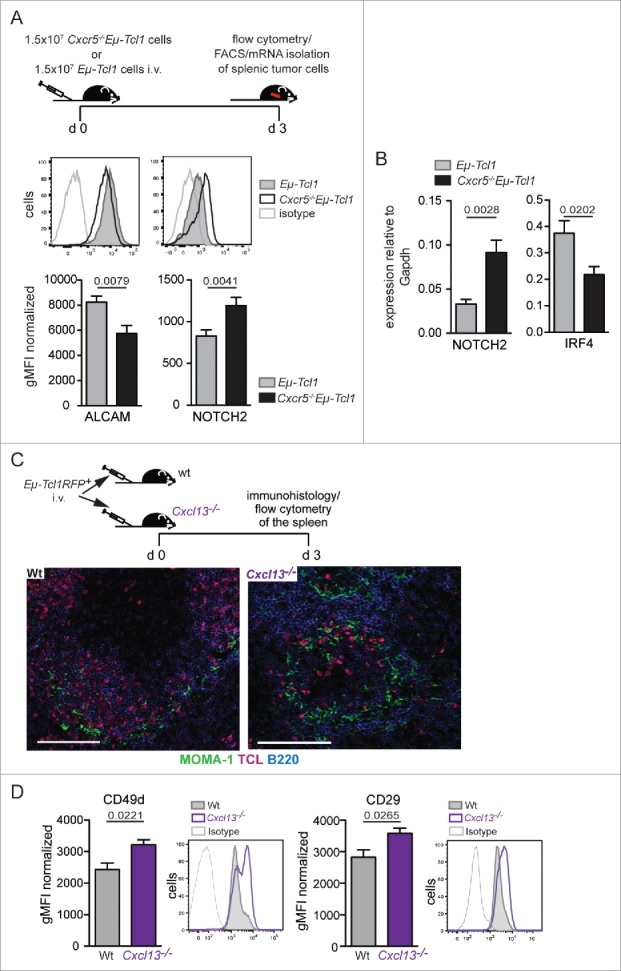
Altered expression levels of ALCAM, CD49d, and NOTCH2 on Eμ-Tcl1 tumor cells in vivo is dependent on the microenvironmental context. (A–B) 1.5 × 107 Eμ-Tcl1 or Cxcr5−/−Eμ-Tcl1 cells were transferred i.v. into wt mice. After 3 d, (A) cells were re-isolated and surface expression of ALCAM and NOTCH2 was determined. Representative histograms show ALCAM and NOTCH2 expression on Eμ-Tcl1 (solid gray) or Cxcr5−/−Eμ-Tcl1 (black line) cells. Bars represent mean expression levels ± SEM of three independent experiments with total n = 8–9 mice per group. (B) Quantitative RT-PCR of NOTCH2 and IRF4 expression in sorted CD5+CD19+ tumor cells was performed. Transcript expression was normalized to Gapdh. Error bars indicate mean ± SEM of five independent experiments (n = 8–9 mice per group). (C) 1 × 106 RFP+Eμ-Tcl1 cells were transferred in wt (n = 5) and Cxcl13−/− mice (n = 5) and re-isolated from spleens after 3 d. Representative splenic sections were stained for TCL (pink), B220 (B cells, blue), and MOMA-1 (MMMs, green). Scale bars, 100 μm. (D) Surface expression of both VLA-4 subunits (CD49d and CD29) was assessed on RFP+ tumor cells. Means ± SEM of two independent experiments are shown. Representative histograms show CD49d and CD29 expression on RFP±Eμ-Tcl1 cells derived from Wt (solid gray) or Cxcl13−/− (black line) mice. p values were determined by the Mann-Whitney test.
To exclude that CXCR5-deficiency itself rather than the microenvironment caused altered expression of adhesion molecules, we transferred Eμ-Tcl1 cells into wt and Cxcl13−/− mice. In Cxcl13−/− mice, B cells fail to home to the B cell follicle whose formation is impaired.34 Three days after transfer, Eμ-Tcl1 cells homed to the B cell follicle in wt recipients, whereas in Cxcl13−/− mice tumor cells were found in the MZ (Fig. 6C). Eμ-Tcl1 cells isolated from Cxcl13−/− mice exhibited higher VLA-4 expression compared with cells from wt mice (Fig. 6D). Thus, leukemia cells exposed to a MZ environment upregulated expression of VLA-4 compared with tumor cells exposed to the follicular microenvironment.
Combined CXCL13 and VLA-4 inhibition reduces tumor growth
To test if VLA-4 is functionally important for the retention of Cxcr5−/−Eμ-Tcl1 cells in the MZ, we administered an inhibitory anti-CD49d Ab in a short-term adoptive transfer experiment. A significant fraction of Cxcr5−/−Eμ-Tcl1 cells was displaced from the MZ to the RP (Fig. 7A and Fig. S5). Thus, VLA-4 is crucial for the retention of Cxcr5−/−Eμ-Tcl1 leukemia cells within the MZ. Next, Eµ-Tcl1 leukemia cells were transferred into wt recipients, which were then treated with an inhibitory anti-VLA4 Ab, an inhibitory anti-CXCL13 Ab, a combination of both, or with an isotype control over 3 weeks (Fig. 7B). Anti-VLA-4 Ab treatment alone did not interfere with follicular tumor cell homing (Fig. 7C), but their overall frequency was reduced (Fig. 7D). Anti-CXCL13 Ab treatment alone caused exclusion of Eµ-Tcl1 leukemia cells from the B cell follicle and a reduction in tumor growth, as described before.5 Most importantly, mice treated with a combination of anti-CXL13 and anti-VLA4 Abs showed the lowest leukemia progression (Figs. 7C and D), suggesting that when leukemic cells were not only excluded from the follicle but additionally from the MZ, tumor expansion can be further reduced.
Figure 7.
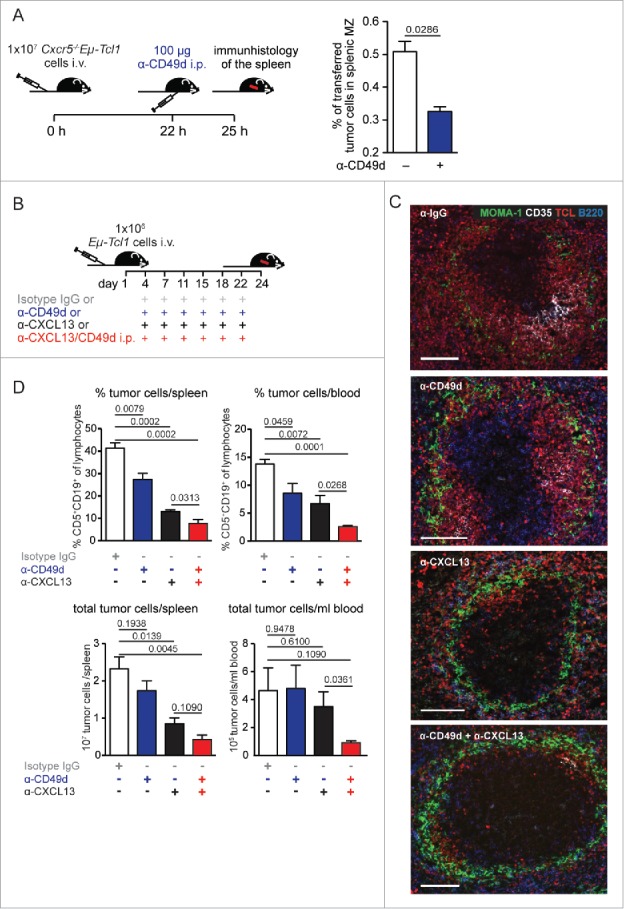
Integrin VLA-4 mediates retention of Cxcr5−/−Eμ-Tcl1 leukemia cells in the splenic MZ. (A) Cxcr5−/−Eμ-Tcl1 cells (1 × 107) were transferred in wt recipients (n = 4). 22 h later, mice were treated with an anti-CD49d Ab. After 3 h, the fraction of transferred cells in splenic MZ and RP was counted (for each spleen four different layers with 3 pictures/layer were counted). MZ was defined immunohistologically as outside of the MOMA-1+ ring and B220+. Mean ± SEM are depicted. p values were calculated with the Mann-Whitney U test. (B–D) 1 × 106 Eμ-Tcl1 cells were transferred i.v. in wt recipients treated with an IgG control, or inhibitory Abs against CXCL13, CD49d, or both (n = 3–5 mice/group and experiment) twice per week for 3 weeks from day 4 on. (C) Representative splenic sections were stained for TCL (red), B220 (B cells, blue), MOMA-1 (MMMs, green), and CD35 (FDCs, white). Scale bar, 100 μm. (D) Tumor progression was analyzed 24 d after transfer. Mean ± SEM of one representative out of three independent experiments are shown. p values calculated with the Student's t test are depicted.
Discussion
Distinct microanatomical environments in both, BM and SLOs, serve as protective niches and sites of leukemic cell proliferation.3,6,35
We recently showed that CXCR5-deletion displaced Eµ-Tcl1 leukemic cells from the B cell follicle into the MZ of the spleen.5 Notably, human SMZL, which is also positioned extra-follicularly, exhibits lower expression of CXCR5 and reduced migratory responses toward CXCL13 compared with follicular-located lymphoma.36
In this study, we now elucidate what cellular and molecular factors provided by the MZ may account for leukemic cell retention and expansion. We provide experimental evidence that the MZ has a direct impact on the leukemic cell phenotype. Using unbiased genome-wide expression arrays, we found that genes involved in the development of MZ B cells and the formation of SMZL were upregulated in Cxcr5−/−Eµ-Tcl1 cells as well as homing receptors and adhesion molecules mediating retention of MZ B cells. The adhesion molecule CD49d, which is upregulated in Cxcr5−/−Eµ-Tcl1 cells, is crucially involved in MZ B cell retention.13 In human SMZL, higher expression levels of CD49d have been described compared with human CLL cells.37
We additionally found enhanced expression of NOTCH2. Activation of the NOTCH2 pathway has been linked to B cell retention in the MZ33 and constitutive expression of active NOTCH2 gives rise to increased numbers of MZ versus follicular B cells.38
ALCAM, which is downregulated in MZ versus follicular B cells,33,39 was also downregulated on Cxcr5−/−Eµ-Tcl1 compared with follicular-located Eµ-Tcl1 cells. In support of the gene expression data, differences in surface expression levels between leukemic cells derived from Cxcr5−/−Eµ-Tcl1 or Eµ-Tcl1 mice were confirmed for CD49d, ALCAM, and NOTCH2. Similar to the genetic deletion of CXCR5, functional ablation of the CXCL13/CXCR5 signaling axis induced upregulation of CD49d and the accumulation of leukemia cells in the MZ.
These results raised the question what compartmental factors and molecular pathways in the stromal MZ were capable of inducing phenotypical changes. Antigenic stimulation through immune receptors such as BCR and TLR, has been postulated to be involved in the development of CLL and SMZL,40-42 in addition expression of functional TLRs and signaling molecules were described in CLL,28,29,43 and SMZL.7 Mutations in genes that are activated through BCR signaling or toll-like and interleukin signaling were associated with constitutive activation of TLR-signaling and a higher histological score in SMZL.44,45
Antibody-mediated BCR stimulation, a stimulus provided in B cell follicles, induced upregulation of ALCAM in a probably BTK- and PKC-dependent manner, whereas a PKC-dependent downregulation of CD49d occurred. Hence, strong in vitro activation of the BCR enhanced the expression of surface molecules that were also exhibited by leukemia cells localized in the follicle in vivo. In contrast, Eµ-Tcl1 cells exposed to a milieu mimicking the MZ microenvironment, as characterized by the presence of bacterial antigens, exhibited upregulated NOTCH2 and CD49d surface expression. Thus, this inducible phenotype recapitulated the expression pattern of MZ-localized Cxcr5−/−Eµ-Tcl1 cells in vivo.
Recently, a study in IRF4-deficient mice showed that replacement of benign follicular B cells into the MZ was associated with enhanced NOTCH2 expression.33 The authors concluded that IRF4 controls the positioning of B cells in SLOs by regulating NOTCH2 expression. However, a causal molecular link of how IRF4 regulates NOTCH2 expression was not provided. Here, we demonstrate instead that external stimuli in the MZ were sufficient to alter expression of NOTCH2 and adhesion molecules. These results were further strengthened by our data showing that surface expression of NOTCH2 and adhesion molecules was upregulated on primary human CLL cells upon TLR stimulation. Notably, activating NOTCH2 mutations have been frequently found in patients with SMZL.11,46 Because MZ-positioned Cxcr5−/−Eµ-Tcl1 cells concomitantly downregulated IRF4 and upregulated NOTCH2, we propose an alternative explanation for dysregulated NOTCH2 expression. IRF4 does not necessarily act upstream of NOTCH2, but could influence indirectly the localization of B cells in the MZ by altering expression levels of homing receptors. Within the MZ, stimulatory factors may then be provided that alter NOTCH2 activity and subsequently, expression of adhesion molecules that facilitate B cell retention in the MZ.
Collectively, our results indicate that the splenic MZ can modulate expression of surface molecules on Eµ-Tcl1 cells and by that, impact on their further retention therein.
Splenic MZ B cells rapidly respond to blood-borne antigens after sensing conserved microbial molecular signatures via TLRs.47 Human B-CLL and SMZL cells also express a wide range of TLRs and their stimulation leads to activation and proliferation of tumor cells.7,28,29 An IRAK1/4 kinase-specific inhibitor blocked TLR-mediated pro-survival effects and it was suggested that this pathway represents a putative therapeutic target.7 In the Eµ-Tcl1 mouse model, unabated TLR-mediated stimulation caused accelerated leukemia progression.48 We show that Eµ-Tcl1 cells expressed TLR2, TLR4, and TLR9 and upon ligand-specific stimulation, surface expression of NOTCH2, ALCAM, and CD49d was altered. Importantly, TLR6/2-ligand induced changes in ALCAM and CD49d expression were effectively reversed by an IRAK1/4 kinase inhibitor. We conclude that TLR stimulation induces the upregulation of adhesion molecules, which facilitates retention of leukemic cells in a growth-promoting niche. Additionally, within the MZ Cxcr5−/−Eµ-Tcl1 leukemic cells recapitulate some features of human SMZL cells, including susceptibility toward TLR stimulation, upregulation of transcription factors such as NOTCH211 and PAX5,10 upregulation of CD49d37 and downregulation of CXCR5 expression.36
In vitro studies showed that the BTK inhibitor Ibrutinib targeted BCR- and chemokine-controlled adhesion and migration49 and induced a partially VLA-4-dependent adhesion defect.19 In a TCL1 adoptive transfer model, treatment with Ibrutinib caused an increase in circulating leukemia cells, probably due to emigration of the leukemia cells from SLOs.50 These studies provided an explanation for treatment-induced lymphocytosis and suggested a role for Ibrutinib in disrupting CD49d-dependent prosurvival signals. Here, we show that treatment with Ibrutinib substantially reduced ALCAM and NOTCH2 expression levels on Eµ-Tcl1 and primary human CLL cells which could essentially contribute to reduced adhesion of CLL cells. Upon transfer of Eµ-Tcl1 cells in immunocompetent recipients, pharmacological inhibition of VLA-4 reduced leukemia growth and was most effective in a combined anti-CXCL13 and anti-VLA-4 application.
Our data indicate that MZ niche-specific cellular and molecular interactions shape the phenotype of leukemic cells, foremost the upregulation of NOTCH2 and adhesion molecules. These microenvironmental signals gain pathogenic significance because they promote leukemic cell retention and disease progression independent from tumor-inherent genetic aberrations.
Materials and methods
Transgenic mice
Eμ-Tcl1, Cxcr5−/−, and Cxcr5−/−Eμ-Tcl1 transgenic mice on a C57BL/6 background were generated as described.51,52 RFP+C57BL/6 mice were obtained from H.J. Fehling (University Clinics Ulm, Germany) and crossed with Eμ-Tcl1 mice to generate RFP+Eμ-Tcl1 mice. Congenic C57BL/6 Ly5.1 mice were obtained from Charles River (Sulzfeld, Germany). Cxcl13−/− mice were obtained from Jackson Laboratory (Bar Habor, ME; USA). All animal studies were performed according to institutional and Berlin State guidelines.
Generation of primary Eμ-Tcl1 leukemia cells for adoptive cell transfer
Spleen-derived CD5+CD19+ leukemia cell suspensions (tumor cell load > 80%) were prepared by tissue homogenization and depletion of red blood cells. Mice were challenged intravenously (i.v.) with 1 × 106 tumor cells in long-term and 1–2 × 107 tumor cells in short-term transfer experiments.
Chemotaxis assay
Chemotaxis assays were performed in 5-μm-pore transwell plates (Corning) for 4 h at 37°C, as described previously.53 Sphingosine 1-phosphate (S1P) (Sigma) was used at a concentration of 1 nM, 10 nM, and 100 nM.
In vivo inhibition of S1P/S1P1–3,5 receptor signaling
Thirteen hours after adoptive tumor cell transfer, mice were treated i.p. with 1 mg/kg body weight FTY720 (Cayman Chemical), a S1P1, S1P3, S1P4, and S1P5 agonist. Three hours later mice were killed and peripheral blood and spleens were further analyzed by flow cytometry and immunohistology.
Ex-vivo cell labeling
Splenic leukemia cells derived from diseased Eμ-Tcl1 mice or derived from diseased Cxcr5−/−Eµ-Tcl1 mice were labeled with 5 μM SNARF-1 (Molecular Probes) in PBS/ 2%FBS for 15 min at room temperature, respectively.
In vivo treatment with antibodies
Tumor challenged mice were injected intraperitoneally (i.p.) over 3 weeks twice weekly with 80–100 µg LE/AF purified rat anti-mouse CD49d (clone R1-2; SouthernBiotech), 50 µg rat anti-mouse CXCL13 (clone # 143614) and rat IgG control antibody (Ab) (R&D Systems).
Cell lines
The murine BM stromal cell line M2-10B4 (ATCC-CRL-1972) was obtained from ATCC (Braunschweig, Germany) in 2010. The cells were passaged 2–3 times over 3 weeks and aliquots were frozen in liquid nitrogen. All experiments were performed with these aliquots.
Patient CLL blood samples
Peripheral blood from six treatment-naive CLL patients was purified over a Ficoll gradient. The study was conducted according to the Declaration of Helsinki and in accordance with local ethical guidelines; written informed consent of all patients was obtained.
Statistical analysis
Results are expressed as arithmetic means ± SEM if not otherwise indicated. Values of p < 0.05 were considered statistically significant, as determined by the unpaired Mann-Whitney U test, the unpaired or paired Student's t test, or the Wilcoxon signed rank test where appropriate.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Carlo Croce (Columbus University, OH, USA) for providing us with essential reagents and Kerstin Krüger, Branka Kampfrath and Heike Schwede (all from the Max-Delbrück-Center for Molecular Medicine, MDC, 13125 Berlin, Germany) for excellent technical assistance.
Funding
This work was funded by a grant from the German Research Foundation (DFG; grant number 1502/4–1 and 11302/9–1) to U.E.H. and A.R.
Author contributions
Conception and design: U.E. Höpken, V. Stache; Development of methodology: V. Stache, U.E. Höpken; Analysis and interpretation of data: V. Stache, L. Verlaat, M. Gätjen, K. Heinig, J. Westermann, A. Rehm, U.E. Höpken. Acquisition and managed patients: J. Westermann; Writing the manuscript: U.E. Höpken, V. Stache; All authors reviewed the manuscript; Study supervision: U.E. Höpken, A. Rehm.
References
- 1.Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol 2012; 30:565-610; PMID:22224767; https://doi.org/ 10.1146/annurev-immunol-020711-075027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopken UE, Rehm A. Homeostatic chemokines guide lymphoma cells to tumor growth-promoting niches within secondary lymphoid organs. J Mol Med (Berl) 2012; 90:1237-45; PMID:22577036; https://doi.org/ 10.1007/s00109-012-0906-z [DOI] [PubMed] [Google Scholar]
- 3.Burger JA, Gribben JG. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: Insight into disease biology and new targeted therapies. Semin Cancer Biol 2014; 24:71-81; PMID:24018164; https://doi.org/ 10.1016/j.semcancer.2013.08.011 [DOI] [PubMed] [Google Scholar]
- 4.Gatjen M, Brand F, Grau M, Gerlach K, Kettritz R, Westermann J, Anagnostopoulos I, Lenz P, Lenz G, Höpken UE et al.. Splenic marginal zone granulocytes acquire an accentuated neutrophil B-Cell helper phenotype in chronic lymphocytic leukemia. Cancer Res 2016; 76:5253-65; PMID:27488528; https://doi.org/ 10.1158/0008-5472.CAN-15-3486 [DOI] [PubMed] [Google Scholar]
- 5.Heinig K, Gätjen M, Grau M, Stache V, Anagnostopoulos I, Gerlach K, Niesner RA, Cseresnyes Z, Hauser AE, Lenz P et al.. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia B-cell activation and proliferation. Cancer Discov 2014; 4:1448-65; PMID:25252690; https://doi.org/ 10.1158/2159-8290.CD-14-0096 [DOI] [PubMed] [Google Scholar]
- 6.Caligaris-Cappio F, Bertilaccio MT, Scielzo C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin Cancer Biol 2014; 24:43-8; PMID:23831274; https://doi.org/ 10.1016/j.semcancer.2013.06.010 [DOI] [PubMed] [Google Scholar]
- 7.Fonte E, Agathangelidis A, Reverberi D, Ntoufa S, Scarfò L, Ranghetti P, Cutrona G, Tedeschi A, Xochelli A, Caligaris-Cappio F et al.. Toll-like receptor stimulation in splenic marginal zone lymphoma can modulate cell signaling, activation and proliferation. Haematologica 2015; 100:1460-8; PMID:26294727; https://doi.org/ 10.3324/haematol.2014.119933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, Proia RL, Cyster JG. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol 2004; 5:713-20; PMID:15184895; https://doi.org/ 10.1038/ni1083 [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Beaty N, Chen S, Qi CF, Masiuk M, Shin DM, Morse HC 3rd. The CXCR7 chemokine receptor promotes B-cell retention in the splenic marginal zone and serves as a sink for CXCL12. Blood 2012; 119:465-8; PMID:22110250; https://doi.org/ 10.1182/blood-2011-03-343608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison AM, Jäger U, Chott A, Schebesta M, OA Haas, Busslinger M. Deregulated PAX-5 transcription from a translocated IgH promoter in marginal zone lymphoma. Blood 1998; 92:3865-78; PMID:9808580. [PubMed] [Google Scholar]
- 11.Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, Huebner-Chan DR, Bailey NG, Yang DT, Bhagat G et al.. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med 2012; 209:1553-65; PMID:22891276; https://doi.org/ 10.1084/jem.20120910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K et al.. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 2003; 18:675-85; PMID:12753744; https://doi.org/ 10.1016/S1074-7613(03)00111-0 [DOI] [PubMed] [Google Scholar]
- 13.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science 2002; 297:409-12; PMID:12130787; https://doi.org/ 10.1126/science.1071632 [DOI] [PubMed] [Google Scholar]
- 14.Hemler ME. VLA proteins in the integrin family: Structures, functions, and their role on leukocytes. Annu Rev Immunol 1990; 8:365-400; PMID:2188667; https://doi.org/ 10.1146/annurev.iy.08.040190.002053 [DOI] [PubMed] [Google Scholar]
- 15.Baalasubramanian S, Harris CL, Donev RM, Mizuno M, Omidvar N, Song W-C, Morgan BP. CD59a is the primary regulator of membrane attack complex assembly in the mouse. J Immunol 2004; 173:3684-92; PMID:15356114; https://doi.org/ 10.4049/jimmunol.173.6.3684 [DOI] [PubMed] [Google Scholar]
- 16.Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, Haqqani AS, Kreymborg K, Krug S, Moumdjian R et al.. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol 2008; 9:137-45; PMID:18157132; https://doi.org/ 10.1038/ni1551 [DOI] [PubMed] [Google Scholar]
- 17.Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, Sivina M, Wierda WG, Estrov Z, Keating MJ et al.. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: Development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009; 114:4441-50; PMID:19762485; https://doi.org/ 10.1182/blood-2009-07-233718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer 2014; 14:219-32; PMID:24658273; https://doi.org/ 10.1038/nrc3702 [DOI] [PubMed] [Google Scholar]
- 19.Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with Ibrutinib inhibits BTK- and VLA-4-Dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin Cancer Res 2015; 21:4642-51; PMID:26089373; https://doi.org/ 10.1158/1078-0432.CCR-15-0781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene 2000; 19:5651-61; PMID:11114746; https://doi.org/ 10.1038/sj.onc.1203958 [DOI] [PubMed] [Google Scholar]
- 21.de Gorter DJJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, Pals ST, Spaargaren M. Bruton's tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 2007; 26:93-104; PMID:17239630; https://doi.org/ 10.1016/j.immuni.2006.11.012 [DOI] [PubMed] [Google Scholar]
- 22.Takahashi-Tezuka M, Hibi M, Fujitani Y, Fukada T, Yamaguchi T, Hirano T. Tec tyrosine kinase links the cytokine receptors to PI-3 kinase probably through JAK. Oncogene 1997; 14:2273-82; PMID:9178903; https://doi.org/ 10.1038/sj.onc.1201071 [DOI] [PubMed] [Google Scholar]
- 23.Brunner C, Avots A, Kreth HW, Serfling E, Schuster V. Bruton's tyrosine kinase is activated upon CD40 stimulation in human B lymphocytes. Immunobiology 2002; 206:432-40; PMID:12437073; https://doi.org/ 10.1078/0171-2985-00192 [DOI] [PubMed] [Google Scholar]
- 24.Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, Walch E, Wirth T, O'Neill LA. Bruton's tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem 2003; 278:26258-64; PMID:12724322; https://doi.org/ 10.1074/jbc.M301484200 [DOI] [PubMed] [Google Scholar]
- 25.Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, Brennan FM, Webster D, Foxwell BM. Bruton's tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol 2006; 176:3635-41; PMID:16517732; https://doi.org/ 10.4049/jimmunol.176.6.3635 [DOI] [PubMed] [Google Scholar]
- 26.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499-511; PMID:15229469; https://doi.org/ 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- 27.Asehnoune K, Strassheim D, Mitra S, Yeol Kim J, Abraham E. Involvement of PKCalpha/beta in TLR4 and TLR2 dependent activation of NF-kappaB. Cell Signal 2005; 17:385-94; PMID:15567069; https://doi.org/ 10.1016/j.cellsig.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 28.Spaner DE, Masellis A. Toll-like receptor agonists in the treatment of chronic lymphocytic leukemia. Leukemia 2006; 21:53-60; PMID:17066089; https://doi.org/ 10.1038/sj.leu.2404456 [DOI] [PubMed] [Google Scholar]
- 29.Muzio M, Scielzo C, Bertilaccio MTS, Frenquelli M, Ghia P, Caligaris-Cappio F. Expression and function of toll like receptors in chronic lymphocytic leukaemia cells. Br J Haematol 2009; 144:507-16; PMID:19036098; https://doi.org/ 10.1111/j.1365-2141.2008.07475.x [DOI] [PubMed] [Google Scholar]
- 30.Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, Gibellini F, Njuguna N, Lee E, Stennett L et al.. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011; 117:563-74; PMID:20940416; https://doi.org/ 10.1182/blood-2010-05-284984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulian P, Shanafelt TD, Fegan C, Zucchetto A, Cro L, Nuc¨kel H, Baldini L, Kurtova AV, Ferrajoli A, Burger JA et al.. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J Clin Oncol 2014; 32:897-904; PMID:24516016; https://doi.org/ 10.1200/JCO.2013.50.8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dal Bo M, Bulian P, Bomben R, Zucchetto A, Rossi FM, Pozzo F, Tissino E, Benedetti D, Bittolo T, Nanni P et al.. CD49d prevails over the novel recurrent mutations as independent prognosticator of overall survival in chronic lymphocytic leukemia. Leukemia 2016; 30:2011-8; PMID:27109509; https://doi.org/ 10.1038/leu.2016.88 [DOI] [PubMed] [Google Scholar]
- 33.Simonetti G, Carette A, Silva K, Wang H, De Silva NS, Heise N, Siebel CW, Shlomchik MJ, Klein U. IRF4 controls the positioning of mature B cells in the lymphoid microenvironments by regulating NOTCH2 expression and activity. J Exp Med 2013; 210:2887-902; PMID:24323359; https://doi.org/ 10.1084/jem.20131026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000; 406:309-14; PMID:10917533; https://doi.org/ 10.1038/35018581 [DOI] [PubMed] [Google Scholar]
- 35.Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, Murphy EJ, Koduru P, Ferrarini M, Zupo S et al.. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest 2005; 115:755-64; PMID:15711642; https://doi.org/ 10.1172/JCI23409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trentin L, Cabrelle A, Facco M, Carollo D, Miorin M, Tosoni A, Pizzo P, Binotto G, Nicolardi L, Zambello R et al.. Homeostatic chemokines drive migration of malignant B cells in patients with non-Hodgkin lymphomas. Blood 2004; 104:502-8; PMID:15001469; https://doi.org/ 10.1182/blood-2003-09-3103 [DOI] [PubMed] [Google Scholar]
- 37.Csanaky G, Matutes E, Vass JA, Morilla R, Catovsky D. Adhesion receptors on peripheral blood leukemic B cells. A comparative study on B cell chronic lymphocytic leukemia and related lymphoma/leukemias. Leukemia 1997; 11:408-15; PMID:9067581; https://doi.org/ 10.1038/sj.leu.2400582 [DOI] [PubMed] [Google Scholar]
- 38.Hampel F, Ehrenberg S, Hojer C, Draeseke A, Marschall-Schroter G, Kuhn R, Mack B, Gires O, Vahl CJ, Schmidt-Supprian M et al.. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood 2011; 118:6321-31; PMID:21795747; https://doi.org/ 10.1182/blood-2010-12-325944 [DOI] [PubMed] [Google Scholar]
- 39.Kin NW, Crawford DM, Liu J, Behrens TW, Kearney JF. DNA microarray gene expression profile of marginal zone versus Follicular B cells and idiotype positive marginal Zone B Cells before and after Immunization with Streptococcus pneumoniae. J Immunol 2008; 180:6663-74; PMID:18453586; https://doi.org/ 10.4049/jimmunol.180.10.6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagner M, Oelsner M, Moore A, Gotte F, Kuhn PH, Haferlach T, Fiegl M, Bogner C, Baxter EJ, Peschel C et al.. Integration of innate into adaptive immune responses in ZAP-70-positive chronic lymphocytic leukemia. Blood 2016; 127:436-48; PMID:26508782; https://doi.org/ 10.1182/blood-2015-05-646935 [DOI] [PubMed] [Google Scholar]
- 41.Isaza-Correa JM, Liang Z, van den Berg A, Diepstra A, Visser L. Toll-like receptors in the pathogenesis of human B cell malignancies. J Hematol Oncol 2014; 7:57; PMID:25112836; https://doi.org/ 10.1186/s13045-014-0057-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan Q, Huang Y, Watkins AJ, Kocialkowski S, Zeng N, Hamoudi RA, Isaacson PG, de Leval L, Wotherspoon A, Du MQ. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica 2012; 97:595-8; PMID:22102703; https://doi.org/ 10.3324/haematol.2011.054080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muzio M, Fonte E, Caligaris-Cappio F. Toll-like receptors in chronic lymphocytic leukemia. Mediterr J Hematol Infect Dis 2012; 4:e2012055; PMID:22973499; https://doi.org/ 10.4084/mjhid.2012.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Troen G, Warsame A, Delabie J. CD79B and MYD88 mutations in splenic marginal zone lymphoma. ISRN Oncol 2013; 2013:252318; PMID:23378931; https://doi.org/ 10.1155/2013/252318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez-Lopez A, Curiel-Olmo S, Mollejo M, Cereceda L, Martinez N, Montes-Moreno S, Almaraz C, Revert JB, Piris MA. MYD88 (L265P) somatic mutation in marginal zone B-cell lymphoma. Am J Surg Pathol 2015; 39:644-51; PMID:25723115; https://doi.org/ 10.1097/PAS.0000000000000411 [DOI] [PubMed] [Google Scholar]
- 46.Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Famà R et al.. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med 2012; 209:1537-51; PMID:22891273; https://doi.org/ 10.1084/jem.20120904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerutti A, Cols M, Puga I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol 2013; 13:118-32; PMID:23348416; https://doi.org/ 10.1038/nri3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertilaccio MTS, Simonetti G, Dagklis A, Rocchi M, Rodriguez TV, Apollonio B, Mantovani A, Ponzoni M, Ghia P, Garlanda C et al.. Lack of TIR8/SIGIRR triggers progression of chronic lymphocytic leukemia in mouse models. Blood 2011; 118:660-9; PMID:21652674; https://doi.org/ 10.1182/blood-2011-01-329870 [DOI] [PubMed] [Google Scholar]
- 49.de Rooij MFM, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012; 119:2590-4; PMID:22279054; https://doi.org/ 10.1182/blood-2011-11-390989 [DOI] [PubMed] [Google Scholar]
- 50.Ponader S, Chen S-S, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O'Brien S, Chiorazzi N, Burger JA. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012; 119:1182-9; PMID:22180443; https://doi.org/ 10.1182/blood-2011-10-386417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, Russo G, Hardy RR, Croce CM. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A 2002; 99:6955-60; PMID:12011454; https://doi.org/ 10.1073/pnas.102181599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Förster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 1996; 87:1037-47; PMID:8978608; https://doi.org/ 10.1016/S0092-8674(00)81798-5 [DOI] [PubMed] [Google Scholar]
- 53.Hopken UE, Foss HD, Meyer D, Hinz M, Leder K, Stein H, Lipp M. Up-regulation of the chemokine receptor CCR7 in classical but not in lymphocyte-predominant Hodgkin disease correlates with distinct dissemination of neoplastic cells in lymphoid organs. Blood 2002; 99:1109-16; PMID:11830455; https://doi.org/ 10.1182/blood.V99.4.1109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.