

Abstract
While the formation of colloidal aggregates leads to artifacts in early drug discovery, their composition makes them attractive as nanoparticle formulations for targeted drug delivery as the entire nanoparticle is composed of drug. The typical transient stability of colloidal aggregates has inhibited exploiting this property. To overcome this limitation, we investigated a series of proteins to stabilize colloidal aggregates of the chemotherapeutic, fulvestrant, including the following: bovine serum albumin, a generic human immunoglobulin G, and trastuzumab, a therapeutic human epidermal growth factor receptor 2 antibody. Protein coronas reduced colloid size to <300 nm and improved their stability to over 48 h in both buffered saline and media containing serum protein. Unlike colloids stabilized with other proteins, trastuzumab-fulvestrant colloids were taken up by HER2 overexpressing cells and were cytotoxic. This new targeted formulation reimagines antibody–drug conjugates, delivering mM concentrations of drug to a cell.
Keywords: self-assembly, colloids, protein corona, drug delivery, cell targeting
Graphical Abstract
INTRODUCTION
Since their discovery, colloidal drug aggregates have been associated with artifacts in screening assays.1 In enzyme2 and cell-surface receptor assays,3 they lead to false-positive hits due to protein adsorption and inactivation on the colloid surface. Furthermore, colloidal aggregates lead to false-negative results in cell-viability assays due to an inability to cross the cell membrane, which inherently limits their efficacy.4 The formation of colloidal particles has been reported for multiple compound classes of organic compounds and even for therapeutic drugs, including several anticancer chemotherapeutics.5 Governed by a critical aggregation concentration, above which these compounds spontaneously self-assemble into amorphous particles, colloidal drug aggregates have several unique properties including their propensity for protein adsorption and detergent-reversible formation.6 Intriguingly, while colloids are undesirable in screening assays, they are attractive as nanoparticle formulations. Composed entirely of drug molecules, they overcome the low loadings typically encountered with nanoparticle systems.7,8
A number of nanoparticle drug formulations are being investigated for chemotherapeutic delivery. To address the issue of poor drug loading, drugs are being chemically modified to enhance their self-assembly.9–13 While these methods have been used successfully in preclinical studies, the need to chemically modify compounds complicates their translation to the clinic, as they become new chemical entities. Exploiting drugs that self-assemble without modification would be an advantage in this respect.
Many drugs form colloidal drug aggregates in biologically relevant environments, including cell culture media and simulated gastrointestinal fluids.5,14–16 Several of these drugs aggregate at micromolar or sub-micromolar concentrations, including chemotherapeutics such as the estrogen receptor (ER) antagonist fulvestrant.5 However, the colloids formed are often polydisperse and precipitate over several hours. Excipients, including polymers and even other colloid-forming compounds, such as azo dyes, can control the size and stability of these colloidal drug aggregates.17–19 We hypothesized that proteins themselves might be useful as stabilizing excipients due to their strong interactions with colloidal surfaces.20 Protein excipients have been successfully used to stabilize other nanoparticle drug dispersions, such as Abraxane, a formulation of the chemotherapeutic paclitaxel that is stabilized by human serum albumin.21 In addition to stabilizing colloidal drug aggregates, proteins can also confer functionality. For example, the adsorption of antibodies onto the surface of drug nanocrystals has been shown to promote selective uptake by target cells.22–24
Here we investigate the use of proteins to both stabilize colloidal drug aggregates and to target them to specific cell populations. We demonstrate that the formation of a protein corona on the colloid controls the size of drug colloids in a concentration-dependent manner and improves their stability in many conditions, including serum-containing media. Antibody-based coronas lead to cellular targeting of colloids, thereby enhancing uptake by target cells and the efficacy of these formulations.
RESULTS
Fulvestrant is a potent chemotherapeutic that forms transiently stable colloidal aggregates in buffer. Three different proteins were evaluated for their ability to stabilize fulvestrant colloids: bovine serum albumin (BSA), human immunoglobulin G (IgG), and trastuzumab, a clinical human epidermal growth factor receptor 2 (HER2) antibody.
As with many other self-assembled particles, colloidal drug aggregate size and stability are influenced by ionic strength.1 In water, fulvestrant colloids typically have nanometer-scale diameters (<200 nm) and narrow size distributions (<0.15 as measured by dynamic light scattering, DLS); however, when formulated in buffer, their diameters are greater than 1 μm (Supporting Information Figure S1).
We hypothesized that proteins would form coronas on the colloid surface, thereby yielding stable colloids of uniform size in buffered solutions. Colloids were first formulated in water to obtain initial diameters < 200 nm, followed by the simultaneous addition of protein and buffer salts. All three proteins controlled colloid size in a concentration-dependent manner, with a sigmoidal relationship consistent with saturation binding (Figure 1A). At low protein concentrations, colloid diameters were greater than 1 μm. As each protein concentration increased, colloid diameters decreased. Each of the three proteins stabilized fulvestrant colloids over different concentration ranges; BSA controlled colloidal size at the lowest concentration, where as little as 25 nM was sufficient to achieve colloidal diameters < 300 nm. IgG and trastuzumab required at least 1 μM and 5 μM, respectively, to reduce colloid size to similar diameters. To confirm the presence of a protein corona, colloids were centrifuged and proteins found in the resulting pelleted material were separated using gel electrophoresis, using this previously described method.2 Proteins were associated with the pelleted colloids only, indicating that they had formed a tightly bound corona at the particle surface (Figure S2). Additionally, the use of surface-sensitive techniques, such as X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (TOF-SIMS), confirmed that proteins were bound to the colloid surface (Figure S3). The control and stability conferred by protein coronas was not limited to colloidal drug aggregates of fulvestrant alone and was also observed for colloid-forming drugs such as sorafenib, vemurafenib, and chlorotrianisene (Figure S4).
Figure 1.

(A) Proteins control fulvestrant colloid size in a concentration-dependent manner. Size measured after a 4 h incubation by DLS. Proteins improve the stability of colloids during incubation in buffer salts at 37 °C as indicated by stability in (B) size and (C) scattering intensity by DLS. Colloids of fulvestrant alone precipitate over the 48 h incubation resulting in reduced scattering intensity. All formulations are 50 μM fulvestrant and 1% DMSO in PBS. For panels B and C [BSA] = 100 nM; [IgG] = 1 μM; [trastuzumab] = 3.5 μM. (n = 3, mean ± SD).
We next studied the stability of the protein–fulvestrant formulations in solution using dynamic light scattering, in order to identify which protein formulation was sufficiently stable. Minimal changes in hydrodynamic diameters were observed for all three protein-stabilized formulations over a 48 h incubation at 37 °C, with all diameters within 100 nm of the initial value (Figure 1B). Conversely, nonstabilized “bare” fulvestrant colloids maintained a large diameter, but a reduction in scattering intensity by 2 orders of magnitude was observed over 48 h due to precipitation of larger aggregates, reflecting their instability in the absence of proteins, as is typical of nonstabilized colloidal aggregates (Figure 1C). Conversely, protein-stabilized formulations maintained high scattering intensities, indicating that colloids were present and stable in buffered solutions over at least 48 h at 37 °C (Figure 1C).
We then evaluated the ability of protein coronas to stabilize fulvestrant colloids in serum-containing media. Since the high concentration and variety of proteins in serum results in a high background signal in DLS, we used transmission electron microscopy (TEM) and fast protein liquid chromatography (FPLC) to study colloidal stability. Significant morphological differences were observed by transmission electron microscopy (TEM) after incubation in 5% serum. Nonstabilized fulvestrant formulations appeared as large nonuniform aggregates, whereas protein-stabilized colloids maintained a spherical morphology of distinct particles (Figures 2A,B and S5).
Figure 2.

Protein corona formulation improves the stability of fulvestrant colloids in serum-containing media. (A) Nonstabilized and (B) trastuzumab-stabilized colloids show distinct morphologies after a 4 h incubation in 5% serum-containing media as shown by TEM. (C) Size exclusion chromatography traces show separation of BSA-stabilized colloids (blue, FRET fluorescence) from serum proteins (pink, absorbance at 280 nm). (D) After incubation in 20% serum, both BSA and trastuzumab-stabilized colloids maintain FRET fluorescence over 48 h, demonstrating their stability over this time frame. Colloids were formulated at 50 μM fulvestrant and 1% DMSO in all cases. (n = 3; mean ± SD; scale bar represents 100 nm).
To study the stability of these formulations in higher serum concentrations (20%), size exclusion chromatography was used to separate intact colloids from serum proteins (Figure 2C). Co-formulations of fulvestrant colloids with a FRET pair consisting of cholesterol derivatives of BODIPY FL (FRET donor) and BODIPY 542/563 (FRET acceptor) provided a measure of intact colloids (Figure S6). These dyes have previously been used to study self-assembled particles25 and were chosen for this study due to their physical and even structural similarity to fulvestrant. A high FRET signal, due to incorporation of these dyes within the colloids, corroborated their amorphous nature and correlated with the presence of intact particles, where exclusion of the dyes from the crystal lattice, due to precipitation, resulted in a low FRET signal (Figure S6).17,26,27 In serum-containing media, both BSA and trastuzumab-stabilized colloids had little dissociation over 48 h as indicated by the relatively constant fluorescence intensity of the colloid fraction (Figure 2D). The increase in fluorescence over the first few hours can be attributed to particle coalescence until equilibrium was reached. With this improved colloid stability, additional functionality can now be provided by adsorbed antibodies.
With colloidal formulations that were stable in serum, we investigated whether the antibody corona would lead to selective uptake by target cells. Previous studies showed that colloidal drug aggregates cannot diffuse across intact cell membranes.4 We hypothesized that colloids loaded with a targeting antibody would be selectively internalized through receptor-mediated endocytosis. We investigated the potential for colloids formulated with trastuzumab, an antibody against HER2, which is overexpressed in 25% of breast cancers,28,29 to selectively deliver fulvestrant, an estrogen receptor antagonist.30
We first evaluated the cellular uptake of trastuzumab-stabilized fulvestrant colloids by confocal laser scanning microscopy. Co-formulation with a BODIPY dye aided in direct visualization of colloids after exposure to cells for 3 h in 5% serum. The trastuzumab-stabilized colloids (green, Figure 3A) were clearly internalized by SKOV-3 cells that overexpress HER2, whereas the control IgG-stabilized colloids showed no uptake in the same cell line (Figure 3B), indicating trastuzumab-mediated uptake of fulvestrant colloids. Quantification of colloid uptake by flow cytometry showed that trastuzumab-stabilized colloids had a 10-fold increase in uptake by HER2 overexpressing cells compared to IgG-stabilized colloids (Figure S7). Furthermore, preincubation with free trastuzumab significantly reduced this uptake. Consistent with a HER2-mediated uptake mechanism, neither the trastuzumabnor the IgG-stabilized formulations were taken up by HER2 low-expressing MDA-MB-231 cells (Figure 3C,D, respectively).
Figure 3.
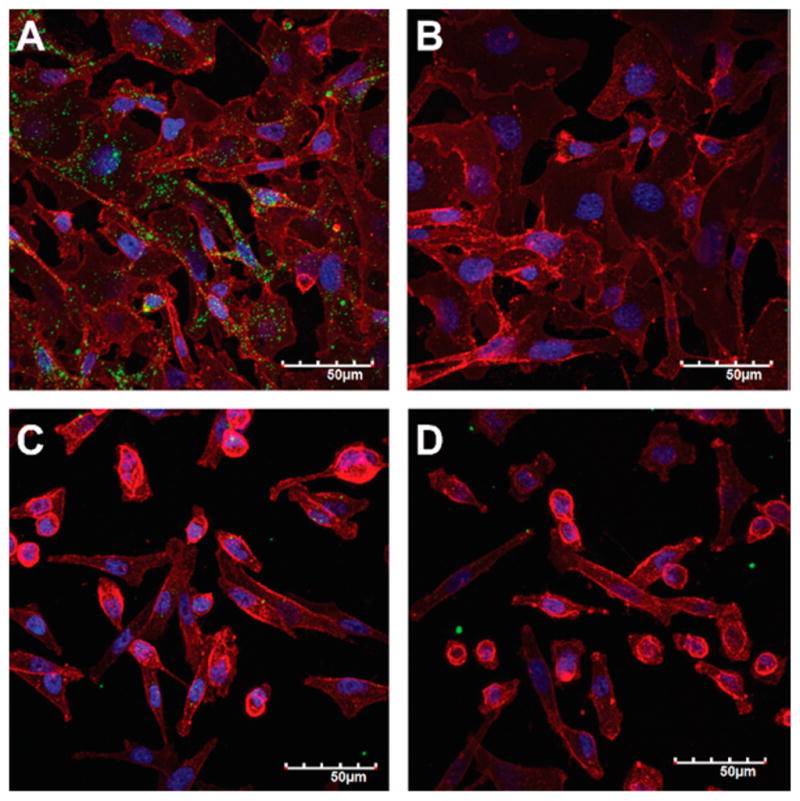
Trastuzumab corona increases uptake of colloids by HER2 overexpressing cells. (A) Trastuzumab-modified colloids (BODIPY, green), but not (B) IgG-modified colloids, are taken up by HER2 overexpressing SKOV-3 cells (blue, Hoechst for cell nuclei; red, wheat germ agglutinin for cell membranes) after a 3 h incubation. Neither (C) trastuzumab-modified nor (D) IgG-modified colloids are taken up by MDA-MB-231 cells that are HER2 low-expressing. Formulations used are 50 μM fulvestrant, 1% DMSO, and 3.5 μM antibody in 5% serum. Scale bars represent 50 μm. Representative confocal microscope images of at least three biological repeats.
Subsequent cell uptake studies revealed that trastuzumab-stabilized colloids are localized to endolysosomal compartments even 24 h after exposure (Figure 4). Trafficking to the lysosomal compartment was indicated by the co-localization of colloid fluorescence (green) with that of a lysosomal dextran marker (red). Lysosomal accumulation is only observed for trastuzumab-modified colloids (Figure 4A) and not for IgG-modified colloids (Figure 4B), consistent with the selective internalization of the former (Figure 3). Unexpectedly, the protein-formulated colloids appear to be mostly stable even in low-pH environments of the endolysosomal pathway (Figure S8).31
Figure 4.

Co-localization of internalized colloids with lysosomal compartments of SKOV-3 cells. (A) Trastuzumab-modified colloids (BODIPY, green) co-localize with lysosomes (Dextran-647, red) after 24 h. (B) IgG-modified colloids have minimal cell uptake. Hoechst (blue) is used to stain cell nuclei. Formulations used are 50 μM fulvestrant, 1% DMSO, and 3.5 μM antibody in 5% serum. Cells were pulsed with colloidal formulations for 3 h followed by chase with full media for 21 h. Representative confocal microscope images of at least three biological repeats. Scale bars represent 20 μm.
We next investigated whether these targeted colloidal formulations would improve drug efficacy relative to non-targeted colloids. Since trastuzumab-stabilized fulvestrant colloids showed selective uptake by HER2 overexpressing cells, we wondered whether they would increase efficacy against fulvestrant-sensitive BT-474 cells, which overexpress both HER2 and ER.32 Fulvestrant colloids co-formulated with trastuzumab significantly reduced viability compared to controls, while colloids stabilized with a nontargeted antibody did not show the same effect (Figure 5). Nonstabilized colloidal fulvestrant formulations had a minimal effect on cell viability. High amounts of trastuzumab were required to stabilize colloids of fulvestrant compared to the other proteins investigated and, as a result, even the trastuzumab-only controls showed a significant reduction in cell viability relative to a 1% DMSO vehicle control (Figure 5). When these formulations were incubated with fulvestrant-sensitive cells that have low expression of HER2 (MCF-7), no differences between targeted and nontargeted colloidal formulations were observed (Figure S9). We note that whereas the targeted colloids significantly reduced cell viability versus the untargeted colloids, the difference did not reflect the substantially higher amount of colloids internalized by the cells. This perhaps reflects the integrity of the colloids long after internalization. This contrasts with the expected cytotoxicity observed for monomeric formulations of fulvestrant (Figures S9 and S10).
Figure 5.

Cell viability after incubation with protein corona formulations. Fulvestrant colloids targeted with a trastuzumab corona reduce cell viability. BT-474 cells were exposed to 50 μM fulvestrant formulations with trastuzumab or IgG (3.5 μM) for 24 h followed by fresh media for an additional 48 h. All formulations used were 50 μM fulvestrant with 1% DMSO in 5% serum. Cell viability is represented as a percentage of 1% DMSO vehicle control. *, p < 0.05; **, p < 0.01. (n = 4 biological replicates; mean ± SD).
DISCUSSION
Two key results emerge from this study. First, colloidal drug aggregates may be stabilized by protein adsorption, converting them from polydisperse particles prone to precipitation into more monodisperse species with multiday stability. Second, colloids can be co-formulated with proteins that are themselves active, such as the anti-HER2 antibody trastuzumab, and can be used to target colloids to specific cell types. This antibody colloidal drug formulation is specifically internalized by target cells, increasing the toxicity of the colloids versus colloids without the targeting antibody and relative to nontarget cells. These antibody–colloidal drug formulations are analogous to antibody–drug conjugates, but deliver orders of magnitude more drug per active antibody while maintaining the drug in an inactive form prior to cell internalization.
Proteins form coronas on nanoparticle formulations upon exposure to biological media.33,34 Both the nature of the protein and colloidal surface determine the strength of this interaction, and as a result, several factors may contribute to the differences in interaction between fulvestrant colloids and the three proteins studied here.35–37 Many studies have shown that proteins readily bind to hydrophobic colloidal surfaces. Albumin, for example, can bind to a number of colloidal particles and, in most cases, improves their stability.15,38–40 The diversity in chemical groups and specifically the hydrophobic pockets of BSA could account for the superior ability of albumin to stabilize the hydrophobic fulvestrant colloids compared to the antibodies investigated here.36,41–44 Differences in post-translational modifications, such as glycosylation, can change the properties of proteins and could explain the differences observed between IgG and trastuzumab.45,46 The properties of the colloids also influence interactions with proteins. For example, the ζ potential of the colloidal surface dictates the strength of electrostatic interactions.35 Although we have focused on fulvestrant colloids in this study, we observed the same ability of proteins to stabilize colloidal drug aggregates of two other anticancer chemotherapeutics, namely, sorafenib and vemurafenib.
The concentration of the protein is an important factor in colloid stabilization; at low protein concentrations, colloidal particles agglomerate with one another, perhaps due to changes in electrostatic interactions and bridging effects.39 The colloidal diameters observed in this study at low protein concentrations are in fact larger than those of fulvestrant colloids alone. At higher protein concentrations, however, the protein corona increases colloidal stability by saturating the surface, resulting in repulsive steric and entropic forces that arise from the displacement of surface-bound water molecules upon protein adsorption.39,47,48 Both forces favor intercolloid repulsion and lead to colloidal stability.
Importantly, protein coronas stabilize colloids upon serum incubation. Indeed, some stabilization was even observed for the bare fulvestrant colloids in serum-containing media, reflecting some corona formation by serum proteins themselves. This is consistent with previous studies, which have found that colloidal drug aggregates can persist in these conditions over 24 h.5 However, the differences between the morphologies of bare and protein-stabilized colloids reflect the superior stability conferred by controlled corona formation. The observation that trastuzumab-modified colloids preferentially targeted HER2 overexpressing cells indicates that the antibody is stably bound to the colloid surface, even after exposure to serum, and that a substantial amount of antibody is oriented such that the Fab region can interact with the target receptor. These results suggest that it should be possible to find other protein–colloid pairs that can optimally stabilize monodisperse colloidal drug aggregates for targeted delivery and improved efficacy.
While we have shown that protein adsorption can stabilize colloidal drug aggregates, it is likely that differences in protein and colloid surface characteristics will demand optimization for different protein–colloid combinations. The formulation of fulvestrant and trastuzumab investigated here is clinically relevant for breast cancer patients with HER2- and ER-positive tumors.29 Combinations of other colloid-forming chemotherapeutics and targeting ligands may prove useful for other cancers. Additionally, although we have shown that trastuzumab-stabilized colloids are specifically internalized by HER2-expressing cells and decrease cell viability, the current protein-stabilized fulvestrant colloids seem to persist in the endolysosomal pathway, limiting their efficacy.49
CONCLUSIONS
Colloidal drug aggregates, typically thought of as a nuisance artifact of early drug discovery, can be stabilized by complexation with proteins and targeted for selective cell uptake with functional antibodies. Since the colloidal aggregates act as both the active agent and the vehicle, antibody-stabilized colloidal drug formulations may address key limitations of nanoparticle formulations, namely, their poor drug loading and necessity for massive amounts of nanoparticle material in the formulation. Many chemotherapeutics form colloidal aggregates,5,14,19 and protein corona formation may reveal opportunities to convert what has been considered a weakness into an opportunity for targeted delivery. In the past decade, both antibody–drug conjugates and nanoparticle formulations have emerged as promising avenues for targeted drug delivery. We demonstrated specific uptake by target cancer cells in vitro, yet, like many other nanoparticle formulations, the stabilized fulvestrant colloids will likely be nonspecifically internalized to some extent by phagocytic cells in vivo.50,51 Notwithstanding, what may be thought of as “antibody–colloidal drug conjugates” may deliver many more orders of magnitude of drug molecules per antibody, improving efficacy.
EXPERIMENTAL SECTION
Materials
Fulvestrant (Cat. No. S1191) was purchased from Selleck Chemicals. Sorafenib (Cat. No. HY-10201) and vemurafenib (Cat. No. HY-12057) were purchased from MedChem Express. Cell culture grade DMSO (Cat. No. D2650), bovine serum albumin (Cat. No. A7030), IgG from human serum (Cat. No. I4506), insulin from bovine pancreas (Cat. No. I0516), and RPMI 1640 cell culture media (R8758) were purchased from Sigma-Aldrich. Trastuzumab (Herceptin) was obtained from Roche (Mississauga, Ontario, Canada). McCoy’s 5A (Cat. No. 1660082) and DMEM F12 (Cat. No. 11330032) cell culture media, CholEsteryl BODIPY FL C12 (Cat. No. C3927MP), CholEsteryl BODIPY 542/563 C11 (Cat. No. C12680), Hoechst 33342 (Cat. No. H1399), wheat germ agglutinin Alexa Fluor 647 conjugate (Cat. No. W32466), dextran Alexa Fluor 647 conjugates (Cat. No. D22914), and PrestoBlue cell viability reagent (A12361) were purchased from Thermo Fisher Scientific. Cell lines SKOV-3 (Cat. No. HTB-77), MDA-MB-231 (Cat. No. HTB-26), BT-474 (Cat. No. HTB-20), and MCF-7 (Cat. No. HTB-22) were purchased from ATCC. Charcoal-stripped fetal bovine serum (Cat. No. 080750) and Hank’s balanced salt solution (Cat. No. 311515) were purchased from Wisent Bioproducts. Polysorbate 80 (HX2) was purchased from NOF America Corp.
Colloid Formation
Stock solutions of each colloid-forming compound were prepared at 5 mM in DMSO. Colloid formation occurred after the rapid addition of double-distilled water (865 μL) to drug stock solution (10 μL). After colloid formation, proteins of interest (25 μL) and 10X PBS (100 μL) were added simultaneously to the colloid solution. Final drug concentration was 50 μM; DMSO was kept to 1% (v/v), and protein concentrations ranged from 5 nM to 5 μM. For experiments including serum, charcoal-stripped fetal bovine serum was added after colloid formation to a concentration of 5% (v/v). Formulations of colloids that include CholEsteryl BODIPY FL C12, CholEsteryl BODIPY 542/563 C11, or both were prepared by inclusion of the fluorophores into the compound stock solutions in DMSO. Final total concentration of fluorophore was 500 nM. For the stability study in buffers of different pH, citric acid was added to formulations to adjust pH to the desired value.
Dynamic Light Scattering
Colloid diameters, polydispersity and normalized scattering intensity were measured using a DynaPro Plate Reader II with a laser width optimized by the manufacturer for colloidal particle detection (Wyatt Technologies). Operating conditions were 60 mW laser at 830 nm and detector angle of 158°. Samples were measured in a 96-well format with 100 μL and 20 acquisitions per sample.
Colloid Centrifugation and Gel Electrophoresis
Colloids were formulated as described above and pelleted by centrifugation at 16000g for 1 h at 4 °C. Proteins in pellet and supernatant were reduced by addition of loading dye containing 2-mercaptoethanol and boiled for 5 min. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Proteins were identified by staining with Coomassie Blue G-250.
Transmission Electron Microscopy
Colloid formulations (5 μL) were deposited onto a freshly glow-discharged 400 mesh carbon coated copper TEM grid (Ted Pella, Inc.) and allowed to adhere for 5 min. Excess liquid was removed with filter paper, followed by a quick wash with double-distilled water (5 μL). Particles were then stained with 1% ammonium molybdate (w/v, pH 7, 5 μL) for 30 s. Stain was removed and samples imaged using a Hitachi H-7000 microscope operating at 75 kV. Images were captured using an Advanced Microscopy Techniques (AMT) XR-60 CCD camera with typical magnifications between 30000× and 100000×. Images were analyzed using ImageJ 64 software and processed with Photoshop.
XPS and TOF-SIMS
Colloids formulated with or without BSA were deposited on a silicon wafer, and excess liquid was evaporated under vacuum. Surface analysis by XPS was carried out using an Escalab 250Xi XPS spectrometer (ThermoFisher Scientific, East Grinstead, U.K.) and a monochromatized Al Kα X-ray source. Samples were cleaned using a 4000 eV high cluster size Ar cluster source, and a nominal spot size of 400 × 400 μm2 was analyzed. Charge compensation was applied using the combined low-energy e−/Ar+ flood gun with the binding energy scale shifted to place the main C 1s peak (C–C) to 285.0. Both survey (pass energy, 100 eV) and high-resolution (pass energy, 30 eV) spectra were obtained. All data collection and analysis were performed using Avantage v.5.957 software.
Negative polarity TOF-SIMS spectra were obtained using an Ion-ToF V spectrometer (ION-TOF GmbH, Muenster, Germany). Spectra were obtained using a 60 keV Bi3+2 cluster primary ion source. A depth profile was obtained in an interlaced, dual source mode run under high spatial resolution conditions.52 An Ar cluster source was used to generate the sputter crater (5000 eV, 4 nA, 100 × 100 μm2), and spectra were obtained from an area of 20 × 20 μm2 centered in the sputter crater. The mass scale was calibrated to standard peaks found in all spectra.
In Vitro Serum Stability
Fulvestrant colloid stability in serum-containing media was assessed using fast protein liquid chromatography (FPLC), as previously described.53,54 Fluorescent colloids were formulated of 50 μM fulvestrant, 875 nM BODIPY FL C12, and 125 nM BODIPY 542/563 C11. Protein concentrations were 100 nM BSS and 3.5 μM trastuzumab. Colloids were incubated in 20% charcoal-stripped FBS, 10 UI/mL penicillin, and 10 μg/mL streptomycin. At selected time points, 500 μL of sample was separated on a Superdex 200 gel filtration column at a flow rate of 1.5 mL/min with PBS as the mobile phase. The FRET signal was measured at excitation wavelength of 490 nm and emission wavelength of 575 nm. Integration of colloid peak area was performed using GraphPad software version 6.0.
Cell Culture
All cells were maintained at 37 °C in 5% CO2 in appropriate cell culture media supplemented with 10% FBS, 10 UI/mL penicillin, and 10 μg/mL streptomycin. RPMI 1640 medium was used for culture of MDA-MB-231 and BT-474 cells. McCoy’s 5A medium was used for SKOV-3 cells and DMEM-F12 was used for MCF-7. Media for MCF-7 cells were additionally supplemental with 10 μg/mL of insulin.
Confocal Microscopy
Fluorescent fulvestrant colloids were prepared as described above with 500 nM BODPY 542/563 C11. Trastuzumab or IgG (3.5 μM) was added to the formulation and incubated for 10 min prior to addition of charcoal-stripped FBS (5% final concentration). SKOV-3 and MDA-MB-231 were seeded at 10,000 cells/well and 7,500 cells/well, respectively, in 16-well glass chamber slides. Cells were incubated with appropriate formulations for 3 h at 37 °C at 5% CO2. Formulations were then removed and either replaced with fresh media for an additional 24 h or with 4% PFA solution for cell fixation. Following fixation cell membranes were stained with wheat germ agglutinin Alexa Fluor 647 conjugate (5 μg/mL) as per the manufacturer protocol and counterstained with Hoechst. Cell lysosomes were visualized by incubating cells with Alexa Fluor 647 conjugated dextran (10 000 g/mol) overnight at 0.05 mg/mL prior to incubation with colloidal formulations and imaged under live-cell imaging conditions. Cells were imaged on an Olympus FV1000 confocal microscope at 60× magnification. Excitation and emission wavelengths are as follows: for Hoechst, excitation at 405 nm, emission at 460 nm; for colloids, excitation at 543 nm, emission at 572 nm; for WGA-647 and dextran-647, excitation at 633 nm, emission at 668 nm. Z-stacks of each field of view were obtained at 1 μm step size and compressed into a single image.
Flow Cytometry
BT-474 cells were seeded at 50,000 cells/well in 24-well plates. Fluorescent colloids were formulated as above and incubated with cells for 3 h at 37 °C at 5% CO2. After the incubation period, cells were washed with media and cells detached using accutase. Cells were counterstained with propidium iodide for exclusion of dead cells. Cell fluorescence was analyzed using a BD Accuri C6 flow cytometer with excitation wavelength of 488 nm and emission filters of 533/30 nm (BODIPY) and 585/40 nm (propidium iodide). Data were analyzed using FlowJo software, and the mean fluorescence intensity of the live-cell fraction was measured for three biological replicates.
Cell-Viability Study
BT-474 and MCF-7 cells were seeded at 10,000 cells/well in 96-well plates and allowed to adhere overnight in 10% charcoal-stripped FBS. Formulations were prepared as described above. Monomeric formulations were prepared by the addition of ultrapure polysorbate 80 to a final concentration of 0.015% (v/v). Cells were incubated with formulations for 24 h followed by replacement with fresh media for an additional 48 h. Cell viability was assessed using Presto Blue viability assay according to manufacturer’s protocols. Cell viability is reported as a percentage of the vehicle controls.
Acknowledgments
This work was supported by grants from the U.S. National Institutes of General Medical Sciences (Grant GM71630 to B.K.S and M.S.S.) and the Canadian Cancer Society Research Institute (to M.S.S. and B.K.S.). A.N.G. is supported by a Natural Sciences and Engineering Research Council (NSERC) Postgraduate Research Scholarship, and C.K.M. is supported, in part, by an NSERC postdoctoral fellowship. We thank S. Boyle and B. Calvieri from the University of Toronto Microscopy Imaging Laboratory for assistance with TEM imaging, Dr. R. Sodhi from the Ontario Centre for the Characterization of Advanced Materials for XPS and TOF-SIMS analysis, Dr. P. Gilbert and S. Davoudi for use and assistance with their flow cytometer, and members of the Shoichet laboratories for thoughtful discussion.
ABBREVIATIONS
- BSA
bovine serum albumin
- CLSM
confocal laser scanning microscopy
- ER
estrogen receptor
- FBS
fetal bovine serum
- FPLC
fast protein liquid chromatography
- FRET
Forster resonance energy transfer
- HER2
human epidermal growth factor receptor 2
- IgG
immunoglobulin G
- PBS
phosphate buffered saline
- TEM
transmission electron microscopy
- TOF-SIMS
time-of-flight secondary ion mass spectrometry
- XPS
X-ray photoelectron spectroscopy
Footnotes
Notes
The authors declare the following competing financial interest(s): We have submitted a provisional patent on this topic of antibody-drug colloids.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.6b15987.
Additional characterization of formulations, XPS and TOF-SIMS analysis, flow cytometry analysis, pH stability, and cytotoxicity data (PDF)
References
- 1.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J Med Chem. 2002;45(8):1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 2.McGovern SL, Helfand BT, Feng B, Shoichet BK. A Specific Mechanism of Nonspecific Inhibition. J Med Chem. 2003;46(20):4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 3.Sassano MF, Doak AK, Roth BL, Shoichet BK. Colloidal Aggregation Causes Inhibition of G Protein-Coupled Receptors. J Med Chem. 2013;56(6):2406–2414. doi: 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owen SC, Doak AK, Ganesh AN, Nedyalkova L, McLaughlin CK, Shoichet BK, Shoichet MS. Colloidal Drug Formulations Can Explain “Bell-Shaped” Concentration-Response Curves. ACS Chem Biol. 2014;9(3):777–784. doi: 10.1021/cb4007584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK. Colloidal Aggregation Affects the Efficacy of Anticancer Drugs in Cell Culture. ACS Chem Biol. 2012;7(8):1429–1435. doi: 10.1021/cb300189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coan KE, Shoichet BK. Stoichiometry and Physical Chemistry of Promiscuous Aggregate-Based Inhibitors. J Am Chem Soc. 2008;130(29):9606–9612. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park K. Facing the Truth About Nanotechnology in Drug Delivery. ACS Nano. 2013;7(9):7442–7447. doi: 10.1021/nn404501g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the Barriers in Micellar Drug Delivery: Loading Efficiency, in Vivo Stability, and Micelle-Cell Interaction. Expert Opin Drug Delivery. 2010;7(1):49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 9.Gaudin A, Yemisci M, Eroglu H, Lepetre-Mouelhi S, Turkoglu OF, Donmez-Demir B, Caban S, Sargon MF, Garcia-Argote S, Pieters G, Loreau O, Rousseau B, Tagit O, Hildebrandt N, Le Dantec Y, Mougin J, Valetti S, Chacun H, Nicolas V, Desmaele D, Andrieux K, Capan Y, Dalkara T, Couvreur P. Squalenoyl Adenosine Nanoparticles Provide Neuroprotection after Stroke and Spinal Cord Injury. Nat Nanotechnol. 2014;9(12):1054–1062. doi: 10.1038/nnano.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maksimenko A, Dosio F, Mougin J, Ferrero A, Wack S, Reddy LH, Weyn AA, Lepeltier E, Bourgaux C, Stella B, Cattel L, Couvreur P. A Unique Squalenoylated and Nonpegylated Doxorubicin Nanomedicine with Systemic Long-Circulating Properties and Anticancer Activity. Proc Natl Acad Sci U S A. 2014;111(2):E217–226. doi: 10.1073/pnas.1313459110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang P, Wang D, Su Y, Huang W, Zhou Y, Cui D, Zhu X, Yan D. Combination of Small Molecule Prodrug and Nanodrug Delivery: Amphiphilic Drug-Drug Conjugate for Cancer Therapy. J Am Chem Soc. 2014;136(33):11748–11756. doi: 10.1021/ja505212y. [DOI] [PubMed] [Google Scholar]
- 12.Ma W, Cheetham AG, Cui H. Building Nanostructures with Drugs. Nano Today. 2016;11(1):13–30. doi: 10.1016/j.nantod.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Addio SM, Prud’homme RK. Controlling Drug Nanoparticle Formation by Rapid Precipitation. Adv Drug Delivery Rev. 2011;63(6):417–426. doi: 10.1016/j.addr.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Doak AK, Wille H, Prusiner SB, Shoichet BK. Colloid Formation by Drugs in Simulated Intestinal Fluid. J Med Chem. 2010;53(10):4259–4265. doi: 10.1021/jm100254w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coan KE, Shoichet BK. Stability and Equilibria of Promiscuous Aggregates in High Protein Milieus. Mol BioSyst. 2007;3(3):208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 16.Frenkel YV, Clark AD, Jr, Das K, Wang Y-H, Lewi PJ, Janssen PAJ, Arnold E. Concentration and Ph Dependent Aggregation of Hydrophobic Drug Molecules and Relevance to Oral Bioavailability. J Med Chem. 2005;48(6):1974–1983. doi: 10.1021/jm049439i. [DOI] [PubMed] [Google Scholar]
- 17.Ilevbare GA, Taylor LS. Liquid-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst Growth Des. 2013;13(4):1497–1509. [Google Scholar]
- 18.Ilevbare GA, Liu H, Pereira J, Edgar KJ, Taylor LS. Influence of Additives on the Properties of Nanodroplets Formed in Highly Supersaturated Aqueous Solutions of Ritonavir. Mol Pharmaceutics. 2013;10(9):3392–3403. doi: 10.1021/mp400228x. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin CK, Duan D, Ganesh AN, Torosyan H, Shoichet BK, Shoichet MS. Stable Colloidal Drug Aggregates Catch and Release Active Enzymes. ACS Chem Biol. 2016;11(4):992–1000. doi: 10.1021/acschembio.5b00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoichet BK. Interpreting Steep Dose-Response Curves in Early Inhibitor Discovery. J Med Chem. 2006;49(25):7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 21.Stirland DL, Nichols JW, Miura S, Bae YH. Mind the Gap: A Survey of How Cancer Drug Carriers Are Susceptible to the Gap between Research and Practice. J Controlled Release. 2013;172(3):1045–1064. doi: 10.1016/j.jconrel.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barua S, Yoo JW, Kolhar P, Wakankar A, Gokarn YR, Mitragotri S. Particle Shape Enhances Specificity of Antibody-Displaying Nanoparticles. Proc Natl Acad Sci U S A. 2013;110(9):3270–3275. doi: 10.1073/pnas.1216893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barua S, Mitragotri S. Synergistic Targeting of Cell Membrane, Cytoplasm, and Nucleus of Cancer Cells Using Rod-Shaped Nanoparticles. ACS Nano. 2013;7(11):9558–9570. doi: 10.1021/nn403913k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cortez C, Tomaskovic-Crook E, Johnston AP, Scott AM, Nice EC, Heath JK, Caruso F. Influence of Size, Surface, Cell Line, and Kinetic Properties on the Specific Binding of A33 Antigen-Targeted Multilayered Particles and Capsules to Colorectal Cancer Cells. ACS Nano. 2007;1(2):93–102. doi: 10.1021/nn700060m. [DOI] [PubMed] [Google Scholar]
- 25.Gaudin A, Tagit O, Sobot D, Lepetre-Mouelhi S, Mougin J, Martens TF, Braeckmans K, Nicolas V, Desmaele D, de Smedt SC, Hildebrandt N, Couvreur P, Andrieux K. Transport Mechanisms of Squalenoyl-Adenosine Nanoparticles across the Blood-Brain Barrier. Chem Mater. 2015;27(10):3636–3647. [Google Scholar]
- 26.Zhao CL, Winnik MA, Riess G, Croucher MD. Fluorescence Probe Techniques Used to Study Micelle Formation in Water-Soluble Block Copolymers. Langmuir. 1990;6(2):514–516. [Google Scholar]
- 27.Wilhelm M, Zhao CL, Wang YC, Xu RL, Winnik MA, Mura JL, Riess G, Croucher MD. Polymer Micelle Formation 03. Poly(Styrene-Ethylene Oxide) Block Copolymer Micelle Formation in Water - a Fluorescence Probe Study. Macromolecules. 1991;24(5):1033–1040. [Google Scholar]
- 28.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the Her-2/Neu Oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 29.Rimawi MF, Schiff R, Osborne CK. Targeting Her2 for the Treatment of Breast Cancer. Annu Rev Med. 2015;66(1):111–128. doi: 10.1146/annurev-med-042513-015127. [DOI] [PubMed] [Google Scholar]
- 30.Wakeling AE, Dukes M, Bowler J. A Potent Specific Pure Antiestrogen with Clincal Potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- 31.Maxfield FR, McGraw TE. Endocytic Recycling. Nat Rev Mol Cell Biol. 2004;5(2):121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 32.Holliday D, Speirs V. Choosing the Right Cell Line for Breast Cancer Research. Breast Cancer Res. 2011;13(4):215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat Nanotechnol. 2012;7(12):779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 34.Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Protein-Nanoparticle Interactions: Opportunities and Challenges. Chem Rev. 2011;111(9):5610–5637. doi: 10.1021/cr100440g. [DOI] [PubMed] [Google Scholar]
- 35.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. Proc Natl Acad Sci U S A. 2008;105(38):14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindman S, Lynch I, Thulin E, Nilsson H, Dawson KA, Linse S. Systematic Investigation of the Thermodynamics of Hsa Adsorption to N-Iso-Propylacrylamide/N-Tert-Butylacrylamide Copolymer Nanoparticles. Effects of Particle Size and Hydrophobicity. Nano Lett. 2007;7(4):914–920. doi: 10.1021/nl062743+. [DOI] [PubMed] [Google Scholar]
- 37.Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, Dawson KA, Linse S. Understanding the Nanoparticle-Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc Natl Acad Sci U S A. 2007;104(7):2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Baldelli Bombelli F, Dawson KA. Physical-Chemical Aspects of Protein Corona: Relevance to in Vitro and in Vivo Biological Impacts of Nanoparticles. J Am Chem Soc. 2011;133(8):2525–2534. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- 39.Moerz ST, Kraegeloh A, Chanana M, Kraus T. Formation Mechanism for Stable Hybrid Clusters of Proteins and Nanoparticles. ACS Nano. 2015;9(7):6696–6705. doi: 10.1021/acsnano.5b01043. [DOI] [PubMed] [Google Scholar]
- 40.Brewer SH, Glomm WR, Johnson MC, Knag MK, Franzen S. Probing Bsa Binding to Citrate-Coated Gold Nanoparticles and Surfaces. Langmuir. 2005;21(20):9303–9307. doi: 10.1021/la050588t. [DOI] [PubMed] [Google Scholar]
- 41.Wertz CF, Santore MM. Effect of Surface Hydrophobicity on Adsorption and Relaxation Kinetics of Albumin and Fibrinogen: Single-Species and Competitive Behavior. Langmuir. 2001;17(10):3006–3016. [Google Scholar]
- 42.Jeyachandran YL, Mielczarski E, Rai B, Mielczarski JA. Quantitative and Qualitative Evaluation of Adsorption/Desorption of Bovine Serum Albumin on Hydrophilic and Hydrophobic Surfaces. Langmuir. 2009;25(19):11614–11620. doi: 10.1021/la901453a. [DOI] [PubMed] [Google Scholar]
- 43.Kim J, Somorjai GA. Molecular Packing of Lysozyme, Fibrinogen, and Bovine Serum Albumin on Hydrophilic and Hydrophobic Surfaces Studied by Infrared–Visible Sum Frequency Generation and Fluorescence Microscopy. J Am Chem Soc. 2003;125(10):3150–3158. doi: 10.1021/ja028987n. [DOI] [PubMed] [Google Scholar]
- 44.He XM, Carter DC. Atomic Structure and Chemistry of Human Serum Albumin. Nature. 1992;358(6383):209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 45.Walsh G, Jefferis R. Post-Translational Modifications in the Context of Therapeutic Proteins. Nat Biotechnol. 2006;24(10):1241–1252. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- 46.Beck A, Wagner-Rousset E, Bussat M-C, Lokteff M, Klinguer-Hamour C, Haeuw J-F, Goetsch L, Wurch T, Van Dorsselaer A, Corvaia N. Trends in Glycosylation, Glycoanalysis and Glycoengineering of Therapeutic Antibodies and Fc-Fusion Proteins. Curr Pharm Biotechnol. 2008;9(6):482–501. doi: 10.2174/138920108786786411. [DOI] [PubMed] [Google Scholar]
- 47.Gebauer JS, Malissek M, Simon S, Knauer SK, Maskos M, Stauber RH, Peukert W, Treuel L. Impact of the Nanoparticle–Protein Corona on Colloidal Stability and Protein Structure. Langmuir. 2012;28(25):9673–9679. doi: 10.1021/la301104a. [DOI] [PubMed] [Google Scholar]
- 48.Au KM, Armes SP. Heterocoagulation as a Facile Route to Prepare Stable Serum Albumin-Nanoparticle Conjugates for Biomedical Applications: Synthetic Protocols and Mechanistic Insights. ACS Nano. 2012;6(9):8261–8279. doi: 10.1021/nn302968j. [DOI] [PubMed] [Google Scholar]
- 49.Fuhrmann K, Gauthier MA, Leroux JC. Targeting of Injectable Drug Nanocrystals. Mol Pharmaceutics. 2014;11(6):1762–1771. doi: 10.1021/mp5001247. [DOI] [PubMed] [Google Scholar]
- 50.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 51.Yan Y, Gause KT, Kamphuis MMJ, Ang C-S, O’Brien-Simpson NM, Lenzo JC, Reynolds EC, Nice EC, Caruso F. Differential Roles of the Protein Corona in the Cellular Uptake of Nanoporous Polymer Particles by Monocyte and Macrophage Cell Lines. ACS Nano. 2013;7(12):10960–10970. doi: 10.1021/nn404481f. [DOI] [PubMed] [Google Scholar]
- 52.Sodhi RNS. Time-of-Flight Secondary Ion Mass Spectrometry (Tof-Sims):-Versatility in Chemical and Imaging Surface Analysis. Analyst. 2004;129(6):483–487. doi: 10.1039/b402607c. [DOI] [PubMed] [Google Scholar]
- 53.Zhao X, Poon Z, Engler AC, Bonner DK, Hammond PT. Enhanced Stability of Polymeric Micelles Based on Postfunctionalized Poly(Ethylene Glycol)-B-Poly(Gamma-Propargyl L-Glutamate): The Substituent Effect. Biomacromolecules. 2012;13(5):1315–1322. doi: 10.1021/bm201873u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Logie J, Owen SC, McLaughlin CK, Shoichet MS. Peg-Graft Density Controls Polymeric Nanoparticle Micelle Stability. Chem Mater. 2014;26(9):2847–2855. [Google Scholar]