

ABSTRACT
Macroautophagy is regarded as a nonspecific bulk degradation process of cytoplasmic material within the lysosome. However, the process has mainly been studied by nonspecific bulk degradation assays using radiolabeling. In the present study we monitor protein turnover and degradation by global, unbiased approaches relying on quantitative mass spectrometry-based proteomics. Macroautophagy is induced by rapamycin treatment, and by amino acid and glucose starvation in differentially, metabolically labeled cells. Protein dynamics are linked to image-based models of autophagosome turnover. Depending on the inducing stimulus, protein as well as organelle turnover differ. Amino acid starvation-induced macroautophagy leads to selective degradation of proteins important for protein translation. Thus, protein dynamics reflect cellular conditions in the respective treatment indicating stimulus-specific pathways in stress-induced macroautophagy.
KEYWORDS: autophagy, degradation, mass spectrometry, protein turnover, proteomics, SILAC
Introduction
The 2 main protein degradation routes in eukaryotic cells are the ubiquitin-proteasome system (UPS) and the autophagosomal-lysosomal pathway. The UPS is mainly responsible for the degradation of cytosolic proteins, whereas the autophagosomal-lysosomal system is considered to be responsible for the bulk degradation of organelles and long-lived proteins.1 Autophagy has been linked inter alia to innate and adaptive immunity,2 as well as to several diseases, including cancers and neurodegenerative disorders.3,4 Autophagy comprises several discrete lysosomal degradation pathways. In chaperone-mediated autophagy (CMA) cytosolic proteins translocate directly across the lysosomal membrane.5 Microautophagy is characterized by lysosomal membrane invagination or protrusion delivering portions of cytoplasm directly into lysosomes.1 The hallmarks of macroautophagy are specialized double-membrane organelles, phagophores, that enclose parts of the cytoplasm destined for recycling, presumably in a nonspecific, bulk manner; the phagophores mature into autophagosomes that fuse with the lysosome. Macroautophagic flux, describing protein degradation via macroautophagy, is generally measured by western blot analysis of single proteins, microscopy techniques addressing autophagosome formation/degradation, or “long-lived protein degradation” assays using radioisotope tracers.1 These techniques monitor single proteins, whole organelles, or bulk protein degradation, but are unable to identify the degradation rates of multiple individual proteins.
In contrast to classical cell/molecular biologic assays, quantitative mass spectrometry (MS)-based proteomics allows the proteome-wide analysis of protein turnover.6 The abundance of a protein, A, at a given time t, can be calculated by the simplified formula:
where ks is the 0th-order rate constant of protein synthesis and kd the 1st-order rate constant of protein degradation.7 Protein turnover is defined as the difference between the rates of protein synthesis and degradation. In contrast to image-based readouts of protein turnover that use ectopically expressed, fluorescent-tagged fusion proteins,8,9 MS-based proteomics allows the analysis of protein turnover,10-12 synthesis,13 and degradation of endogenous proteins.14-16 Protein degradation rates have so far been determined assuming steady-state conditions using the change in relative isotope abundance (RIA) as a measure to calculate degradation constants,11 measuring absolute protein levels,17 or relatively quantifying protein abundances of starved and nonstarved organisms.15 These approaches rely on metabolic pulse labeling or saturation labeling, respectively.
To globally analyze protein turnover in differently induced macroautophagy we have used classical pulse-labeling strategies based on stable isotope labeling by amino acids in cell culture (SILAC).16,18 In addition, a new, straightforward method for the proteome-wide analysis of relative protein degradation rates under nonsteady-state conditions termed pulse-chase SILAC (pcSILAC) was developed. Quantitative MS-based proteomics data are combined with mathematical models based on image analyses of autophagosomes and autolysosomes to outline the influences of specific stimuli on macroautophagy-dependent protein turnover.
Results
Induction of functional macroautophagy
As model systems we used MCF7 breast cancer cell lines that express fluorescently tagged versions of the rat autophagosome-associated protein MAP1LC3B/LC3B (microtubule- associated protein 1 light chain 3 β).19,20 Cells expressing eGFP-LC3B were used to monitor the induction of macroautophagy by 3 well characterized stimuli, rapamycin (Rap) treatment, amino acid (AA) and glucose (Glc) starvation.21 Translocation of eGFP-LC3B into dotted structures is commonly assayed to follow macroautophagy induction.1 In untreated control cells (Ctrl) eGFP-LC3B was found diffusely in the cytosol and nucleus. As expected, after 24 h treatment the number of eGFP-LC3B puncta increased indicating an accumulation of autophagosomes (Fig. 1A–B).
Figure 1.

Induction of macroautophagy. (A) Accumulation of autophagosomes. MCF7-eGFP-LC3B cells were left untreated, AA, Glc starved, or treated with Rap (100 nM) for 24 h. Stimulus-dependent accumulation of autophagosomes was visualized by translocation of eGFP-LC3B into dotted structures. Nuclei were stained with DAPI. Scale bar: 10 µm. (B) Quantification of autophagosomes. Autophagosomes were counted in a minimum of 50 cells as shown in (A). Average autophagosome numbers ± SD are depicted. (C) Macroautophagic flux analysis using eGFP-LC3B-II. Cells were treated as in (A) and, in addition, ConA (2 nM) was added as indicated. Samples were normalized to cell number and eGFP-LC3B-II bands of ConA-treated samples were analyzed relative to respective untreated samples by western blot. Right panel depicts quantification (n = 3). (D) Macroautophagic flux analysis using SQSTM1. Samples were treated and analyzed as in (C). (E) Inhibition of MTOR. Phosphorylation levels of RPS6KB1 were monitored using a Thr389 phosphosite-specifc antibody (upper lane). Lower lane shows loading control. Right panel depicts quantification (n = 3). *: p < 0.05; **: p < 0.01; ***: p < 0.001, unpaired T test.
Elevated autophagosome numbers may either be due to an increased formation, a blockage of degradation, or a combination of both. To analyze macroautophagic flux we quantified LC3B-II, the membrane-bound form of LC3B, as well as SQSTM1 (sequestosome 1), the prototypic autophagy receptor,22 by western blot. Macroautophagy was induced for 24 h in the presence or absence of concanamycin A (ConA), an inhibitor of the lysosomal vacuolar-type H+-ATPase that blocks lysosomal acidification and, by this, degradation. In 24 h housekeeping proteins commonly used as loading controls in western blots, such as ACTB (actin β) or GAPDH (glyceraldehyde-3-phosphate dehydrogenase), also got degraded (see Table S4). Therefore samples were normalized to cell numbers. The 3 treatments led to an increased accumulation of LC3B-II (Fig. 1C) and SQSTM1 (Fig. 1D) in the presence of ConA as compared with the respective control cells. AA starvation led to the highest flux, followed by Rap treatment. Glc starvation had only a minor influence. The blockage of the kinase MTOR (mechanistic target of rapamycin) is thought to be mandatory for the induction of classical macroautophagy. All 3 stimuli inhibited MTOR kinase activity as judged by the phosphorylation level of an MTOR substrate, RPS6KB1 (ribosomal protein S6 kinase B1) (Fig. 1E). Thus, all 3 stimuli induced functional macroautophagy in MCF7 cells, glucose starvation having the weakest effects.
Stimulus-specific autophagosomal turnover
To study potential influences of stress stimuli on organellar turnover of autophagosomes, i.e. turnover kinetics and subcellular localizations of organelles, we used MCF7 cells stably expressing mRFP and GFP tandem fluorescent-tagged LC3B.20 GFP is sensitive to an acidic pH, whereas mRFP is not. Thus, autophagosomes appear as yellow puncta, whereas autolysosomes appear red (Fig. 2A). Using an image-based readout, organelle turnover can be analyzed. Cells were treated as mentioned above using Rap or AA and Glc starvation and analyzed 2, 12 and 24 h after macroautophagy induction. Cells were fixed and 15 z-stacks per image were recorded. Using a local threshold feature detector we built a bag-of-visual-words model in which each time point for each condition is represented by the composition of different types of objects. The major components of variation among all samples were found by principal component analysis (PCA) and the average trajectories of the 3 conditions were plotted in the resulting PCA space (Fig. 2B). The 3 conditions showed step-wise differences in the spatial distribution of LC3B, the most intense being AA starvation, followed by Rap treatment, and Glc starvation; this is in agreement with the autophagy flux data presented in Figure 1 and was also confirmed by live cell imaging over 24 h (Fig. S1).
Figure 2.
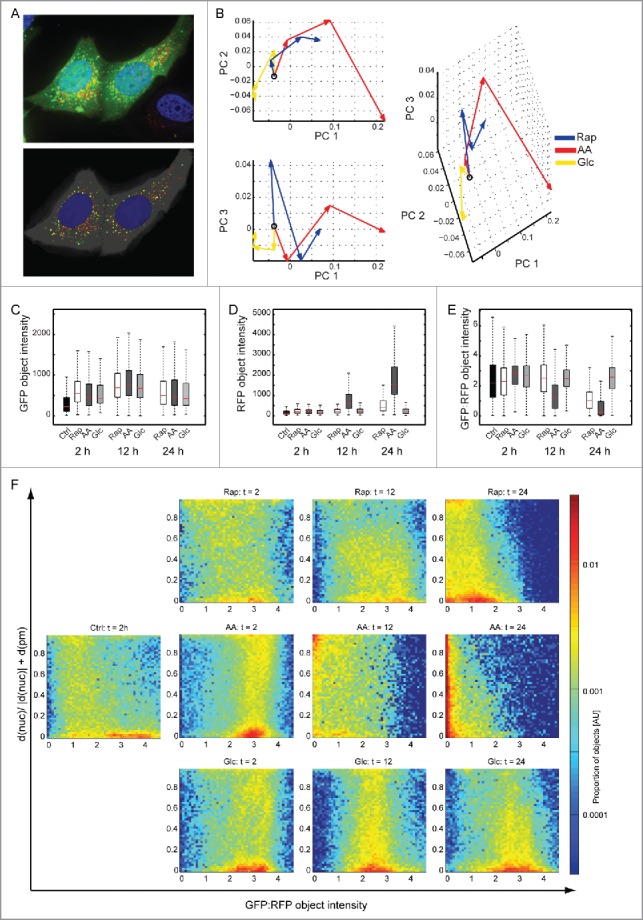
Autophagosome turnover analyzed by fluorescence microscopy. (A) Original and segmented images. MCF7-mRFP-GFP-LC3B cells were treated as in Figure 1 and images of GFP (green), RFP (red) and DAPI (blue) were acquired after 2, 12 and 24 h. An example of 15 merged z-stacks (100x) is shown (upper panel). The lower panel shows the segmented objects. (B) Principal component analysis. The lines for each condition represent the trajectory connecting the average values at each time point (the open circle indicates the untreated condition). AA starvation had the most pronounced effects, followed by Rap treatment and Glc starvation. (C) Accumulation of GFP. All treatments led to an accumulation of GFP as indicated by object intensity (KS test p < 0.001 with Bonferroni correction). (D) Accumulation of RFP. Whereas AA starvation led to strong accumulation of RFP intensity, Rap had only a moderate effect. Glc starvation did not change RFP intensity (KS test p < 0.001 with Bonferroni correction). (E) Autophagosome turnover. The ratio of GFP:RFP object intensity can be used as a surrogate for autophagosome turnover. AA starvation and Rap treatment led to increased turnover. Glc starvation had no effect (KS test p < 0.001 with Bonferroni correction). (F) Autophagosome localization. Autophagosome localization and turnover T between plasma (pm) and nuclear (nuc) membrane is depicted using GFP:RFP ratios. A 2D histogram of the proportion of LC3B-containing organelles with a given localization and GFP:RFP ratio is shown on a log scale. The Y-axis depicts the distance between nuclear and plasma membrane, 0 being at the nuclear membrane and 1 at the plasma membrane. Color indicates the proportion of organelles, with red indicating a high proportion (see color scale). AU, arbitrary units.
To further analyze the nature of the changes, we used a cell segmentation and protein object detection algorithm to measure several aspects of the protein location pattern: spatial distribution of protein objects, relative proportion of GFP to RFP, protein object counts, and size. GFP object intensities increased in all treatments compared with control cells (Fig. 2C, KS test p < 0.001 Bonferroni corrected). RFP object intensities exhibited a drastic increase after 12 h AA starvation and 24 h Rap treatment compared with control (Fig. 2D, KS test p < 0.001 Bonferroni corrected). Hence, GFP:RFP object intensities, which are a measure for autophagosome turnover, showed clear effects for AA and Rap treatment and hardly any effect for Glc starvation (Fig. 2E), as was expected from western blot experiments. In AA starvation, increased GFP intensity was based on increased autophagosome numbers, whereas increased RFP intensity was due to fewer, but larger, autolysosomes (Fig. S2). The spatial distribution of protein objects relative to the plasma membrane and the outside border of the nucleus was also analyzed (Fig. 2F). Under control conditions, autophagosomes were located near the nucleus (population on the lower right), and a low frequency of autolysosomes was observed spread between the nuclear and the plasma membrane. Under AA starvation 2 populations of autophagosomes could be observed, one close to the nucleus and another one in the periphery. After 12 h both were turned over to autolysosomes and after 24 h autolysosomes appeared to move closer to the nucleus. Under Rap treatment the 2 populations were also observable, although the peripheral population was less pronounced. Autophagosomes and autolysosomes appeared more heterogeneous. As expected, under Glc starvation only autophagosomes could be observed, which appeared to move toward the nucleus at 24 h. Thus, these data imply stimulus-specific turnover and localization of autophagosomes/autolysosomes. To study if these observations are mirrored by stimulus-specific protein turnover by macroautophagy we designed quantitative MS-based proteomics approaches directly measuring autophagy-dependent protein synthesis, turnover and degradation.
Decreased protein turnover in functional macroautophagy
To study protein turnover in macroautophagy, defined as the net change of protein abundance, i.e. as the difference between protein synthesis and degradation, we performed SILAC-based pulse-labeling experiments. Amino acid pools and recycling rates may influence quantification accuracy in pulse labeling experiments, especially in vivo. In our experimental set-up this was not the case. During pulse labeling, RIAs were on average approx. 90% and incorporation rates were constant (Fig. S3). Cells were fully SILAC labeled by “heavy” AA, followed by 7-h and 24-h pulses in “light” AA (Fig. 3A). During the pulse-phase cells were either left untreated (Ctrl) or treated as outlined above. To study the influence of pharmacological macroautophagy inhibition on protein turnover, we also included treatments plus addition of 3-methyladenine (3-MA). Turnover rates were determined by quantifying “light:heavy” peptide ratios. As treatments influence the cell cycle, doubling times were determined for all conditions and used to calculate proliferation-independent turnover rates. Rap and Glc starvation led to a decreased cell proliferation, and AA starvation led to a cell cycle arrest (Table S1). Whereas, we did not identify global changes in protein turnover after 7 h of treatment (Fig. 3B), 24 h of treatment led to overall changes in proteome turnover for all experimental conditions (Fig. 3C, Table S2, Fig. S4). All 3 treatments led to a decrease in protein turnover compared with control, on average by 15.1%, 8.2%, and 12.5% for AA, Glc starvation and Rap treatment, respectively (Fig. 3C), which is at least partly due to a dramatic decrease in protein synthesis (Fig. S5). Interestingly, inhibition of macroautophagy by 3-MA further decreased protein turnover in all conditions to a similar extent (Fig. S6).
Figure 3.
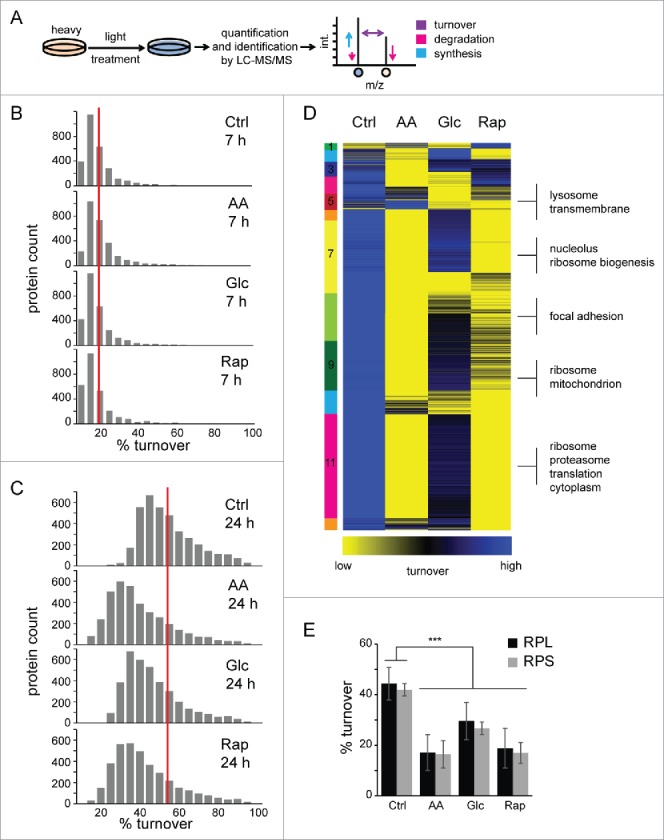
Protein turnover in macroautophagy. (A) Experimental outline to determine protein turnover changes in macroautophagy. Cells were fully labeled with “heavy” SILAC AA before 7-h or 24-h pulses under different treatments, and control conditions were performed with “light” SILAC AA. Cells were lysed, proteins separated by SDS-PAGE and in-gel digested with trypsin. Resulting peptide mixtures were analyzed by LC-MS/MS. Peptide ratios of light:heavy represent turnover values of respective proteins during 7 h or 24 h and reflect synthesis as well as degradation changes. (B) Histograms of protein turnover rates in 7-h stimulations. SILAC ratios were normalized by cell growth to determine proliferation-independent protein turnover rates. Rates were binned and upper values are indicated. The red line indicates the average turnover rate under control conditions (17%) in 7 h. (C) Histograms of protein turnover rates in 24-h stimulations. Data were analyzed as in (B). The red line indicates the average turnover rate under control conditions (52%) in 24 h. (D) k-means cluster analysis of protein turnover values. SILAC ratios representing turnover rates of 2 biologic replicates each were averaged, log2 transformed and z-score normalized. Clusters are indicated by numbers. Selected, significant enriched GO terms and keywords are indicated (BH corrected p < 0.05). (E) Turnover rates of ribosomal proteins decrease during active macroautophagy. Average turnover rates of 46 proteins of the 60S large ribosomal subunit (RPL) and 27 proteins of the 40S small ribosomal subunit (RPS) are depicted. Error bars indicate standard deviations. *: p < 0.05; **: p < 0.01; ***: p < 0.001, unpaired T test.
Under control conditions, proteins exhibit a skewed distribution of turnover rates, similar to that described in yeast,23 with an average 50% protein turnover rate of approximately 23 h (Fig. 3C), which correlates well with recent MS-based proteomics studies (Fig. S7).11,17 Mitochondrial and metabolic proteins exhibited the lowest turnover rates under control conditions, followed by ribosomal proteins, plasma membrane and membrane proteins as analyzed by gene ontology (GO) enrichment. Comparing 24-h stimuli by cluster analysis it became evident that AA starvation was most distinct from the control, followed by Rap treatment (Fig. 3D, Table S3). In AA starvation and Rap treatment translation-associated and ribosomal proteins of the large and small subunit showed the most dramatic decrease in protein turnover (Fig. 3E), whereas proteins related to focal adhesions and lysosomes were affected in Glc starvation. Lysosomal proteins were surprisingly stable in Rap-treated and AA-starved cells, which might indicate their importance for macroautophagy. Thus, inhibition of MTOR leads to a general decrease of protein turnover, defined as the net change of protein abundance, at least partially through a drop in protein synthesis. Distinct macroautophagy-inducing stimuli appear to exhibit differential effects on protein turnover reflecting the nature of the respective stimulus.
Degradation of protein translation machinery by amino acid starvation-induced macroautophagy
To study specific protein degradation under macroautophagy-stimulating conditions, we have developed a SILAC-based degradation assay. In pulse-chase (pc) SILAC, cells are pulse-labeled with 2 different forms of “medium” and “heavy” amino acids (Fig. 4A, Fig. S8). Both, “medium” and “heavy” cells undergo a chase period in “light” medium, “medium” cells under control conditions and “heavy” cells under macroautophagy-inducing conditions, respectively. Afterwards cells are mixed, normalized to cell numbers using the “proteomic ruler” approach and intensity differences between peptides from “medium” and “heavy” cells can be readily analyzed.24 This setup allows the determination of “heavy” protein degradation during the chase period relative to “medium” control conditions. Using pcSILAC, data comparable to the long-lived protein degradation assay using radioisotope labeling can be generated (Fig. S9).1,25 However, in contrast to radioisotope labeling, pcSILAC yields a wealth of data.
Figure 4.

Protein degradation by macroautophagy analyzed by pcSILAC. (A) pcSILAC protein degradation assay. Cells are combined 1:1, lysed, separated by SDS-PAGE and proteins in-gel digested with trypsin. Peptides are analyzed by LC-MS/MS. Relative protein degradation rates are determined by quantifying “medium” to “heavy” signal intensities of corresponding peptides. (B) Correlation of pcSILAC data and western blot analysis. Cells were treated as in (A) and protein synthesis was inhibited with 1 µM cycloheximide. Protein abundance was analyzed by western blot (Fig. S11). SILAC and western blot ratios of treated versus control cells were log2 transformed and plotted. (C-D) Degradation of autophagy marker proteins. pcSILAC data of the 2 autophagy marker proteins MAP1LC3B and SQSTM1 (C) is compared with protein degradation analysis by western blot (D) using cycloheximide-treated cells (n = 4 for LC3 and n = 3 for SQSTM1). The red line indicates degradation under control conditions. 3MA blocks macroautophagy-dependent protein degradation. Error bars represent standard deviations of replicate measurements, or peptide variability in the case of single identifications. (E) k-means cluster analysis of protein degradation values. SILAC ratios representing degradation rates of 2 biologic replicates each were averaged, log2 transformed and z-score normalized. Clusters are indicated by numbers. Selected, significant enriched GO terms and keywords are indicated (BH corrected p < 0.05). *: p < 0.05; **: p < 0.01; ***: p < 0.001, unpaired t test.
In 2 biological replicates each, 3,871 proteins were identified and quantified (false discovery rate <1% on the peptide and protein level) giving rise to 1,747 degradation rates of individual proteins being detected in all conditions (Fig. S10, Table S4). pcSILAC data correlated well with quantitative western blots of cycloheximide-treated cells (Fig. 4B; Fig. S11). Cycloheximide inhibits protein synthesis allowing determination of protein degradation. Autophagy receptors have been identified that target proteins to phagophores and get degraded via macroautophagy.26 We analyzed the degradation of the autophagosomal marker MAP1LC3B/LC3B and the autophagy receptor SQSTM1 by pcSILAC (Fig. 4C) and western blot (Fig. 4D). As expected all 3 stimuli led to the degradation of the 2 proteins, both methods showing similar results. AA starvation had the biggest effect followed by Rap treatment and Glc starvation. Importantly, 3-MA was able to block degradation identifying macroautophagy as the main degradation pathway. Western blot analysis of SQSTM1 degradation under Glc starvation gave contradicting results, which likely reflect technical limitations.
To get a global impression of degradation differences we analyzed data by k means clustering (Fig. 4E); this yielded 10 clusters of similar size. AA starvation had the strongest effect followed by Rap treatment and Glc starvation. Comparison with 3-MA-treated samples highlights that stimuli not only induced macroautophagy-dependent protein degradation; e.g., in cluster 2 the degradation of several proteins in AA starvation could not be blocked by the addition of 3-MA. This cluster contained no clearly enriched protein set. Cluster 8 behaved similar for Rap treatment. Cluster 7 contained enzymes involved in carbohydrate metabolism which were degraded under Glc starvation and which could not be blocked by 3-MA. Interestingly several of these enzymes, such as PKM (pyruvate kinase, muscle), are CMA substrates.27 Also, we observed the 3-MA nonresponsive degradation of HK2 (hexokinase 2), a newly identified, Glc starvation-dependent CMA target (cluster 1, Table S5).28 Importantly, we identified the 3 clusters 5, 6, and 10, which contained proteins that were degraded by AA starvation-induced macroautophagy and which were blocked by the addition of 3-MA. These clusters contained proteins involved in protein translation, mRNA processing, and mitochondrial homeostasis, respectively (Table S5).
As could be shown in the yeast Saccharomyces cerevisiae that ribosomes can be selectively degraded under starvation conditions by macroautophagy,29 we focused on the protein translation machinery in our further analysis. AA starvation not only led to degradation of large and small ribosomal subunits by macroautophagy (Fig. 5A–B), but the EIF3 (eukaryotic translation initiation factor 3) complex was also selectively degraded under AA starvation conditions (Fig. 5C). Importantly, the increased degradation in AA starvation cannot be simply explained by a general increase in protein degradation and autophagic flux compared with the other 2 stimuli. Normalization to the respective SQSTM1 degradation indicated that degradation of protein translation machinery was selectively increased in AA starvation conditions (Fig. 5D). The majority of tRNA ligase family members was also degraded by AA starvation-induced macroautophagy (Fig. 5E). However, LARS and IARS (leucyl- and isoleucyl-tRNA synthetase) appeared to be spared from degradation. Interestingly the former has been shown to be a critical AA sensor upstream of MTOR.30 The apparent exclusion of LARS from degradation by AA starvation-induced macroautophagy led to an accumulation of the protein under starvation conditions compared with other tRNA ligases as detected by quantitative MS and western blot analyses (Fig. 5F).
Figure 5.
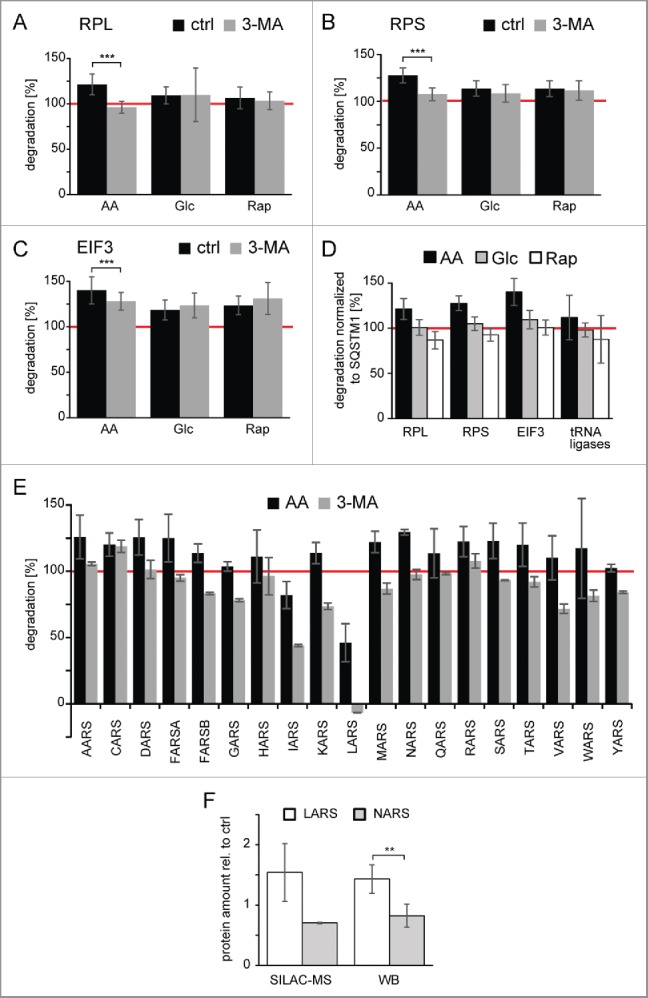
Degradation of protein translation machinery by amino acid-starvation induced macroautophagy. (A) Degradation of the large ribosomal subunit (RPL). Shown is the average degradation value of 39 components of the RPL. Only in the case of AA starvation is 3-MA able to block degradation. Error bars indicate standard deviation. (B) Degradation of the small ribosomal subunit (RPS). Shown is the average degradation value of 27 components of the RPS. Especially in the case of AA starvation 3-MA is able to block degradation. Error bars indicate standard deviation. (C) Degradation of the EIF3 (eukaryotic translation initiation factor 3). Shown is the average degradation value of 11 components of the EIF3 complex. Only in the case of AA starvation is 3-MA able to partially block degradation. Error bars indicate standard deviation. (D) Degradation relative to SQSTM1. Degradation rates (panel A-C, and E) relatively normalized to respective SQSTM1 degradation are depicted. Error bars indicate standard deviation. (E) Degradation of tRNA ligases by AA starvation-induced macroautophagy. Shown are average degradation values of tRNA ligases (n = 2). The amino acid sensor LARS, and to a minor extent IARS, are spared from degradation. Error bars indicate standard deviations. The red line indicates degradation under control conditions. (F) Quantification of protein abundances of LARS and NARS. Protein abundances of LARS and NARS after 24 h of amino acid starvation were quantified relative to untreated control conditions using SILAC-based MS (n = 2) and western blot (WB) analysis (n = 5). Whereas NARS abundance decreases relative to control cells, LARS abundance increases. *: p < 0.05; **: p < 0.01; ***: p < 0.001, unpaired t test.
Discussion
In the current study we globally analyzed protein turnover and degradation as well as autophagosomal dynamics by a complementary set of analytical techniques generating a data resource for the autophagy research community. Quantitative MS-based proteomics data indicated stimulus-specific protein degradation by macroautophagy. Image-based, time-resolved analyses highlighted stimulus-specific dynamics of autophagosomes and autolysosomes. Thus, large-scale analyses outlined discrete cellular responses in stress-induced macroautophagy. Whereas basal macroautophagy may indeed be a nonspecific, bulk degradation pathway, stress-induced macroautophagy clearly reflected specific, cellular conditions.
Protein turnover by quantitative MS has been studied in diverse biologic settings, recently, and data presented here correlate well with the respective studies.11,17 In addition to MS-based approaches large-scale image-based protein turnover studies using ectopically expressed fusion-proteins have been performed.8,9 Compared with these, MS data presented in this investigation did not correlate. However, compared with each other, imaging studies show also near random correlation. Overexpression of tagged fusion proteins may alter intrinsic protein properties, which may influence respective turnover analyses.
To our knowledge, this is the first comprehensive study of endogenous protein turnover in active macroautophagy using a global MS-based proteomics approach. Although automated global analyses may lead in single cases to wrongly assigned peptide signals in the case of low abundant and/or co-eluting peptides, this is often reflected by higher quantification errors. Thus, respective data can be investigated manually and checked for consistency. Interestingly, protein turnover, defined as the net change of protein abundance, i.e. as the difference between protein synthesis and degradation, dropped regardless of the inducing stimulus, which may be explained by a decrease in protein synthesis through inactivation of MTOR.31,32 For example, in AA starvation, protein translation is impaired due to a lack of protein building blocks, which is highlighted by a cell cycle arrest and a dramatic decrease in protein synthesis. Hence, ribosomes are no longer needed in large quantities, are no longer synthesized, and, consequently, exhibit a pronounced decrease in turnover. In Rap treatment, under nutrient-rich conditions, protein turnover also decreased. The drop of protein turnover despite increased degradation by functional macroautophagy indicates the dominant influence of protein synthesis on turnover measurements in the chosen experimental conditions. The lack of identifying global stimulus-specific changes in protein turnover after 7 h of treatment likely reflects a combination of influences of the chosen biologic system and technical limitations. Shorter labeling periods lead to less label incorporation making accurate quantification by MS more challenging. In addition, MCF7 cells show low metabolic rates as highlighted by the doubling time of 36.5 h, again leading to less label incorporation. Currently, we are establishing new cell line models and labeling protocols to study short-term responses. Interestingly, 3-MA appears to interfere with pulse labeling strategies. However, if this is due to interference with protein synthesis or with label incorporation is unclear.
As the direct effects of protein degradation by macroautophagy cannot be easily deduced from pulse-labeling experiments under nonsteady-state conditions, we designed a pulse-chase-labeling strategy using stable isotopes, pcSILAC, to directly study the effects of macroautophagy. In pcSILAC relative degradation differences compared with control conditions are analyzed and different treatments can be compared without the need to assume steady-state conditions and to perform time course experiments.10,15 Similar labeling approaches have recently been taken, e.g. to study protein palmitoylation.33 Compared to the commonly used radioisotope labeling experiments, or fluorescent approaches,34 MS-based approaches yield generally larger errors. However, proteome-wide degradation rates can be readily analyzed allowing simultaneous, detailed analyses of numerous proteins. We used 3 well-established macroautophagy-inducing conditions to study their effects on protein degradation by macroautophagy. Although we cannot exclude other degradative routes influencing our results,27 previous studies showed that on a global scale CMA can be neglected in MCF7 cells during long-term starvation,21 and that proteasome abundance as well as activity decreases during active macroautophagy.35 In addition, 3-MA blocked protein degradation. Thus, we conclude that macroautophagy was the main mechanism responsible for the observed phenotypes.
Glc starvation did not globally alter degradation rates. The weak global effects in Glc-starved cells were underlined by only minor differences in autophagosome turnover as analyzed by imaging. In contrast, AA starvation led to an approximately 10-fold increase in autophagosomal turnover, followed by a 2-fold increase by Rap treatment. Additionally, image analyses highlighted altered numbers, sizes, and localizations of autophagosomes and autolysosomes depending on the inducing stimulus. We monitored an efficient, perinuclear degradation of autophagosomes under nutrient deprivation, which is in agreement with lysosomal clustering around the nucleus under these conditions.36 This population seemed to expand under long-term treatments. In addition, a second peripheral autophagosome population was observed. As images differ substantially over the entire timeframe the observed differences in organellar dynamics cannot simply be explained by altered, stimulus-dependent kinetics. Rather, it appears that the characteristics of the organelles and their cellular localization are “unique” to each stimulus, possibly influencing target selection. The overexpression of LC3B may influence these results, and analyses of other LC3 isoforms may differ. However, as images were used to relatively compare stimuli in the same cell line, observed differences reflect differential, stimulus-specific cellular responses. In addition, image data, flux analyses by western blot, and proteomics data are in agreement. Differences on organellar level as analyzed by the first 2 bulk approaches, western blot and imaging, were relatively big compared with relative protein degradation measurements by MS-based proteomics indicating that the protein content per autophagosome has to be rather small.
In AA starvation, proteins involved in translation, such as ribosomal proteins and translation initiation factors, exhibited pronounced degradation. It has been observed in RAS-driven cancer cells that macroautophagy in response to acute amino acid starvation has specific effects on cellular proteomes.37 Whereas in the respective study ribosomes appeared stable within 5 h of starvation, we found that in long-term starvation for 24 h ribosomes are specifically degraded by macroautophagy. These observations are in agreement with the finding that organelles are degraded at later time points, whereas cytosolic proteins are degraded early in starvation.21 Also, the stimulus-specific proteomic composition of autophagosomes underlines the dominant effect of the inducing stimulus on the cargo targeted for autophagosomal degradation.35 The fact that amino acids sensors such as LARS appear to be spared from degradation further indicates that stress-induced macroautophagy is specific,30 either by explicitly targeting proteins to phagophores, or by specifically preventing this from happening.
Taken together, using a combination of quantitative MS-based proteomics data and image-based modeling we outline stimulus-specific differences in protein and organelle turnover in macroautophagy. A new, versatile, and generic MS-based assay termed pcSILAC was developed for the assessment of individual, relative protein degradation rates on a proteomic scale without the need to assume steady-state conditions or measure absolute protein abundances. Using this assay stimulus-dependent degradation differences by macroautophagy were outlined by monitoring more than 1,000 proteins. Whereas Glc starvation differed profoundly from the other 2 stimuli and had effects on single proteins, AA starvation and Rap treatment exhibited stronger outcomes, with AA starvation specifically targeting protein translation components for degradation. Among others, one open question is if organelles are degraded as complete structures or if sorting events lead to selective degradation of substructures.38 In the future the analysis of signaling events that are responsible for stimulus-specific autophagosomal target selection may shed more light onto the selective nature of stress-induced macroautophagy. Our data imply that these signaling events are not limited to a subset of cargo proteins, but rather that a substantial part of the protein population is selectively targeted to phagophores.
Materials and methods
Cell culture
MCF7-eGFP-LC3 cells were cultured in DMEM (Gibco, Thermo Fisher, 21969035) containing 4.5 g/l glucose, sodium pyruvate, 3.7 g/l NaHCO3, supplemented with penicillin/streptomycin (100 U/ml, 100 µg/ml) (PAA, P06–07100), glutamine (PAN, P04–80100), 10% fetal calf serum (Gibco, Thermo Fisher, 10270–106).35 Cell populations were labeled with either “medium” AA: L-arginine-13C6–14N4 (Arg6; EURISO-TOP GmbH, CLM-2265-H-1) and L-lysine-2H4 (Lys4; EURISO-TOP GmbH, DLM-2640–1), or “heavy” AA: L-arginine-13C6–15N4 (Arg10; EURISO-TOP GmbH, CNLM-539-H-1) and L-lysine-13C6–15N2 (Lys8; EURISO-TOP GmbH, CNLM-291-H-1) (10-cm cell culture dishes; ∼50% confluent after treatment). SILAC-DMEM (Thermo Fisher, 89985) supplemented with proline (Sigma, 81709), penicillin/streptomycin (100 U/ml, 100 µg/ml), glutamine, and 10% dialyzed fetal calf serum (Gibco, Thermo Fisher, 26400–044) was used during labeling. For pulse-chase experiments cell populations labeled with Arg10 and Lys8 (pulse) were subsequently treated for 24 h (chase) after washing twice with DPBS (Genaxxon, C4219.0500). Rapamycin (Sigma-Aldrich, R0395) was used in a concentration of 100 nM, and 3-methylalanine 10 mM (Sigma-Aldrich, M9281). HBSS (Pan Biotech, 14025050) contained calcium, magnesium, 0.37 g/l NaHCO3 and 1 g glucose per liter supplemented with penicillin/streptomycin (100 U/ml, 100 µg/ml). Cycloheximide (C6255) was from Sigma Aldrich. All treatments were performed at 37°C and 5% CO2. The cells were trypsinzed and 3 aliquots of each sample were used for normalization by cell counting.
MS sample preparation and antibodies
Harvested cells were centrifuged for 5 min at 2000 rcf at 4°C and subsequently lysed in SDS-loading buffer. Benzonase (Merck, 1.01695.0001) was added before samples were reduced with DTT (100 mM; Sigma-Aldrich, 43817) and alkylated using iodoacetamide (5.5 mM; Sigma-Aldrich, I1149). Protein mixtures were separated using SDS-PAGE (4–12% Bis-Tris gradient gel, NuPAGE; Invitrogen, Thermo Fisher, 1 mm, 10 well, NP0321BOX), gel lanes were cut into 10 slices according to their protein content, samples in-gel digested using trypsin (Promega, V5113), and resulting peptide mixtures were STAGE tipped.
The following antibodies were from Cell Signaling Technologt: RPS6KB1 phospho-Thr389 (9206), RPS6KB1 (9202), SQSTM1 (5114). Antibodies against NARS (asparaginyl-tRNA synthetase) and LARS were from Abcam: anti-NARS (ab53473) and anti-LARS (ab204558). Antibodies from Santa Cruz Biotechnology were: anti-GFP antibody (sc-9996), anti-ACTB (sc-47778), anti-ATP2A1/SERCA1 (sc-30110), anti-HSPA5 (sc-166490), anti-CAT (sc-69762), anti-EEA1 (sc-137130), anti-TFRC (sc-32272), and anti-HSPA9 (sc-13967).
Mass spectrometry
Mass spectrometric measurements were performed on a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a nanoelectrospray ion source (Proxeon Biosystems, Odense, Denmark) and coupled to an Agilent 1200 nanoflow-HPLC (Agilent Technologies GmbH, Waldbronn, Germany). HPLC-column tips (fused silica) with 75-µm inner diameter (New Objective, FS360–75–10-N-5-C25) were self packed with ReproSil Pure C18-AQ 3 µm (Dr. Maisch, r13.aq) to a length of 20 cm. Samples were applied directly onto the column without a precolumn. A gradient of A (0.5% acetic acid [high purity, LGC Standards GmbH, HPA-0050-B010] in Milipore water) and B (0.5% acetic acid in 80% ACN [LC-MS Optigrade, Wako, SO-9340-B025]) with increasing organic proportion was used for peptide separation (loading of sample with 2% B; separation ramp: from 10% to 30% B within 80 min). The flow rate was 250 nl/min and for sample application 500 nl/min. The mass spectrometer was operated in the data-dependent mode and switched automatically between MS (max. of 1 × 106 ions) and MS/MS. Each MS scan was followed by a maximum of 5 MS/MS scans in the linear ion trap using a collision energy of 35% and a target value of 5,000. Parent ions with charge states from z = 1 and unassigned charge states were excluded for fragmentation. The mass range for MS was m/z = 370 to 2000. The resolution was set to 60,000. Mass-spectrometric parameters were as follows: spray voltage 2.3 kV; no sheath and auxiliary gas flow; ion-transfer tube temperature 125°C.
Identification of proteins and protein ratio assignment using MaxQuant and data analysis
The MS raw data files were uploaded into the MaxQuant software version 1.4.1.2, which performs peak detection, SILAC-pair detection, generates peak lists of mass error corrected peptides and performs database searches using the following parameters: UniProt human decoy database, August 2015, containing common contaminants such as keratins and enzymes used for in-gel digestion was used, carbamidomethyl-cysteine was set as fixed modification, methionine oxidation, protein amino-terminal acetylation formation of N-pyroglutamate and glutamine, were set as variable modifications.39 Double or triple SILAC were chosen as quantification mode. Three missed cleavages were allowed, enzyme specificity was trypsin/P, and the MS/MS tolerance was set to 0.5 Da. Peak lists were searched by Andromeda for peptide identification. The average mass precision of identified peptides was in general less than 1 ppm after recalibration. Peptide lists were further used by MaxQuant to identify and relatively quantify proteins using the following parameters: peptide and protein false discovery rates were set to 0.01, maximum peptide posterior error probability (PEP) was set to 1, minimum peptide length was set to 7, minimum number peptides for identification and quantification of proteins was set to one, which must be unique and identified proteins have been requantified.
In pcSILAC experiments H/M SILAC-ratios of the MaxQuant output were used for data analysis. The SILAC-ratios were normalized to cell count using the average ratios of identified histone proteins.24 Functional enrichment analysis was performed using Perseus and significant enrichment was determined using the Fisher's Exact Test (BH corrected p value < 0.05).40
Microscopy
For confocal imaging, cells were grown on coverslips as described above and rinsed 3 times with DPBS before they were fixed in 4% paraformaldehyde for 10 min. Coverslips were mounted on glass slides using ProLong Gold antifade reagent (Invitrogen, Thermo Fisher, P36930) and imaged using a Zeiss LSM 510 Meta laser scanning confocal microscope and ZEN 2008 Software (Carl Zeiss MicroImaging GmbH, Esslingen, Germany). Each experiment was performed as 2 biologic replicates and 4 pictures were taken per experiment. Autophagosomes were identified by green fluorescence of eGFP-fused LC3B. Cell nuclei were visualized by DAPI staining contained in Pro Long Gold.
Widefield imaging was performed on paraformaldehyde-fixed cells on the Axio Cell Observer microscope (Carl Zeiss MicroImaging GmbH) equipped with the Axiocam Rev.3 CCD camera using a 100x oil objective. Images were taken as 15 z-stacks with a spacing of 0.5 µm for 3 channels (ch1: DAPI, ch2: GFP, ch3: RFP). Images were deconvolved using the Huygens Essential software (Scientific Volume Imaging, Hilversum, Netherlands).
Time-lapse live-cell imaging was performed on the Biostation IM (Nikon, Tokyo, Japan) using a 20x objective. For this, cells were cultivated in glass bottom micro-dishes (Ibidi GmbH, 80136) at 37°C and 5% CO2.
Image analysis
Local feature models
For high resolution (100x) images, regions of connected pixels with single local maxima, which we call objects, were found by adaptive thresholding. Using k-means clustering, objects were grouped into object types using 3D-Subcellular Object Features.41 After choosing the number of object type clusters that gave the lowest AIC, each image was represented by concatenating the vectors that describe the proportion of each object type in each of the RFP and GFP channels. The first 3 principal components of the average vectors for each condition and time point were calculated. The same process as above was performed using only the GFP channel of the lower resolution (20x) time-series images.
Protein object model
Individual nuclei were found in the DAPI channel with region-based active contour segmentation and their positions used for seeded watershed segmentation on the summed GFP, RFP and DAPI channels to determine the boundaries between all cells in the image.42 Cells with a nucleus touching the border of the image were discarded and not used in the analysis. The cell boundary for each of the remaining cells was refined using active contour segmentation on each cellular region with the combined channels. For each channel, objects were detected within each cell by active thresholding (to find regions of local maxima) starting at the Ridler-Calvard intensity threshold for each cell. The resulting image was ANDed with the highest 98% magnitude responses after convolving the image with a 3×3×3 pixel Laplacian of Gaussian filter (to find punctate regions). Finally, the positions of each object relative to the nucleus and cell boundaries, and the object intensities in each channel were calculated.
Statistical analyses
To analyze if changes in abundance/turnover of specific proteins/protein groups were significant unpaired Student t test with equal sample variance were performed. Input data were quantified western blots (minimally 3 biological replicates) or MS data of protein turnover and degradation rates. In enrichment analyses using proteomic input data Fishers' exact tests were performed and p values were corrected for multiple testing using the BH approach.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- AA
amino acid
- BH
Benjamini Hochberg
- CMA
chaperone-mediated autophagy
- ConA
concanamycin A
- Glc
glucose
- GO
gene ontology
- KS
Kolmogorov Smirnov
- MS
mass spectrometry
- PCA
principal component analysis
- Rap
rapamycin
- RIA
relative isotope abundance
- SD
standard deviation
- SILAC
stable isotope labeling by amino acids in cell culture
- UPS
ubiquitin-proteasome system
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank R. Nitschke and colleagues from the LIC for microscopy support, and M. Selbach for data sharing.
Funding
The research leading to these results received funding from the Swiss National Science Foundation, grant 31003A‐166482/1, from the Excellence Initiative of the German Federal and State Governments through FRIAS, from TRANSAUTOPHAGY (COST Action CA15138), from the Federal Ministry of Education and Research through GerontoSys II – NephAge (031 5896 A), by a grant from the Danish Natural Sciences Research Council (KR), the Alexander von Humboldt Foundation (RFM, JD), and by National Institutes of Health grant EB009403 and GM103712.
References
- [1].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; https://doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, et al.. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6:367-80; PMID:19837376; https://doi.org/ 10.1016/j.chom.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; https://doi.org/ 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; https://doi.org/ 10.1038/nrd3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24:92-104; PMID:24281265; https://doi.org/ 10.1038/cr.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hinkson IV, Elias JE. The dynamic state of protein turnover: It's about time. Trends Cell Biol 2011; 21:293-303; PMID:21474317; https://doi.org/ 10.1016/j.tcb.2011.02.002 [DOI] [PubMed] [Google Scholar]
- [7].Li Q. Advances in protein turnover analysis at the global level and biological insights. Mass Spectrom Rev 2010; 29:717-36; PMID:19757418; https://doi.org/ 10.1002/mas.20261 [DOI] [PubMed] [Google Scholar]
- [8].Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, Cohen L, Mayo A, Alon U. Proteome half-life dynamics in living human cells. Science 2011; 331:764-68; PMID:21233346; https://doi.org/ 10.1126/science.1199784 [DOI] [PubMed] [Google Scholar]
- [9].Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science 2008; 322:918-23; PMID:18988847; https://doi.org/ 10.1126/science.1160489 [DOI] [PubMed] [Google Scholar]
- [10].Pratt JM, Petty J, Riba-Garcia I, Robertson DH, Gaskell SJ, Oliver SG, Beynon RJ. Dynamics of protein turnover, a missing dimension in proteomics. Mol Cell Proteomics 2002; 1:579-91; PMID:12376573; https://doi.org/ 10.1074/mcp.M200046-MCP200 [DOI] [PubMed] [Google Scholar]
- [11].Boisvert FM, Ahmad Y, Gierlinski M, Charriere F, Lamond D, Scott M, Barton G, Lamond AI. A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol Cell Proteomics 2012; 11:M111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Doherty MK, Beynon RJ. Protein turnover on the scale of the proteome. Expert Rev Proteomics 2006; 3:97-110; PMID:16445354; https://doi.org/ 10.1586/14789450.3.1.97 [DOI] [PubMed] [Google Scholar]
- [13].Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008; 455:58-63; PMID:18668040; https://doi.org/ 10.1038/nature07228 [DOI] [PubMed] [Google Scholar]
- [14].Larance M, Ahmad Y, Kirkwood KJ, Ly T, Lamond AI. Global subcellular characterization of protein degradation using quantitative proteomics. Mol Cell Proteomics 2013; 12:638-50; PMID:23242552; https://doi.org/ 10.1074/mcp.M112.024547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Larance M, Pourkarimi E, Wang B, Brenes Murillo A, Kent R, Lamond AI, Gartner A. Global Proteomics Analysis of the Response to Starvation in C. elegans. Mol Cell Proteomics 2015; 14:1989-2001; PMID:25963834; https://doi.org/ 10.1074/mcp.M114.044289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Engelke R, Becker AC, Dengjel J. The degradative inventory of the cell: proteomic insights. Antioxid Redox Signal 2012; 17:803-12; PMID:22074050; https://doi.org/ 10.1089/ars.2011.4393 [DOI] [PubMed] [Google Scholar]
- [17].Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature 2011; 473:337-42; PMID:21593866; https://doi.org/ 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- [18].Zimmermann AC, Zarei M, Eiselein S, Dengjel J. Quantitative proteomics for the analysis of spatio-temporal protein dynamics during autophagy. Autophagy 2010; 6:1009-1016; PMID:20603599; https://doi.org/ 10.4161/auto.6.8.12786 [DOI] [PubMed] [Google Scholar]
- [19].Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al.. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193-205; PMID:17244528; https://doi.org/ 10.1016/j.molcel.2006.12.009 [DOI] [PubMed] [Google Scholar]
- [20].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; https://doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [21].Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, Dengjel J, Andersen JS. Ordered Organelle Degradation during Starvation-induced Autophagy. Mol Cell Proteomics 2008; 7:2419-28; PMID:18687634; https://doi.org/ 10.1074/mcp.M800184-MCP200 [DOI] [PubMed] [Google Scholar]
- [22].Khaminets A, Behl C, Dikic I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol 2016; 26:6-16; PMID:26437584; https://doi.org/ 10.1016/j.tcb.2015.08.010 [DOI] [PubMed] [Google Scholar]
- [23].Belle A, Tanay A, Bitincka L, Shamir R, O'Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A 2006; 103:13004-009; PMID:16916930; https://doi.org/ 10.1073/pnas.0605420103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wisniewski JR, Hein MY, Cox J, Mann M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics 2014; 13:3497-3506; PMID:25225357; https://doi.org/ 10.1074/mcp.M113.037309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol 2009; 452:47-61; PMID:19200875 [DOI] [PubMed] [Google Scholar]
- [26].Khaminets A, Behl C, Dikic I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol 2015; PMID:26437584 [DOI] [PubMed] [Google Scholar]
- [27].Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014; 20:417-32; PMID:25043815; https://doi.org/ 10.1016/j.cmet.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T, et al.. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol 2015; 210:705-16; PMID:26323688; https://doi.org/ 10.1083/jcb.201503044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 2008; 10:602-10; PMID:18391941; https://doi.org/ 10.1038/ncb1723 [DOI] [PubMed] [Google Scholar]
- [30].Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012; 149:410-24; PMID:22424946; https://doi.org/ 10.1016/j.cell.2012.02.044 [DOI] [PubMed] [Google Scholar]
- [31].Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol 2012; 22:274-82; PMID:22444729; https://doi.org/ 10.1016/j.tcb.2012.02.006 [DOI] [PubMed] [Google Scholar]
- [32].Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21-35; PMID:21157483; https://doi.org/ 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. Global profiling of dynamic protein palmitoylation. Nat Methods 2011; 9:84-89; PMID:22056678; https://doi.org/ 10.1038/nmeth.1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang J, Wang J, Ng S, Lin Q, Shen HM. Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy 2014; 10:901-12; PMID:24675368; https://doi.org/ 10.4161/auto.28267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dengjel J, Høyer-Hansen M, Nielsen MO, Eisenberg T, Harder LM, Schandorff S, Farkas T, Kirkegaard T, Becker AC, Schroeder S, et al.. Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol Cell Proteomics 2012; 11:M111; PMID:22311637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al.. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 2011; 13:453-60; PMID:21394080; https://doi.org/ 10.1038/ncb2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, White E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 2014; 55:916-30; PMID:25175026; https://doi.org/ 10.1016/j.molcel.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Abeliovich H, Zarei M, Rigbolt KT, Youle RJ, Dengjel J. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat Commun 2013; 4:2789; PMID:24240771; https://doi.org/ 10.1038/ncomms3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008; 26:1367-72; PMID:19029910; https://doi.org/ 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- [40].Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 2012; 13(Suppl 16):S12; PMID:23176165; https://doi.org/ 10.1186/1471-2105-13-S16-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhao T, Velliste M, Boland MV, Murphy RF. Object type recognition for automated analysis of protein subcellular location. IEEE Trans Image Process 2005; 14:1351-59; PMID:16190470; https://doi.org/ 10.1109/TIP.2005.852456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chan TF, Vese LA. Active contours without edges. IEEE Trans Image Process 2001; 10:266-77; PMID:18249617; https://doi.org/ 10.1109/83.902291 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.