

ABSTRACT
Mitophagy plays pivotal roles in the selective disposal of unwanted mitochondria, and accumulation of damaged mitochondria has been linked to aging-related diseases. However, definitive proof that mitophagy regulates mitochondrial quality in vivo is lacking. It is also largely unclear whether damaged mitochondria are the cause or just the consequence of these diseases. We previously showed that FUNDC1 is a mitophagy receptor that interacts with LC3 to mediate mitophagy in response to hypoxia in cultured cells. We established Fundc1 knockout mouse models and used genetic and biochemical approaches, including a synthetic peptide that blocks the FUNDC1-LC3 interaction, to demonstrate that mitophagy regulates both mitochondrial quantity and quality in vivo in response to hypoxia or hypoxic conditions caused by ischemia-reperfusion (I/R) heart injury. We found that hypoxic mitophagy regulates platelet activities. Furthermore, we found that hypoxic preconditioning induces FUNDC1-dependent mitophagy in platelets and reduces I/R-induced heart injury, suggesting a new strategy to protect cardiac function and fight cardiovascular diseases.
KEYWORDS: heart injury, hypoxic mitophagy, ischemia/reperfusion, mitochondrial quality, mitophagy receptor, platelets
Accumulation of dysfunctional and superfluous mitochondria can be harmful to cells and organisms, and drives aging and aging-related pathogenesis. Elaborate pathways have evolved at both the protein and organellar levels to maintain mitochondrial quality. At the organellar level, the mitochondrial fission-fusion cycle and mitophagy pathways, as well as crosstalk between them, are critical for segregation and removal of damaged or unwanted mitochondria. The 2 major mitophagy pathways in mammalian cells are the PINK1-PARK2/Parkin pathway and receptor-mediated mitophagy. Distinct mitophagy mechanisms are activated by different mitochondrial stresses. Ablation of PARK2 or PINK1 causes impaired cardiac functions in mice. Knockout of the mitophagy receptor BNIP3L/NIX prevents enucleation and removal of mitochondria during the terminal differentiation of erythroid cells, resulting in anemia. However, definitive proof of the functional roles of receptor proteins in mitophagy and mitochondrial quality control under (patho-)physiological conditions is lacking.
We identified FUNDC1 as a novel receptor that mediates mitophagy by interacting with LC3 in response to hypoxia and mitochondrial stress. FUNDC1 is widely expressed in various cells, tissues and organs, especially heart. Under normoxic conditions, FUNDC1 is phosphorylated by SRC kinase and CSNK2/CK2, reducing its affinity for LC3. Under hypoxic conditions, FUNDC1 is dephosphorylated by PGAM5 or other yet-to-be identified phosphatases, which greatly enhances the FUNDC1-LC3 interaction. The fundamental unanswered question is how FUNDC1 regulates mitochondrial quality and mitophagy in (patho-)physiological settings. To address this, we developed germline and tissue-specific Fundc1 knockout mice using the Cre-LoxP system. When wild-type mice are exposed to hypoxia (8% oxygen) for 72 h, biochemical mitophagic hallmarks, including degradation of mitochondrial proteins and SQSTM1/p62, and appearance of LC3-II, are observed in heart, brain, liver, lung, and skeletal muscle. These phenotypes are largely prevented in knockout mice, which shows that mitophagy depends on FUNDC1 under hypoxic conditions.
Platelets may not make mitochondria, but they still have defined mitophagy mechanisms in place. We examined the functional significance of FUNDC1-mediated mitophagy in platelets upon hypoxic shock. Exposure of platelet-specific and wild-type mice to 8% hypoxia results in extensive mitochondrial degradation, which is largely prevented in FUNDC1-deficient platelets. The FUNDC1-LC3 interaction was investigated by in vivo administration of a cell-penetrating peptide (peptide P) encompassing the LIR motif of FUNDC1, which prevents mitochondrial degradation in wild-type platelets but not Fundc1-knockout platelets. Fundc1-null platelets contain an increased mass of dysfunctional mitochondria, as judged by reduced oxygen consumption rate, loss of mitochondrial membrane potential, low ATP production capacity and increased ROS levels. Thus, FUNDC1 mediates mitophagy in vivo and regulates mitochondrial quantity and quality under physiological conditions. We further showed that both mitochondrial quality and functional integrity are crucial for platelet activation. Fundc1-null platelets have impaired mitochondrial quality control and reduced levels of activation, and are insensitive to hypoxia and peptide treatments.
Platelets play key roles in thrombus formation and the pathogenesis of acute myocardial infarction, but the underlying molecular pathways are unclear. In ischemia/reperfusion (I/R) mouse models, platelets initially participate in thrombus formation, which causes coronary artery occlusion. Later, thrombi at the injury site impair the microcirculation, provoking myocardial ischemia and hypoxia. We therefore asked how mitophagy in platelets affects the I/R outcomes. We chose the I/R mouse model because it mimics clinical scenarios like myocardial infarction and other coronary vascular diseases. As expected, I/R triggers mitophagy in platelets from wild-type mice at later stages when the oxygen level becomes low. This response is significantly reduced in Fundc1-specific knockout (Fundc1fl/fl::Pf4Cre+) platelets, as the impaired mitochondria reduce platelet activity, which is negatively correlated with I/R injury. Intriguingly, Fundc1fl/fl::Pf4Cre- mice have larger infarct size and decreased cardiac functions, especially ejection fraction and fractional shortening, than Fundc1fl/fl::Pf4Cre+ mice. Initially, platelets from Fundc1fl/fl::Pf4Cre− mice have high activity and cause infarct injury, and in the late stage of I/R, hypoxic mitophagy leads to extensive mitochondrial degradation that diminishes mitochondrial activity and platelet activation, thus preventing worsening of I/R injury. Thus, we discovered dual roles of platelet mitophagy in regulating platelet activity and I/R injury. Under normal physiological conditions, damaged ‘toxic’ platelet mitochondria are removed, maintaining good mitochondrial quality and platelet activation. Platelets adhere to the injury site, reducing the blood oxygen levels and causing myocardial infarction. Later, vessel occlusion creates a hypoxic environment, which triggers platelet mitophagy and compromises platelet activity, thus preventing the I/R injury from worsening.
Hypoxic preconditioning in clinics reduces I/R-induced-heart injury. However, the mechanism remains unclear. We tested whether hypoxic preconditioning in mice induces mitophagy in platelets and determines the outcome of I/R injury. As expected, hypoxic preconditioning reduces the heart infarct size and maintains heart functions, because hypoxia increases the level of mitophagy in platelets and reduces platelet activation. The preconditioning effect is significantly reduced by the membrane-penetrating peptide encompassing the dephosphorylated LIR motif of FUNDC1.
In conclusion, we demonstrated that FUNDC1-mediated mitophagy regulates both mitochondrial quality and quantity in vivo and plays a causal role in mitochondrial quality control and functional integrity in platelet activation. We suggest that targeting mitophagy by hypoxia or pharmacological compounds may represent a novel approach to fighting cardiovascular diseases (Fig. 1).
Figure 1.
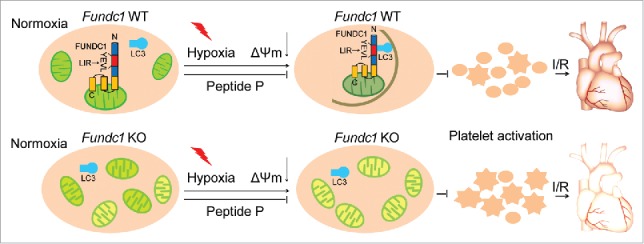
Working model of the protective role of FUNDC1in I/R injury through regulation of mitochondrial quality and platelet activity. The figure illustrates how hypoxia induces mitochondrial autophagy in vivo. Mitophagy occurs in a FUNDC1-dependent manner under stress conditions such as hypoxia and treatment with mitochondrial membrane uncouplers. Prolonged hypoxia causes extensive mitochondrial degradation, impaired platelet activity and reduced ischemia/reperfusion heart injury. Reduced platelet activation has a strong cardioprotective effect in response to I/R injury, and this effect can be blocked by administration of a cell-penetrating unphosphorylated FUNDC1 LIR peptide. When FUNDC1 is ablated, mitophagy is blocked and damaged mitochondria accumulate, resulting in diminished but sustained platelet activity. Under I/R conditions, Fundc1-knockout (KO) platelets with diminished but sustained mitochondrial activity cause more I/R heart injury. Gray curved line in the top middle panel, phagophore membrane; green mitochondrion, healthy mitochondrion; yellow mitochondrion, unhealthy mitochondrion; round platelet, unhealthy platelet; star-shaped platelet, healthy platelet; hearts in the top right panel and bottom right panel, better and poor functional hearts, respectively.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to all laboratory members for useful discussions.
Funding
This research was supported by the Natural Science Foundation of China (31520103904), the National Key Research and Development Program (2016YFA0500201, 2016YFA0100503), Special Fund for Strategic Pilot Technology Chinese Academy of Sciences (QYZDJ-SSW-SMC004) and the Beijing Natural Science Foundation of China (5161002) to QC, the Natural Science Foundation of China (31301130) and China postdoctoral grant (2013 M541041) to WZ.