

ABSTRACT
Inflammation plays a pivotal role in pathophysiological processes of kidney diseases. Macroautophagy/autophagy plays multiple roles in inflammatory responses, and the regulation of inflammation by autophagy has great potential as a treatment for damaged kidneys. A growing body of evidence suggests autophagy protects kidney from versatile kidney inflammatory insults, including those that are acute, chronic, metabolic, and aging-related. It is noteworthy that, in kidney, mitophagy is active, and damaged lysosomes are removed by autophagy. In this mode, autophagy suppresses inflammation to protect the kidney. Systemic inflammation also affects the kidney via pro-inflammatory cytokines and infiltration of inflammatory cells, and autophagy also has a regulatory role in systemic inflammation. This review focuses on the roles of autophagy in kidney diseases and aging through inflammation, and discusses the potential usage of autophagy as an inflammatory modulator for the treatment of kidney diseases.
KEYWORDS: acute kidney injury, aging, autoimmune disease, autophagy, chronic kidney disease, cytokine, diabetes, end-stage kidney disease, inflammation, innate immunity, kidney, lysosome, mitochondria, sepsis
Introduction
The kidney is one of the most complicated organs in the body, physiologically, structurally, and metabolically. Currently, most kidney diseases are incurable, and the number of patients with kidney diseases is increasing and accounts for a significant portion of medical expenses in developed countries. One of the keys for the development of kidney therapy is the regulation of inflammation, since inflammation is the basis of most kidney diseases.1
Autophagy is an intracellular degradation system for cellular homeostasis.2 Autophagy plays key roles in several diseases, including kidney diseases.3 This disease-related role of autophagy is at least partially derived from autophagic regulation of immune systems; autophagy has multiple roles in immune responses, ranging from the innate immune response to the acquired immune response, and going as far as direct pathogen restriction.4,5 Autophagy has been considered as a bulk degradation system as seen under starvation conditions to meet the energy needs of the cell,6 but autophagy can also recognize specific targets selectively.7 Recently, the importance of selective autophagy has been highlighted in disease processes,7 because the targets of selective autophagy include key organelles that are involved in diseases, such as mitochondria (where the process is termed “mitophagy”)7 and lysosomes (termed “lysophagy”).8
A growing number of studies have revealed the role of autophagy in kidney diseases8–19 and aging11,12,14,20 through genetic modifications of autophagy-related genes. The relatively clear phenotypes of kidney-specific autophagy-deficient mice have fascinated researchers in the kidney field, leading them to explore the potential benefits of autophagy modulations for incurable kidney diseases. The fact that mitophagy, a key regulator of inflammation,21 is active in kidney22 will also promote this trend. Multiple immune functions of autophagy9,10 are suggested to play key roles in kidney diseases (Fig. 1).
Figure 1.
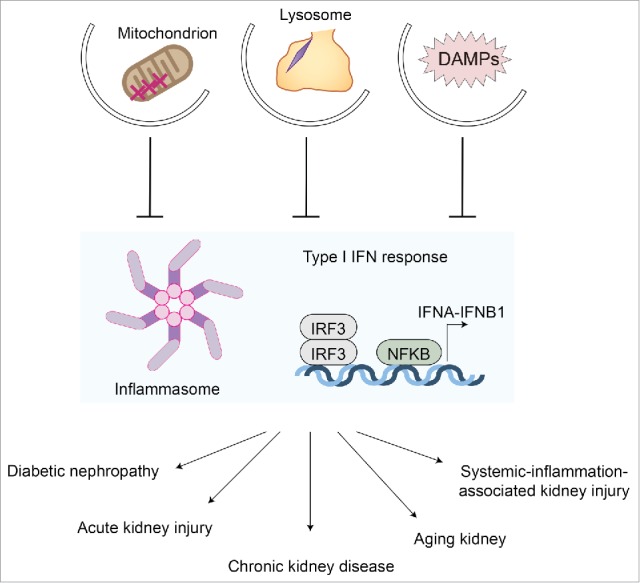
Autophagy and kidney inflammation. Autophagy suppresses excessive inflammatory responses, such as inflammasome activation and type I interferon responses, through the clearance of damage-associated molecular patterns (DAMPs) and damaged mitochondria (mitophagy). Autophagy of damaged lysosomes (lysophagy) also prevents activation of inflammatory responses. Excessive inflammatory responses are the fundamental basis for most kidney diseases, including acute kidney injury, chronic kidney disease, diabetic nephropathy, and the aging process, as well as systemic inflammation-associated kidney injury, which is often seen in sepsis patients. Thus, autophagy protects kidney from these insults via suppression of inflammatory responses. IRF3, interferon regulatory factor 3.
Basic information of kidney and inflammation
Before moving to the role of autophagy in kidney inflammation, we provide basic information about the kidney and inflammation for readers who are unfamiliar with this organ. The kidney maintains whole body homeostasis and comprises the functional unit called the nephron (i.e., a glomerulus plus a subset of tubules). Nephrons eliminate body wastes by glomerular filtration through its 3-layered structure of the capillary (small blood vessels) wall: the fenestrated endothelial cells, basement membrane, and foot processes of podocytes. Kidney tubules maintain volume and content (e.g., ions, glucose, amino acids, etc.) of body fluids through reabsorption.
Chronic kidney disease (CKD), a condition with reduced kidney function—referred to as glomerular filtration ratio (GFR)—is a global health problem with a prevalence of 5–10% of the world population.23 Progression of CKD increases mortality, as well as results in end-stage kidney disease (ESKD) necessitating kidney replacement therapy (dialysis or transplantation). Treatment for CKD, especially ESKD, is cost-prohibitive, and accounts for a significant portion of medical expenses in developed countries. These critical rationales strongly motivate researchers in kidney fields, but most kidney diseases are currently incurable.
Inflammation is a key pathology in kidney diseases.1 A number of acute or chronic insults, such as ischemia, drugs, toxins, metabolism, as well as inflammation in itself, injure kidney tubules. Injuries of the tubules, in turn, provoke inflammatory responses and result in kidney fibrosis. Aging kidney also manifest chronic inflammation of the tubular lesions. The extent of damage to the kidney tubules and subsequent fibrosis due to inflammation is strongly associated with reduced GFR in kidneys.24 Moreover, chronic inflammation is prevalent in patients with CKD, potentially due to chronic infection, or for unknown reasons. Chronic inflammation worsens kidney anemia and kidney function, and provokes malnutrition.25 Glomerular inflammation, called glomerulonephritis, causes proteinuria and worsening of GFR due to nephron loss. Glomerulonephritis often accompanies deposits of immune complexes and damage to the glomerular capillaries.
Autophagy and kidney tubular inflammation
The role of autophagy in tubules, the key determinant of kidney prognosis,24 has mainly been demonstrated in proximal tubules, because injuries of tubules mainly occur in their proximal part. Proximal tubules consume a large amount of oxygen and energy for electrolyte reabsorption.26 Kidney tubules also harbor abundant lysosomes and perform endocytosis actively.27 For these reasons, proximal tubules are rich in intracellular organelles such as mitochondria and lysosomes.
In response to acute pathological stimuli, such as ischemia/reperfusion injury and toxic side effects of drugs such as cisplatin and cyclosporine A, and urinary tract obstruction, autophagy is quickly upregulated in kidney tubules, as assessed by the number of MAP1LC3B/LC3B puncta of GFP-LC3 transgenic mice.12–14,17,28 Functionally, autophagy largely plays protective roles; autophagy protects the kidney from tubular damage and its consequent fibrosis.9,10 The majority of kidney diseases are driven by sterile inflammation,1 and autophagy plays a role in the regulation of endogenous proteins and organelles as discussed below.
1. Mitochondria
Autophagy, be it general or selective (mitophagy), targets damaged and depolarized mitochondria and regulates mitochondrial quality control. Kidney tubules are rich in mitochondria, and reactive oxygen species (ROS) produced in mitochondria trigger an inflammatory response,29 leading to pathogenesis in kidney disease. Acute insults such as ischemia/reperfusion injury and toxic drugs induce autophagy to prevent cell death.12–15,17 In response to these stimuli, autophagy-deficient kidney cells accumulate abnormal mitochondria.12–14,17 This is well-characterized in a cisplatin-induced kidney injury model in vitro.13 Cisplatin, a frequently-used chemotherapeutic drug, induces mitochondrial damage and promotes production of ROS. Autophagy protects kidney tubules from cisplatin through the removal of ROS-producing mitochondria.13 In addition to the removal of the ROS-producing acutely damaged mitochondria, autophagy also controls the quality of mitochondria. Lack of quality control of mitochondria in aging autophagy-deficient mice worsens mitochondrial function,20 a characteristic of the aging process.30
From the metabolic standpoint, mitochondria are the main intracellular energy source through oxidative phosphorylation. The worsening mitochondrial function in autophagy-deficient kidney affects intracellular metabolism.6,17 Reduced mitochondrial respiratory chain activity due to depolarization seen in autophagy-deficient kidney affects the kidney's adaptation to metabolic acidosis,18 a common pathological condition seen in patients with kidney diseases. Failure to cope with metabolic acidosis has been implicated as a pathogenesis of systemic inflammation seen in chronic kidney disease and malnutrition.25 Nowadays, the link between intracellular metabolism and immune systems (immunometabolism) has been emphasized.31 Regulation of mitochondrial metabolism by autophagy may play immunometabolic roles in kidney diseases.
A transgenic mouse with a pH-sensitive fluorescent mitochondrial signal is just established recently for in vivo monitoring of mitophagy.22 The analyses of this mouse, called mito-QC, revealed that the kidney is one of the most active tissues of mitophagy. In the kidney, mitophagy mainly occurs in proximal tubules. This fact strongly suggests the critical role of mitophagy in proximal tubules and thus confirms the protective role of autophagy in these regions.12–14,17
2. Lysosomes
Lysosomal rupture strongly activates inflammation. Crystals such as monosodium urate damage lysosomal membranes. Lysosomal rupture, in turn, activates the inflammasome, which induces the secretion of pro-inflammatory cytokines such as IL1B/interleukin-1β, leading to further enhancement of inflammation.32 Leakage of lysosomal enzymes into the cytosol provokes apoptotic or necrotic cell death or reduced capacity of lysosomal catabolism.
In kidney, hyperuricemia, both acute and chronic, causes lysosomal rupture. Upon chemotherapy, dying hematopoietic malignant cells release a huge amount of uric acid produced through increased purine catabolism. As a result, uric acid becomes oversaturated acutely in the urine, leading to the formation of urate crystals in the kidney.33 Although the affected lesions remain to be elucidated, crystal deposits in proximal tubules are supposed to induce hyperuricemia-induced kidney injury for 2 reasons; (i) proximal tubules have robust urate transporters and (ii) urate crystals can precipitate on the cell surface of kidney epithelial cells.34,35 Chronic hyperuricemia due to gout can also induce kidney inflammation and disease (gout nephropathy) via precipitation of urate crystals. These urinary crystals are endocytosed and delivered to lysosomes, where rupture happens while inducing inflammation.
Autophagy in kidney also protects against lysosomal rupture-induced inflammatory injuries.8,35 Genetic ablation of Atg5 worsens kidney function in a mouse model of acute hyperuricemic kidney injury. Mechanistically, autophagy engulfs damaged lysosomes and sequesters them from the cytosol to prevent cellular injury. Through this process, autophagy also restores lysosomal functions (lysosomal pH and degradation capacity) and biogenesis.
3. Damage-associated molecular patterns (DAMPs)
DAMPs encompass a range of cellular components, both nuclear (such as histones, DNA/RNA, and HMGB1) and cytosolic (mitochondrial DNA, ATP and glycoproteins) ones. DAMPs activate innate immune responses including the inflammasome and type I interferon responses, which in turn trigger macrophages and leukocytes to infiltrate into the kidney interstitium (the extravascular intertubular spaces of the kidney) to promote inflammation.
Autophagy also suppresses the release of DAMPs.5 Endogenous DAMPs are well-known targets for autophagic degradation. The release of mitochondria DNA, which strongly induces inflammasome activation, is suppressed by autophagy.36 Thus, autophagy suppresses inflammation through suppression of DAMPs release by cellular protection and DAMPs degradation. Although it is not examined in depth in the kidney, autophagic control of intracellular protein quality should reduce or prevent releasing DAMPs from this organ.
Additionally, autophagy has a secretory role for DAMPs.5 One of the best-characterized DAMPs secreted by autophagy is IL1B. Whereas basal autophagy suppresses IL1B secretion through clearance of DAMPs such as damaged mitochondria that generate ROS,29,36,37 autophagy promotes IL1B secretion once the inflammasome is activated.38,39 Although the significance is yet to be proven in the kidney, autophagy secretion may play pro-inflammatory roles in kidney disease formation.
In summary, autophagy largely suppresses kidney tubular inflammation through the removal of damaged and malfunctioning mitochondria. Mitophagy is active in kidney tubules to suppress inflammation and subsequent worsening of kidney function. Quality control of mitochondria may also have an immunomodulatory role in kidney via metabolism. Removal of damaged lysosomes also has immunomodulatory effects, and may suppress DAMPs release.5
Autophagy and kidney glomerular inflammation
Inflammation plays a key role in the pathogenesis of glomerulonephritis as mentioned above, but the roles of autophagic regulation of inflammation in glomerular diseases are still unclear.
In glomeruli, the role of autophagy was demonstrated in podocytes and endothelial cells. Podocytes are terminally differentiated cells, and are considered to be poorly regenerative. Thus, essential roles of autophagy in maintenance of podocytes have been expected. GFP-LC3-expressing transgenic mice show a high number of LC3 dots in podocytes.40 Although GFP-LC3 dots may suggest high basal autophagic activity, podocyte-specific Atg5-deficient mice show a mild phenotype of glomerular injury: late onset of slight proteinuria (8–12 mo) and mild glomerulosclerosis (24 mo). The discrepancy between the at-a-glance high activity of autophagy in GFP-LC3 mice and the slow appearance of a phenotype in knockout mice remains to be resolved.41 The study with mito-QC shows mitophagy is active in embryonic glomeruli, but not in adult ones.22 This fact may illuminate a critical role of mitophagy in glomerular development, but not in the maintenance of adult glomeruli.
Interestingly, podocyte-specific deficiency of Pik3c3/Vps34, the class III phosphatidylinositol 3-kinase for initiation of autophagy, leads to severe forms of proteinuria and earlier onset of glomerular sclerosis and death (9 wk).42,43 This phenotype is unlike the ones seen in autophagy-deficient mice or in liver- and heart-specific Pik3c3/Vsp34-deficient mice,44 the latter of which (milder and later-onset of organ dysfunction) are suggested to be due to the blockade of autophagy. Rather, this kidney phenotype is attributed to the disruption of the endosomal pathway.43 The reason for the different phenotypes in tissue-specific Pik3c3/Vps34-deficient mice, autophagic vs nonautophagic ones, awaits to be elucidated.11,40,42,43
In summary, autophagy largely plays a protective role in glomeruli, but unique features of podocytes (i.e., terminal differentiation and active endocytosis) may complicate our understanding of phenotypes seen in mice with genetic ablations of autophagy-related genes. Additionally, the activity status and significance of autophagy in glomerular inflammation still needs to be proven. Inactive mitophagy in adult glomeruli22 may indicate less contribution of mitochondria in glomerular inflammation.
Autophagy, diabetic nephropathy and inflammation
Diabetic nephropathy is one of the major complications of diabetes and diabetic nephropathy is the leading cause of end-stage kidney disease in industrialized countries. The main pathologies of diabetic nephropathy are microvascular injury and loss of podocytes in the glomerulus.45 Inflammation is a worsening factor for the progression of diabetes nephritis.1 Diabetes is associated with increased intracellular ROS,32 an activator of the inflammasome.29 Advanced glycation end products (AGEs), irreversibly-glycated proteins generated under hyperglycemia, are endocytosed by kidney proximal tubules for lysosomal degradation to suppress AGEs-induced inflammation; either overproduction of AGEs or impairment of degradation of AGEs activate inflammation and thus promote diabetic nephropathy.46 Along with the notion that IL1B, the product of inflammasome activation, pivotally worsens glucose tolerance,32 inflammasome activation is recognized as a key pathophysiology of diabetic nephropathy.
Some evidence has shown that deficiency of autophagy worsens diabetic nephropathy in rodent models. For example, endothelial-specific autophagy-deficiency worsens the diabetic phenotype of a streptozotocin-induced diabetes model (type I diabetes model); severe microalbuminuria, endothelial lesions and damage in podocytes are seen in streptozotocin-induced autophagy-deficient mice.19 High-fat diet challenge, which induces hyperglycemia with proteinuria, damages podocytes in podocyte-specific autophagy-deficient mice.47 Autophagy also contributes to the degradation of AGEs,48,49 and thus suppresses inflammation in kidney.48
Though these phenotypes are intriguing, the overall activity of autophagy, either activated or inactivated, in diabetic nephropathy is still controversial.9 Hyperinsulinemia due to hyperglycemia may suppress autophagic activity through MTOR activation.9 This suppressive function of insulin seems to have a tissue-specificity; i.e., insulin suppresses autophagy in muscles whereas amino acids suppress autophagy in liver.50 Conversely, autophagy is also suggested to be induced under hyperglycemia due to the production of ROS or direct cytotoxicity of hyperglycemia.51 Whether over-nutrition status and hyperinsulinemia could suppress autophagy in kidney needs to be determined.
What further modifies the autophagic activity under diabetic condition is the presence of diabetic byproducts and resulting complications. AGEs suppress autophagic/lysosomal degradation activity.48,49 Similar phenomena were reported under hyperlipidemia (high-fat diet challenge),47,52 a common metabolic complication seen in diabetes. Mechanistically, AGEs are suggested to impair lysosomal membrane permeability and function.49 Autophagy, in turn, upregulates lysosomal biogenesis and function via nuclear translocation of TFEB (transcription factor EB),48 a key regulator of lysosomal biogenesis.53 Autophagy-deficient kidney tubular cells show neither TFEB nuclear translocation nor lysosomal biogenesis upon AGEs challenge,48 suggesting a critical role of the autophagy-TFEB axis against AGEs. These byproducts and complications of diabetes may be associated with autophagic regulation of inflammation of the kidney in diabetes.48
In summary, autophagy basically plays a protective role in diabetic nephropathy, but whether autophagy is active or inactive under diabetic conditions is unclear. Inflammation is the key factor in the pathogenesis of diabetic nephropathy, and autophagy may play a protective role against kidney inflammation under diabetic conditions.
Autophagy, systemic inflammation and kidney injury
Autophagy also has a potential to suppress kidney damage from chronic inflammation. Chronic inflammation, a common condition in CKD patients, affects the kidney. Low-grade inflammatory cytokines, such as IL1B, mediate kidney injury though such mechanism as invasion of inflammatory cells, hemodynamic changes, and endothelial dysfunction.54 Secondary amyloidosis, defined by extracellular deposition of SAA (serum amyloid A), an acute-phase protein produced upon chronic inflammation, affects most tissues including the kidney. Additionally, chronic inflammation indirectly affects the kidney though inflammation-related diseases, such as diabetes and cardiovascular diseases.25,55
Recently, the molecular mechanisms of how autophagy suppresses NLRP3 inflammasome activation started to be revealed. One such mechanism is demonstrated in the study of MEFV/TRIM20/Pyrin,56 the risk locus for familial Mediterranean fever (FMF).57 FMF is an autosomal recessive disease characterized by episodes of fever with peritonitis, pleural inflammation, arthritis, and systemic amyloidosis, leading to end-stage kidney disease.57 These episodes seen in FMF are due to the overactivation of the NLRP3 inflammasome and its consequent production of IL1B and SAA/amyloid A.
MEFV suppresses inflammasome activation through using autophagy in the following way:56,58 MEFV is an autophagic receptor that recognizes NLRP3. Once MEFV recognizes NLRP3, autophagic factors such as ULK1 and BECN1/Beclin 1, as well as mammalian Atg8 homologs, are recruited to the MEFV protein complex and activate autophagy. The resultant activation of autophagy mediates specific degradation of NLRP3. The additional fact that MEFV variants with FMF-associated mutations reduce the capacity to degrade NLRP3 through autophagy may suggest autophagic protection against kidney inflammation and amyloidosis. This MEFV-dependent highly specific type of selective autophagy, termed precision autophagy,58 suppresses NLRP3 inflammasome activity and its consequent amyloid depositions.
Acute systemic inflammation, represented by sepsis, induces a cytokine storm and severely affects multiple tissues including the kidney.54 Autophagy harbors the capacity to suppress sepsis-induced kidney injury through regulation of infection5 and through targeting inflammasome and type I interferon responses.56 Thus, autophagy suppresses key innate immune responses to prevent kidney diseases. Conversely, autophagy can also activate a type I IFN response59 and promote IL1B secretion.39 Both pro- and anti-inflammatory roles of autophagy prevent excessive inflammatory responses,5 and proper modifications of autophagy can suppress kidney diseases due to systemic inflammation.
Autoimmune diseases, such as systemic lupus erythematosus, affect the kidney (called lupus nephritis). Glomerular deposition of immune-complexes is characteristically seen in lupus nephritis. A recent study demonstrated that deficiency of LC3-associated phagocytosis (LAP, a process to engage autophagic machinery in phagocytosis) causes systemic lupus erythematosus-like phenomena, characterized by the presence of autoantibody deposited in glomeruli, and systemic inflammation in the mouse kidney: Lyz2/LysM-driven or systemic ablation of LAP-associated genes (Atg5, Atg7, Becn1, Cybb/Nox2, and Rubcn/Rubicon) invoke anti-nuclear antibody production in the blood and deposits of immune complexes in the kidney.60 Thus, autophagy or LAP-associated proteins may regulate the autoimmune response.
In summary, autophagy affects systemic inflammatory responses via regulation of the production of cytokines and autoantibodies, or via direct restriction of pathogens. Since systemic inflammation has direct toxic effects on the kidney, systemic regulations of autophagy have the potentials to benefit the kidney.
Autophagy, kidney aging, and inflammation
Chronic inflammation may also promote kidney senescence.61 Invasive macrophages and lymphocytes, seen in aging kidneys, affect the kidney by reducing its mass mainly through tubular fibrosis and atrophy. Mechanistically, such factors as induction of fibrosis, increased production of ROS, and increased apoptosis, are suggested to connect chronic inflammation to kidney senescence and kidney injuries. Indeed, chronic kidney disease is highly prevalent in the elderly population.61
Autophagy protects the kidney from aging-related inflammatory stresses. Aging stress induces degeneration of the kidney under autophagy-deficient conditions.11,12,14,20 In kidney tubules, autophagy-deficient mice show age-dependent kidney injuries; manifestations of kidney injuries and fibrosis appear starting from a few months after birth,12,14,20 and these manifestations become severe at older age (24 mo).20 Quality control of mitochondria by autophagy seems to play a central role in the protection of kidney from aging stress as mentioned above.12,20 In contrast, podocyte-specific autophagy-deficient mice only show mild forms of glomerulosclerosis at the age of 24 mo,11 suggesting a more critical role of autophagy in kidney tubules.
Future perspectives
This review implicates autophagy and inflammation as the common pathophysiological axes in major kidney diseases. The important roles of autophagy in kidney inflammation are just starting to be recognized. We now know that autophagy is active in kidney, and inflammation plays the key role in disease formation of the kidney. The consistently clear roles of autophagy against rodent kidney diseases are now convincing kidney researchers that the manipulation of autophagy would be a therapeutic option to combat incurable kidney disease. The significance of studying kidney diseases is not in question; currently, most kidney diseases are incurable, and the number of patients with kidney diseases is increasing and occupying a significant portion of medical expenses in developed countries.
Autophagy would surely be a key therapeutic option in inflammatory diseases such as kidney diseases. To regulate the kidney inflammation by autophagy, one major open question here is how we shall modulate autophagy. Modulation of autophagy, both activation and suppression, is enthusiastically pursued these days; these approaches would also benefit kidney disease patients through the modulation of inflammatory responses by autophagy. However, general modulation of autophagy might result in unwanted side effects on the kidney. For example, modulation of autophagy in cancer therapy can trigger worsening of kidney function.16 Rough and bulk modulation of autophagy therefore may provoke unwanted side effects in the kidney.
Another key question is what the targets of autophagy are, and how autophagy recognizes these specific targets, in kidney diseases. Autophagy recognizes damaged organelles and protein aggregates in kidney disease; however, the precise trigger of autophagy is not known in kidney diseases. Which proteins are degraded in kidney diseases by autophagy are scarcely known. Mitochondria are obviously the targets of autophagy in kidney,22 but how autophagy selectively recognizes damaged or depolarized mitochondria, but not the healthy ones, awaits determination. Receptors of mitophagy for kidney diseases have not been identified thus far. Specific targeting of autophagic targets would, of course, potentiate precision medicine. In other words, precise degradation of autophagic targets by precision autophagy58 would benefit patients.
As autophagy has dual roles (both activating and suppressing) in the inflammatory response, the precise mechanistic study of these regulatory processes would also benefit kidney patients. One potential regulatory mechanism may be via metabolism, since autophagy is a metabolic process.6 Modulations of autophagy may potentiate immunometabolic regulations in kidney diseases.
In summary, autophagic regulation of inflammation plays the key protective role in most kidney diseases. The precise role of autophagy in kidney diseases through inflammation awaits clarification for future therapy. Due to the complicated nature of kidney diseases, the number of researchers from outside the kidney field is not big nowadays. But, for further development of this field, we are waiting for researchers outside kidney fields to join us.
Abbreviations
- AGEs
advanced glycation end products
- CKD
chronic kidney disease
- DAMPs
damage-associated molecular patterns
- ESKD
end-stage kidney disease
- FMF
familial Mediterranean fever
- GFR
glomerular filtration ratio
- IL1B
inteleukin 1 β
- LAP
LC3-asssociated phagocytosis
- ROS
reactive oxygen species
- SAA
serum amyloid A
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank past and present laboratory members and collaborators for discussion.
Funding
TK was supported by Nakayama Foundation for Human Science and Akaeda Medical Research Foundation.
References
- [1].Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol 2014; 25:1387-400; PMID: 24762401; https://doi.org/ 10.1681/ASN.2014010117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID: 21801009; https://doi.org/ 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- [3].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID: 22078875; https://doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- [4].Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity 2013; 39:211-27; PMID: 23973220; https://doi.org/ 10.1016/j.immuni.2013.07.017 [DOI] [PubMed] [Google Scholar]
- [5].Deretic V, Kimura T, Timmins G, Moseley P, Chauhan S, Mandell M. Immunologic manifestations of autophagy. J Clin Invest 2015; 125:75-84; PMID: 25654553; https://doi.org/ 10.1172/JCI73945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID: 21127245; https://doi.org/330/6009/1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Randow F, Youle RJ. Self and nonself: How autophagy targets Mitochondria and Bacteria. Cell Host Microbe 2014; 15:403-11; PMID: 24721569; https://doi.org/ 10.1016/j.chom.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al.. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J 2013; 32:2336; PMID: 23921551; https://doi.org/ 10.1038/emboj.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lenoir O, Tharaux PL, Huber TB. Autophagy in kidney disease and aging: Lessons from rodent models. Kidney Int 2016; 90:950; PMID: 27325184; https://doi.org/ 10.1016/j.kint.2016.04.014 [DOI] [PubMed] [Google Scholar]
- [10].Takabatake Y, Kimura T, Takahashi A, Isaka Y. Autophagy and the kidney: Health and disease. Nephrol Dial Transplant 2014; 29:1639-47; PMID: 24520117; https://doi.org/ 10.1093/ndt/gft535 [DOI] [PubMed] [Google Scholar]
- [11].Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, et al.. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 2010; 120:1084-96; PMID: 20200449; https://doi.org/ 10.1172/JCI39492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al.. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902-13; PMID: 21493778; https://doi.org/ 10.1681/ASN.2010070705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, et al.. Autophagy guards against Cisplatin-induced acute kidney injury. Am J Pathol 2012; 180:517-25; PMID: 22265049; https://doi.org/ 10.1016/j.ajpath.2011.11.001 [DOI] [PubMed] [Google Scholar]
- [14].Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, Huber TB. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012; 826:8; PMID: 22617445; https://doi.org/ 10.4161/auto.19419 [DOI] [PubMed] [Google Scholar]
- [15].Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 2012; 82:1271; PMID: 22854643; https://doi.org/ 10.1038/ki.2012.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res 2013; 73:3-7; PMID: 23288916; https://doi.org/ 10.1158/0008-5472.CAN-12-2464 [DOI] [PubMed] [Google Scholar]
- [17].Kimura T, Takahashi A, Takabatake Y, Namba T, Yamamoto T, Kaimori JY, Matsui I, Kitamura H, Niimura F, Matsusaka T, et al.. Autophagy protects kidney proximal tubule epithelial cells from mitochondrial metabolic stress. Autophagy 2013; 9:1876-86; PMID: 24128672; https://doi.org/ 10.4161/auto.25418 [DOI] [PubMed] [Google Scholar]
- [18].Namba T, Takabatake Y, Kimura T, Takahashi A, Yamamoto T, Matsuda J, Kitamura H, Niimura F, Matsusaka T, Iwatani H, et al.. Autophagic clearance of Mitochondria in the kidney copes with Metabolic acidosis. J Am Soc Nephrol 2014; 25:2254; PMID: 24700866; https://doi.org/ 10.1681/ASN.2013090986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lenoir O, Jasiek M, Henique C, Guyonnet L, Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A, et al.. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015; 11:1130-45; PMID: 26039325; https://doi.org/ 10.1080/15548627.2015.1049799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yamamoto T, Takabatake Y, Kimura T, Takahashi A, Namba T, Matsuda J, Minami S, Kaimori JY, Matsui I, Kitamura H, et al.. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016; 12: 801; PMID: 26986194; https://doi.org/ 10.1080/15548627.2016.1159376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333:1109-12; PMID: 21868666; https://doi.org/333/6046/1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG. Mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 2016; 214:333-45; PMID: 27458135; https://doi.org/ 10.1083/jcb.201603039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Levey AS, Atkins R, Coresh J, Cohen EP, Collins AJ, Eckardt KU, Nahas ME, Jaber BL, Jadoul M, Levin A, et al.. Chronic kidney disease as a global public health problem: approaches and initiatives - a position statement from Kidney disease improving global outcomes. Kidney Int 2007; 72:247-59; PMID: 17568785; https://doi.org/ 10.1038/sj.ki.5002343 [DOI] [PubMed] [Google Scholar]
- [24].Mackensen-Haen S, Bader R, Grund KE, Bohle A. Correlations between renal cortical interstitial fibrosis, atrophy of the proximal tubules and impairment of the glomerular filtration rate. Clin Nephrol 1981; 15:167-71; PMID: 7237863 [PubMed] [Google Scholar]
- [25].Kalantar-Zadeh K, Mehrotra R, Fouque D, Kopple JD. Metabolic acidosis and malnutrition-inflammation complex syndrome in chronic renal failure. Semin Dial 2004; 17:455-65; PMID: 15660576; https://doi.org/ 10.1111/j.0894-0959.2004.17606.x [DOI] [PubMed] [Google Scholar]
- [26].Torelli G, Milla E, Faelli A, Costantini S. Energy requirement for sodium reabsorption in the in vivo rabbit kidney. Am J Physiol 1966; 211:576-80; PMID: 5927884 [DOI] [PubMed] [Google Scholar]
- [27].Bomsel M, Prydz K, Parton RG, Gruenberg J, Simons K. Endocytosis in filter-grown Madin-Darby canine kidney cells. J Cell Biol 1989; 109:3243-58; PMID: 2689455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ding Y, Kim S, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 2014; 25:2835-46; PMID: 24854279; https://doi.org/ 10.1681/ASN.2013101068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221-5; PMID: 21124315; https://doi.org/ 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- [30].Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005; 120:483-95; PMID: 15734681; https://doi.org/S0092-8674(05)00109-1 [DOI] [PubMed] [Google Scholar]
- [31].O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016; 16:553-65; PMID: 27396447; https://doi.org/ 10.1038/nri.2016.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: A sensor for metabolic danger?. Science 2010; 327:296-300; PMID: 20075245; https://doi.org/ 10.1126/science.1184003 [DOI] [PubMed] [Google Scholar]
- [33].Waisman J, Mwasi LM, Bluestone R, Klinenberg JR. Acute hyperuricemic nephropathy in rats. An electron microscopic study. Am J Pathol 1975; 81:367-78; PMID: 1190294 [PMC free article] [PubMed] [Google Scholar]
- [34].Koka RM, Huang E, Lieske JC. Adhesion of uric acid crystals to the surface of renal epithelial cells. Am J Physiol Renal Physiol 2000; 278:F989-98; PMID: 10836987 [DOI] [PubMed] [Google Scholar]
- [35].Isaka Y, Takabatake Y, Takahashi A, Saitoh T, Yoshimori T. Hyperuricemia-induced inflammasome and kidney diseases. Nephrol Dial Transplant 2015; 31:890; PMID: 25829326; https://doi.org/ 10.1093/ndt/gfv024 [DOI] [PubMed] [Google Scholar]
- [36].Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al.. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222-30; PMID: 21151103; https://doi.org/ 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID: 18849965; https://doi.org/ 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- [38].Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J 2011; 30:4701-11; PMID: 22068051; https://doi.org/emboj2011398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, Farzam F, et al.. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. Embo J 2017; 36:42-60; PMID: 27932448; https://doi.org/ 10.15252/embj.201695081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID: 14699058; https://doi.org/ 10.1091/mbc.E03-09-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID: 26799652; https://doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen J, Chen MX, Fogo AB, Harris RC, Chen JK. mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. J Am Soc Nephrol 2013; 24:198-207; PMID: 23291473; https://doi.org/ 10.1681/ASN.2012010101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bechtel W, Helmstadter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, et al.. Vps34 deficiency reveals the importance of Endocytosis for Podocyte homeostasis. J Am Soc Nephrol 2013; 24:727; PMID: 23492732; https://doi.org/ 10.1681/ASN.2012070700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, et al.. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A 2012; 109:2003-8; PMID: 22308354; https://doi.org/ 10.1073/pnas.1112848109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Najafian B, Alpers CE, Fogo AB. Pathology of human diabetic nephropathy. Contrib Nephrol 2011; 170:36-47; PMID: 21659756; https://doi.org/ 10.1159/000324942 [DOI] [PubMed] [Google Scholar]
- [46].Tan AL, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol 2007; 27:130-43; PMID: 17418682; https://doi.org/ 10.1016/j.semnephrol.2007.01.006 [DOI] [PubMed] [Google Scholar]
- [47].Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K, et al.. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 2016; 65:755-67; PMID: 26384385; https://doi.org/ 10.2337/db15-0473 [DOI] [PubMed] [Google Scholar]
- [48].Takahashi A, Takabatake Y, Kimura T, Maejima I, Namba T, Yamamoto T, Matsuda J, Minami S, Kaimori JY, Matsui I, et al.. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes 2017; [Epub ahead of print] PMID: 28246295; https://doi.org/ 10.2337/db16-0397 [DOI] [PubMed] [Google Scholar]
- [49].Liu WJ, Shen TT, Chen RH, Wu HL, Wang YJ, Deng JK, Chen QH, Pan Q, Huang Fu CM, Tao JL, et al.. Autophagy-Lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J Biol Chem 2015; 290:20499-510; PMID: 26100632; https://doi.org/ 10.1074/jbc.M115.666354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Naito T, Kuma A, Mizushima N. Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J Biol Chem 2013; 288:21074-81; PMID: 23744068; https://doi.org/ 10.1074/jbc.M113.456228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ma T, Zhu J, Chen X, Zha D, Singhal PC, Ding G. High glucose induces autophagy in podocytes. Exp Cell Res 2013; 319:779-89; PMID: 23384600; https://doi.org/ 10.1016/j.yexcr.2013.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yamamoto T, Takabatake Y, Takahashi A, Kimura T, Namba T, Matsuda J, Minami S, Kaimori JY, Matsui I, Matsusaka T, et al.. High-Fat Diet-Induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J Am Soc Nephrol 2016; [Epub ahead of print] PMID: 27932476; https://doi.org/ 10.1681/ASN.2016070731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14:283-96; https://doi.org/ 10.1038/nrm3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol 2011; 22:999-1006; PMID: 21566052; https://doi.org/ 10.1681/ASN.2010050484 [DOI] [PubMed] [Google Scholar]
- [55].Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296-305; PMID: 15385656; https://doi.org/ 10.1056/NEJMoa041031 [DOI] [PubMed] [Google Scholar]
- [56].Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, Deretic V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol 2015; 210:973-89; PMID: 26347139; https://doi.org/ 10.1083/jcb.201503023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol 2009; 27:621-68; PMID: 19302049; https://doi.org/ 10.1146/annurev.immunol.25.022106.141627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kimura T, Mandell M, Deretic V. Precision autophagy directed by receptor regulators - emerging examples within the TRIM family. J Cell Sci 2016; 129:88; https://doi.org/ 10.1242/jcs.163758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007; 315:1398-401; PMID: 17272685; https://doi.org/ 10.1126/science.1136880 [DOI] [PubMed] [Google Scholar]
- [60].Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, et al.. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016; 533:115; PMID: 27096368; https://doi.org/ 10.1038/nature17950 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [61].Bolignano D, Mattace-Raso F, Sijbrands EJ, Zoccali C. The aging kidney revisited: A systematic review. Ageing Res Rev 2014; 14:65-80; PMID: 24548926; https://doi.org/ 10.1016/j.arr.2014.02.003 [DOI] [PubMed] [Google Scholar]