

ABSTRACT
Glutaminolysis plays a critical role in nutrient sufficiency and cell signaling activation in mammalian cells. Unexpectedly, our recent investigations revealed that the unbalanced activation of glutaminolysis during nutritional restriction causes a particular form of apoptotic cell death, that we termed “glutamoptosis.“ We found that the inhibition of autophagy is a key step to allow glutamoptosis-mediated cell death. Thus, autophagy controls glutamoptosis during nutritional imbalance.
KEYWORDS: α-ketoglutarate, apoptosis, autophagy, glutaminolysis, MTORC1, SQSTM1/p62
Glutamine is the most abundant amino acid in the blood of humans, and a major source of biomass for cells. Glutamine catabolism or glutaminolysis produces α-ketoglutarate (αKG). As the glutaminolitic αKG feeds the tricarboxylic acid (TCA) cycle (a process termed anaplerosis), the metabolism of glutamine is crucial to maintain the bioenergetic status of the cell. The relevance of the anaplerotic role of glutamine has been extensively studied in the context of nutrient sufficiency. However, we and others had previously highlighted the importance of glutaminolysis not only for energetic purposes, but also as a mechanism to inhibit macroautophagy/autophagy. The inhibition of autophagy by glutaminolysis is part of an anabolic program controlled by the serine/threonine protein kinase mechanistic target of rapamycin complex 1 (MTORC1), a master regulator of cell growth. Our recent work has now confirmed the sufficiency of glutaminolysis to sustain the inhibition of autophagy in an MTORC1-dependent manner in the absence of any other amino acid during prolonged times. However, these investigations have also revealed an unexpected connection between the unbalanced induction of glutaminolysis in the absence of other amino acids and cell death. We are naming this type of metabolic cell death as “glutamoptosis,” as it follows a particular apoptotic mechanism.
During glutamoptosis, the production of αKG is induced by the presence of glutamine (as the carbon source) and leucine (as an allosteric activator of glutaminolysis) in the absence of other amino acids. The production of αKG in this condition of nutritional imbalance induces the activation of MTORC1. Surprisingly, and despite the growth promoter functions of both MTORC1 and glutaminolysis, the prolonged activation of MTORC1 during these unbalanced conditions reduces cell viability drastically. The confirmation that this cell-death mechanism is mediated by the anomalous activation of MTORC1 was obtained by the observation that rapamycin treatment, while arresting cell growth, was also able to abrogate cell death.
Further observations allowed us to assign this MTORC1-dependent cell-death phenotype to the capacity of MTORC1 to inhibit autophagy. Indeed, the activation of MTORC1, as observed in other contexts, prevents autophagy initiation. This MTORC1-mediated inhibition of autophagy leads to the accumulation of the autophagic cargo protein SQSTM1/p62 (sequestosome 1), a protein that is degraded during autophagy. SQSTM1/p62 levels normally correlate with nutrient availability: in nutrient-rich conditions the levels of SQSTM1/p62 are high (as autophagy is inhibited), whereas nutrient-deprived conditions induce a decrease in SQSTM1/p62 levels (resulting from the activation of autophagy). However, during glutamoptosis, SQSTM1/p62 levels are unusually high, despite the absence of most amino acids (with the exception of glutamine and leucine). Our observations indicate that this upregulation of SQSTM1/p62 during nutrient restriction is detected by the cell as an anomalous situation that leads to the activation of an atypical cell death program. These results are in line with previous studies in which SQSTM1/p62 was shown to play a positive role in liver injury. During glutamoptosis, SQSTM1/p62 interacts with and activates CASP8 (caspase 8). This interaction requires the upregulation of the pro-apoptotic protein BAX. The exact mechanism by which BAX promotes SQSTM1/p62-CASP8 interaction is not clear. Surprisingly, the release of CYCS (cytochrome c, somatic) out of the mitochondria (typically observed upon BAX activation) does not operate during glutamoptosis. This unexpected result reveals a new yet uncharacterized mechanism of action of BAX during glutamoptosis (Fig. 1).
Figure 1.
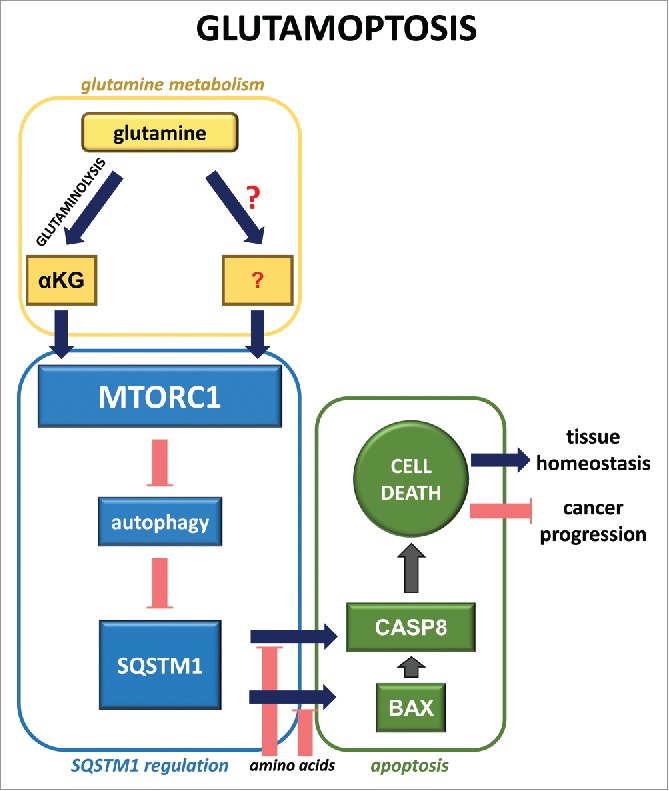
Molecular mechanism of glutamoptosis. During glutamoptosis, the activation of MTORC1 by αKG production through glutaminolysis (and perhaps also by other inputs from glutamine) induces the autophagy-dependent accumulation of SQSTM1/p62. In the absence of other amino acids, SQSTM1/p62 induces CASP8 cleavage in a BAX-dependent manner, leading to cell death.
A number of questions arise upon the discovery of glutamoptosis. First, why is SQSTM1/p62 used by the cell as a sensor to recognize a nutritional imbalance situation? SQSTM1/p62 functions as a receptor protein that interacts with several signaling processes in the cell, including those involving MTORC1, autophagy, NFKB/NF-κB, MYC/c-MYC, and caspases. This strategic position of SQSTM1/p62 might support its role as an integrator of growing signals to detect imbalances not only at the nutritional levels, but also in other stress-inducing situations. Indeed, the interaction between SQSTM1/p62 and CASP8 operates under endoplasmic reticulum stress or during proteasomal inhibition. It is also unclear as to the physiological benefit of undergoing cellular suicide upon the unbalanced activation of glutaminolysis and MTORC1. A potential scenario could be that glutamoptosis is a mechanism to detect microenvironments with nutritional imbalance as a result of a pathological situation. For instance, abnormally high levels of glutamine in liver microenvironments might result from an excess of glutamine secretion from hepatocellular carcinoma cells with an increased activity of GLUL/glutamine synthetase. Additionally, unbalanced high levels of αKG might result as a consequence of metabolic alterations of the TCA cycle, such as mutations in TCA cycle-involved enzymes. Through glutamoptosis, these situations will be recognized as metabolic abnormalities to be eliminated. Finally, one important question to be addressed is the potential involvement of glutamoptosis as a therapeutic approach. Cell death escape is a main hallmark of cancer, and inducing cell death in cancer cells is a major line of translational research to find new therapies. Thus, whether glutamoptosis induction could constitute a new strategy to specifically kill cancer cells in microenvironments particularly rich in glutamine is a relevant question that remains to be answered.
While the physiological relevance of glutamoptosis in other eukaryotic systems is still unclear, our recent work opens the door to applying this newly described cell death-mechanism to remote fields such as nutritional science, aquaculture, or translational research in biomedicine. Certainly, our work extends the recent observations closely linking nutritional inputs with cellular pathologies such as cancer. The involvement of the MTORC1-autophagy-SQSTM1/p62 axis in glutamoptosis is just another confirmation of the central role played by autophagy in the fine control of cellular homeostasis and its potential role in therapy design.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by funds from the following institutions: Institut National de la Santé et de la Recherche Médicale - INSERM, Fondation pour la Recherche Médicale, Conseil Régional d'Aquitaine, Fondation ARC pour la Recherche sur le Cancer, SIRIC-BRIO, and Institut Européen de Chimie et Biologie.