

ABSTRACT
ATF4 (activating transcription factor 4) is an important transcription factor that has many biological functions, while its role in hypothalamic POMC (pro-opiomelanocortin-α) neurons in the regulation of energy homeostasis has not been explored. We recently discovered that mice with an Atf4 deletion specific to POMC neurons (PAKO mice) are lean and have higher energy expenditure. Furthermore, these mice are resistant to high-fat diet (HFD)-induced obesity and obesity-related metabolic disorders. Mechanistically, we found the expression of ATG5 (autophagy-related 5) is upregulated in POMC neurons of PAKO mice, and ATF4 regulates ATG5 expression by binding directly to its promoter. Mice with Atf4 and Atg5 double knockout in POMC neurons have reduced energy expenditure and gain more fat mass compared with PAKO mice under a HFD. Finally, the effect of Atf4 knockout in POMC neurons is possibly mediated by enhanced ATG5-dependent macroautophagy/autophagy and α-melanocyte-stimulating hormone (α-MSH) production in the hypothalamus. Together, this work not only identifies a beneficial role for ATF4 in hypothalamic POMC neurons in the regulation of obesity, but also provides a new potential therapeutic target for obesity and obesity-related metabolic diseases.
KEYWORDS: ATF4, ATG5, autophagy, POMC neurons, obesity
In recent years, obesity has become an increasing health risk worldwide. Obesity is closely associated with nonalcoholic fatty liver diseases, type 2 diabetes, vascular diseases and cancer. Therefore, searching for effective tools or drug targets against obesity is of great importance.
Obesity is caused by the imbalance between food intake and energy expenditure. The hypothalamus in the central nervous system plays an important role in the regulation of energy homeostasis. There are 2 major populations of neurons in the arcuate nucleus of the hypothalamus that are crucial for the regulation of energy homeostasis, one is AGRP (agouti related neuropeptide) neurons, and the other is POMC neurons. Autophagy is a cellular process that functions to degrade damaged cytoplasmic components. Recent studies have demonstrated an important role for autophagy in hypothalamic POMC neurons in the regulation of obesity. ATF4 is an important transcription factor that has previously been shown to regulate autophagy and the expression of some autophagy-related (ATG) regulators; however, whether ATF4 in hypothalamic POMC neurons regulates obesity via autophagy remains largely unknown.
In a recent study, we reported that mice with an Atf4 knockout specific to POMC neurons (PAKO mice) have lower body weight, fat mass component and abdominal fat mass, whereas the lean mass component of PAKO mice is not changed compared with littermate control mice. It is well-known that energy homeostasis is maintained by a balance between food intake and energy expenditure; we found the food intake of PAKO mice is not changed, but the energy expenditure and oxygen consumption are higher in PAKO mice. Under normal conditions, the energy expenditure is mainly caused by physical activity and basal metabolism to maintain body temperature. The physical activity is not changed, while the body temperature is significantly higher in PAKO mice. The increased body temperature in PAKO mice is likely caused by enhanced thermogenesis, as the expression of UCP1 (uncoupling protein 1 [mitochondrial, proton carrier]) is increased in brown adipose tissue of PAKO mice, and serum norepinephrine (NE) that activates the sympathetic nervous system (SNS) is also increased in PAKO mice. Consistent with the increased serum NE levels, lipolysis is also increased in white adipose tissue of PAKO mice. Furthermore, we found that PAKO mice are resistant to HFD-induced body weight gain and fat mass accumulation; HFD-induced INS (insulin) resistance and LEP (leptin) resistance are also ameliorated in PAKO mice.
Mechanistically, we found that the autophagic flux is enhanced by ATF4 inhibition and inhibited by ATF4 overexpression in vivo and in vitro, as demonstrated by the corresponding changes in protein levels of the autophagy markers SQSTM1 and LC3-II, and by an autophagic flux analysis. Moreover, the expression of ATG5 is increased or decreased by ATF4 knockdown or overexpression, respectively, in primary cultured hypothalamic neurons, and ATF4 inhibits the transcription of Atg5 by binding to its promoter. Importantly, mice with Atf4 and Atg5 double knockout in POMC neurons gain more body weight and fat mass, and have lower energy expenditure and body temperature compared with PAKO mice under a HFD. Consistently, the ameliorated INS and LEP sensitivity in PAKO mice are also largely reversed by Atg5 knockout. Finally, the increased autophagy (as shown by the decreased SQSTM1 staining) in POMC neurons of PAKO mice is also largely reversed by Atg5 knockout.
Autophagy mediates POMC precursor cleavage to produce α-MSH, which is a key regulator of energy expenditure via activation of SNS. We found that the α-MSH levels and the expression of POMC precursor cleavage enzymes PCSK2/prohormone convertase 2 and CPE (carboxypeptidase E) are increased in the hypothalamus of PAKO mice, and these increases are also largely reversed by Atg5 knockout.
Taken together, ATF4 in hypothalamic POMC neurons regulates obesity via ATG5-dependent autophagy (Fig. 1). Mice with Atf4 deletion in POMC neurons have increased ATG5 expression and autophagy; the increased autophagy promotes POMC precursor cleavage to produce α-MSH, and α-MSH binds to the melanocortin receptors in the paraventricular nucleus (PVN) of the hypothalamus to activate SNS. Possibly through the activation of SNS, the thermogenesis in brown adipose tissue and lipolysis in white adipose tissue are increased, which leads to fat loss. These findings suggest that ATF4 in the hypothalamic POMC neurons may be a new potential therapeutic target for obesity and obesity-related metabolic diseases.
Figure 1.
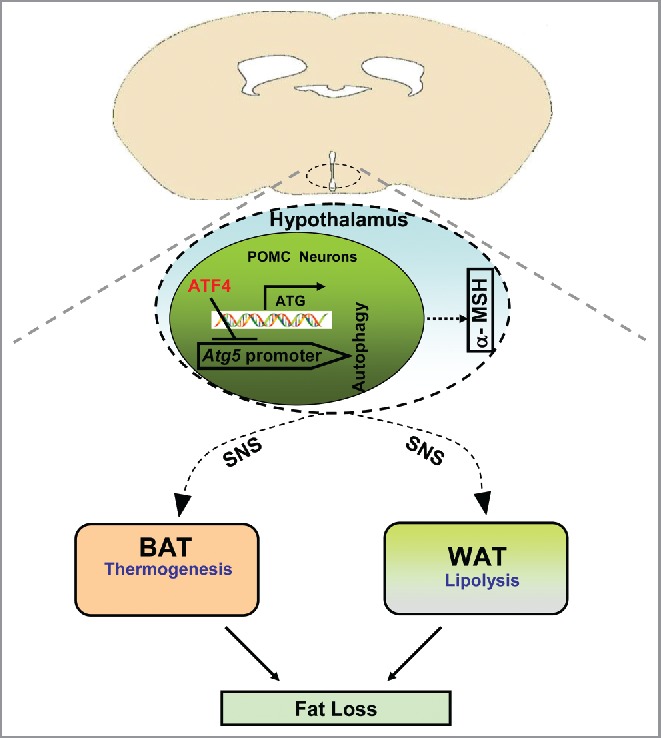
A schematic model of ATF4 in hypothalamic POMC neurons in the regulation of obesity. See the text for details.
Disclosure of potential conflicts of interest
No potential conflicts of interest relevant to this work were reported.
Funding
This work was supported by grants from the NNSF (81325005, 81471076, 81570777, 81390350, 81130076, 31271269, 81400792, 81500622 and 81600623), SHSTC (16JC1404900 and 17XD1404200), International Cooperation Program (Singapore 2014DFG32470) and CAS/SAFEA international partnership program. Feifan Guo was also supported by the One Hundred Talents Program of CAS.